
Abstract
Numerous agents of infections in humans and other mammals are found among fungi that are able to survive extreme environmental conditions and to quickly adapt to novel habitats. Nevertheless, the relationship between opportunistic potential and polyextremotolerance was not yet studied systematically in fungi. Here, the link between polyextremotolerance and opportunistic pathogenicity is shown in a kingdom-wide phylogenetic analysis as a statistically significant co-occurrence of extremotolerance (e.g. osmotolerance and psychrotolerance) and opportunism at the level of fungal orders. In addition to extremotolerance, fungal opportunists share another characteristic—an apparent lack of specialised virulence traits. This is illustrated by a comparative genomic analysis of 20 dothideomycetous and eurotiomycetous black fungi. While the genomes of specialised fungal plant pathogens were significantly enriched in known virulence-associated genes that encode secreted proteases, carbohydrate active enzyme families, polyketide synthases, and non-ribosomal peptide synthetases, no such signatures were observed in human opportunists. Together the presented results have several implications. If infection of human hosts is a side effect of fungal stress tolerance and adaptability, the human body is most likely neither the preferred habitat of such species, nor important for their evolutionary success. This defines opportunism as opposed to pathogenicity, where infection is advantageous for the species’ fitness. Since opportunists are generally incapable of the host-to-host transmission, any host-specific adaptations are likely to be lost with the resolution of the infection, explaining the observed lack of specialised virulence traits. In this scenario opportunistic infections should be seen as an evolutionary dead end and unlikely to lead to true pathogenicity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Fungi form an integral part of biodiversity of many extreme environments. Here, the maintenance of costly stress tolerance and adaptability mechanisms pays off in decreased competition with other microbes. Two modes of adaptation can be recognised in fungi. On the one hand specialised extremophilic and extremotolerant species (“monoextremophilic” and “monoextremotolerant” species) evolved to efficiently cope with a specific stress factor and are limited in their capacity for habitat shifts. Such species can tolerate some of the most extreme conditions on the planet, with one important exception—very few fungi are truly thermophilic. Polyextremotolerant species on the other hand tolerate many different types of stress and are often extremely adaptable (Gostinčar et al. 2010). This makes them good candidates for colonising novel habitats that are considered as suboptimal for microbial growth (Gostinčar et al. 2011). Hot and wet indoor habitats, for example, uncovered a surprising diversity of polyextremotolerant, oligotrophic fungi (Zalar et al. 2011; Hamada and Abe 2010; Lian and de Hoog 2010). The human-shaped environment is additionally characterised by factors such as hygienic measures, chemical pollution, and limited water availability, which all provide novel extreme conditions for specialized microbes and have been suggested to increase the risks of harmful fungus-human interactions (Gostinčar et al. 2011; Robert and Casadevall 2009; Casadevall et al. 2011; Gostinčar et al. 2015).
For fungi to successfully infect a human body, they have to overcome several obstacles, such as high temperature (Robert and Casadevall 2009), low water activity and low pH in case of skin penetration (Elias 2007), oxidative bursts of human phagocytes and severe iron limitation (Hamad 2008; Kumamoto 2008). In the case of the few true fungal pathogens (also named primary pathogens), which can infect healthy individuals, specialised mechanisms to counter the above described immune defences possibly evolved as a response to selection pressures during an infection. The infection potential of these species enhances their fitness and is therefore considered as an essential part of their natural lifestyle. However, such adaptive evolution seems improbable in the case of opportunists, a much longer list of species limited to sporadic infections of often immunocompromised hosts. Compared to the large populations of opportunistic pathogens outside the host the infection events are extremely rare and it is unlikely that they would noticeably contribute to the biological success of the species. Therefore, traits promoting virulence in opportunists have likely evolved for purposes other than survival within the host (Song et al. 2017). Published literature suggests various selection pressures driving the emergence of such pre-existing adaptations (exaptations), among them adaptations to stress encountered outside the mammalian host. These traits would primarily promote the survival of the fungus in the environment, but (as an unintentional side-effect) also allow its establishment in the host (van Burik and Magee 2001; Casadevall 2007). For example, in the prominent pathogen Cryptococcus neoformans the mechanisms that enable its survival during infection are thought to have evolved in response to stress in its primary ecological niche, bird manure (Brown et al. 2007). Several examples of polyextremotolerant fungi that are also opportunistic pathogens are found among dothideomycetous and eurotiomycetous black yeasts, a group of melanised ascomycetous species, which are investigated here in more detail in a comparative genomic analysis.
If opportunistic infections are indeed accidental colonisations enabled by adaptability and stress tolerance, opportunistic potential should be phylogenetically linked to polyextremotolerance. Opportunists should contain few if any traits important in virulence compared to their non-opportunistic relatives. In this study we investigate the distribution of stress tolerant and opportunistic species across all orders of the fungal kingdom, and use a comparative genome analysis of 11 dothideomycetous and 9 eurotiomycetous fungi in search of genomic signatures of extremotolerance and opportunism.
Materials and methods
Phylogenetics
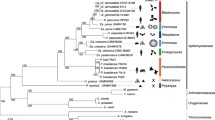
Large subunit ribosomal RNA (LSU) sequences were obtained from GenBank following the Catalogue of Life, the fungal taxonomic tree, the index of fungi and Dictionary of Fungi. The sequences were initially aligned using Clustal X 1.8 (Thompson et al. 1997). The alignment was subsequently checked visually and corrected for obviously misaligned positions to maximize primary sequence homology utilizing BioEdit 7.0.0 (Hall 1999). This final dataset was used for the construction of a phylogenetic tree including 1214 LSU sequences spanning 5 phyla, 40 classes and 141 orders, with Chytridiales as an outgroup. Maximum-likelihood (ML) analyses were performed using RaxML 8.2.9 on the CIPRES Science Gateway portal (Miller et al. 2010). For ML analyses, the rapid bootstrapping (BS) algorithm with the automatic halt option and the default parameters were used. The resulting trees were plotted using FigTree 1.4.2 and MEGA 6.06. Maximum likelihood bootstrap values (BS) equal or greater than 80% are given above each node (Fig. 1).


RAxML phylogenetic tree of fungal 140 orders based on the sequences of large subunits ribosomal RNA (LSU). The order tree was estimated by a maximum likelihood analysis. All branches received at least 80% bootstrap support (numbers above respective branches). Collapsed parts of the tree are shaded in pink and correspond to orders listed in the right panel. The right panel depicts the relative number of species in each order assigned to different categories (grey: rock inhabiting, blue: psychrotolerant, yellow: osmotolerant, red or red asterisk: clinical; green: plant surface). Numbers on the left hand side of the chart indicate the number of medically relevant species and of all species contained within each order (according to the Index Fungorum, catalogue of life). The tree was rooted with Chytridiales as an outgroup
Four approximate ecological categories were plotted on the tree. (1) Surface colonisers, mainly belonging to rock-inhabiting fungi and epiphytes, which are subjected in their natural habitat to varying temperatures, dryness, and solar irradiation. Most are recognizable by compacted phenotypes and survival of external conditions of their habitat during the most hostile season. Lichens, the algal-fungus combination enhancing survival of environmental stress, and lichenicolous fungi, species subjected to similar irradiation and tolerating the antimicrobial toxins produced by lichen thalli, as well as fungi tolerating xenobiotics, were also placed in this category. (2) Osmotolerant/osmophilic fungi, including fungi of the dry fraction of the indoor mycobiome, from desert soil, and other similar habitats. (3) Psychrotolerant/psychrophilic fungi, represented mainly by species from deep ocean waters and polar regions. (4) Clinically relevant fungi covering opportunists and true pathogens; the fungi colonising the healthy mammal body usually without causing infection, such as Malassezia (cutaneous) and Pneumocystis (pulmonary) were also added to this category. These are the species involved in a minimum of ten proven cases of infection and listed in the Atlas of Clinical Fungi (de Hoog et al. 2015). Nearly all are able to grow at 37 °C. The number of strictly thermotolerant/thermophilic fungi never reported from mammalian infection are extremely rare, their number being too low to be included as a separate category for the statistical comparison. A special subcategory of animal-associated commensals, which typically survive without tissue invasion but cause occasional infections, is included in the medical category. (5) Plant pathogens. Plant pathogenicity has been principally defined as causing recurrent disease on specific plant hosts, but this category is difficult to delimit from growth on plant debris and endophytic life styles. We included only those groups where a significant number of species is involved in well-known plant diseases.
Phylogenomics
The proteomes of 11 dothideomycetous and 9 eurotiomycetous fungi, were used in this and other performed comparative genomics analyses. Among the Dothideomycetes, representatives of the orders Dothideales (A. pullulans, A. subglaciale, A. namibiae, A. melanogenum) (Gostinčar et al. 2014), Capnodiales [H. werneckii (Lenassi et al. 2013; Sinha et al. 2017), Baudoinia compniacensis (Ohm et al. 2012), Mycosphaerella graminicola (Goodwin et al. 2011)), Hysteriales (Hysterium pulicare (Ohm et al. 2012)), Pleosporales (Cochliobolus heterostrophus (Ohm et al. 2012)] and Botrysphaeriales [Botryosphaeria dothidea (Grigoriev et al. 2014)] were included. Among the Eurotiomycetes, representatives of the orders Eurotiales [Aspergillus nidulans (Arnaud et al. 2010), A. niger (Pel et al. 2007), A. fumigatus (Cerqueira et al. 2014), Eurotium rubrum (Kis-Papo et al. 2014)], Onygenales [Coccidioides immitis (Black Yeasts Sequencing Project, Broad Institute of Harvard and MIT), Histoplasma capsulatum (Black Yeasts Sequencing Project, Broad Institute of Harvard and MIT)], and Chaetothyriales (E. aquamarina (Black Yeasts Sequencing Project, Broad Institute of Harvard and MIT), E. dermatitidis (Chen et al. 2014), Cladophialophora yegresii [Black Yeasts Sequencing Project, Broad Institute of Harvard and MIT) and Coniosporium apollinis (NCBI BioProject PRJNA245130)], were included. Neurospora crassa (Sordariales, Sordariomycetes) (Galagan et al. 2003) and Sclerotinia sclerotiorum (Helotiales, Leotiomycetes) (Amselem et al. 2011) were used as outgroups. In case of H. werneckii, which contains two nearly identical copies of most genes, one copy from each pair was randomly selected for the phylogenomic analysis. The fungi were assigned to various ecological categories as colour-coded in the tree (Fig. 2).

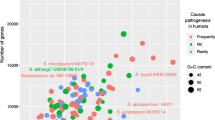
Phylogenetic tree of the selected fungal species. The genomes of the listed species were used here for comparative genomics studies. Fungal lifestyle: blue, extremotolerant; red, animal opportunists; green, plant pathogens. The phylogenetic tree was constructed using PhyML (Guindon et al. 2010), from super alignment of the whole proteomes produced by the Hal pipeline (Robbertse et al. 2011). The branch supports are calculated as approximate Bayes values. Right: Genome sizes and number of predicted proteins
A super alignment of the proteomes was constructed with the Hal pipeline (Robbertse et al. 2011), without allowing for missing data. After removing the poorly aligned positions and positions with gaps using Gblocks 0.91b (Talavera and Castresana 2007), this resulted in a 328 552-amino-acid-long alignment. The maximum number of allowed contiguous non-conserved positions was set to five amino acids, and the minimum length of a block to 15 amino acids. The best protein evolution model was determined using ProtTest 3.2.1 (Darriba et al. 2011). The PhyML 3.1 software (Guindon et al. 2010) was used to generate the species tree with the approximate Bayes branch support values. The LG model of evolution with the ProtTest-estimated a-parameter of the g-distribution of six substitution rate categories (1.155), and the ProtTest-determined proportion of invariable sites (0.215), were used. The tree was calibrated with the r8 s software (Sanderson 2003), by assigning the root of the tree to an arbitrary value of 1.
CAFE analysis
The analysis of the protein family expansion and contraction was performed with the CAFE 3.0 software (Han et al. 2013). Three tables of protein families/clusters were used as input: (i) the numbers of proteins with a specific Pfam domain in each proteome, for each Pfam motif found by the stand-alone Pfam scanner and a database downloaded on 8 July, 2015 (Punta et al. 2012); (ii) the numbers of blast hits in each proteome (e-value cut-off, 1 × e−20) where proteins from the Database of Fungal Virulence Factors [downloaded on 7 July, 2015; (Lu et al. 2012)] were used as queries; (iii) the numbers of blast hits in each proteome (e-value cut-off, 1 × e−20) where proteins from the core set of the Fungal Stress Response Database [received from the authors on 4 March, 2015; (Karanyi et al. 2013)] were used as queries. The resulting list of protein groups with significant predicted expansion/contraction was manually checked with a focus on the branches leading to A. melanogenum, H. werneckii and E. dermatitidis.
Secreted proteins
Predicted secreted proteins were identified by searching through the whole proteomes for signal peptides with SignalP (Petersen et al. 2011), using the default cut-off D-value of 0.43. Proteins containing predicted transmembrane regions using TMHMM (Krogh et al. 2001) were then removed from the output. The functions of putative secreted proteins were predicted by the CAZymes Analysis Toolkit for CAZymes (Park et al. 2010) and MEROPS for proteases (Rawlings et al. 2012). The respective heatmaps were produced with the package heatmap.2 in R (R Development Core Team 2015) from scaled data and using the default parameters. For CAZymes, only families with more than five representatives are shown, and for proteases, only families with more than two representatives are shown.
Polyketide synthases and non-ribosomal peptide synthetases
Polyketide synthases (PKS) were identified using NaPDoS (Ziemert et al. 2012). The ketosynthase domains identified by the programme, together with the representative PKSs for each previously defined PKS cluster (HR-PKSs (lovastatin—AAD39830.1, T toxin—AAB08104.3, fumonisin—AAD43562.2), PR-PKSs (6-MSAS—BAA20102.2), NR-PKSs [group I—XP_681178.1, AGC95321.1; group II—AAD31436.3, CAM35471.1; group III—AAC39471.1, Q03149.2; group IV—Q12397.2, BAE71314.1; group V—XP_664675.1, XP_746435.1; group VI—XP_681652.1, XP_664052.1; group VII—XP_658638.1, XP_658127.1)] and fatty-acid synthase (outgroup, AN9407), were used for the phylogenetic analysis with the PhyML software (Liu et al. 2015; Varga et al. 2003).
The protein representatives of the melanin synthesis pathway according to (Chen et al. 2014) were identified by running local blastp against the proteomes of all of the listed fungi, with A. fumigatus homologues used as queries, and with an e-value cut-off of 1 × e−20.
Non-ribosomal peptide synthetases (NRPSs) were identified by analysing the whole proteomes with a stand-alone version of antiSMASH, using the default parameters (Blin et al. 2013). Adenylation (A) domains as determined by the programme, together with A domains from previously characterised NRPSs from A. nidulans, A. fumigatus and C. heterostrophus were used for the phylogenetic analysis (Cramer et al. 2006; von Döhren 2009; O’Hanlon et al. 2012; Condon et al. 2013; Bushley and Turgeon 2010) (A. nidulans, A. fumigatus: SidC [XP_753088.1], SidD [XP_748662.1], GliP [XP_750855.1], FtmA [XP_747187.1], Pes3 [XP_753380.1], PesL [XP_751084.1], Pes1 [XP_752404.1], ACVS [XP_660225.1], and C. heterostrophus: NPS2 [AAX09984.1], NPS10 [AAX09992.1], NPS4 [AAX09986.1], NPS6 [AAX09988.1]) and several adenosine monophosphate (AMP)-binding proteins (outgroup; ACS [XP_751720.1], AAL [AAG53991.2], LCFAL [XP_753087.1], AAR [XP_751705.1]. As one NRPS can contain several A domains, all of them were included in the analysis, but the numbers discussed here were given for the abundance of proteins in the species, and not for the number of A domains.
The phylogenies of keto-synthase (in PKSs) and adenylation (in NRPSs) domains were estimated by first aligning the protein sequences with the MAFFT software in the ‘-auto’ mode (Katoh and Toh 2008). Then the model of protein evolution, the a-parameter, and the proportion of invariable sites were estimated using ProtTest 3.2.1 (Darriba et al. 2011). Finally, the trees were generated with the PhyML 3.1 software (Guindon et al. 2010), and the branch supports were calculated as approximate Bayes values.
Statistics
T test implemented in The Gnumeric (http://www.gnumeric.org/), an open-source spreadsheet programme (Keeling and Pavur 2011), was used to test the significance of differences between the abundances of selected protein families for the following pairs of species groups: extremotolerant versus non-extremotolerant, plant and animal opportunists versus non-opportunists, Eurotiomycetes versus Dothideomycetes. Significant differences (P < 0.05) are reported and/or indicated in the figures with an asterisk (*). For phylogeny, statistical analyses were performed using the SPSS statistical software, version 20 (SPSS Inc., Chicago, IL, USA). The significance of the associations among fungal presence and the environmental variables and the strength of the association was determined using Chi square test (χ2). Chi square was used for examining the ecological characteristics correlation between medical fungi and surface colonisers, between medical relevance and general extremotolerance, between medical fungi and osmotolerance, and between medical relevance and psychrotolerance. Differences with P < 0.05 were considered to be statistically significant.
Results
Distribution of extremotolerance and pathogenicity in the fungal kingdom
An LSU tree was reconstructed on the basis of representatives of 140 fungal orders or ordinal groups, distinguished according to recent taxonomy of the fungal kingdom (Fig. 1). The backbone of the tree remained largely unresolved, which interfered with classification above the ordinal level. Most of the orders, however, were statistically supported (bootstrap > 80%), particularly in basal taxonomic lineages. In the Basidiomycota a number of groups were discernible of which the Ustilaginomycotina were in ancestral position to the Ascomycota, and Pucciniomycotina and Agaricomycotina formed recognisable clusters. The lower fungi were all found at large phylogenetic distances from each other. The adjacent clusters of Ascomycota were generally closer together. Some groups were found in unexpected positions, such as the family Ajellomycetaceae, currently classified in the Onygenales but in our tree forming a distinct cluster. The small orders Calosphaeriales and Togniniales were united under the overarching order Diaporthales. Protoventuria was clearly different from Venturiales.
We subsequently plotted broad ecological categories [surface colonisers (1), osmotolerant/osmophilic fungi (2), psychrotolerant/psychrophilic fungi (3), clinically relevant fungi covering primary pathogens, opportunistic pathogens and mammal colonisers (4), plant pathogens (5)], expressed as the number of species with a particular ecology, relative to the number of species described in that order. Orders containing lichenised fungi (1) showed a preponderance of rock-inhabiting lifestyles. Lichens are known to be highly diverse (Schoch et al. 2009) and are found in Arthoniales and adjacent orders of Arthoniomycetes, in Lecanoromycetes and in Ostropomycetes. Non-lichenised rock-inhabiting fungi are associated to e.g. Verrucariales and Chaetothyriales. Other types of extremotolerant ecology (2, 3) are more difficult to define and categorise over the entire fungal kingdom. Rock-inhabiting lifestyles involve multiple stress factors such as limited nutrient availability, and osmotic and temperature stress, which are linked to survival under arid conditions. Desert-fungi (3) are common e.g. among Sordariales and Eurotiales. These fungi are also prevalent in the dry fraction of indoor fungal biomes. For this reason, we separated osmotolerance (2) from rock-inhabiting lifestyles and analysed data in combination as well as separate. An entirely different type of osmotolerance is found in numerous yeasts (Saccharomycetales) which live in flowers and fruit juices. In ocean water, basidiomycetous yeasts tend to be prevalent. The latter category is further characterized by low temperature, which otherwise is noted in the small groups of polar fungi. Plant-associated lifestyles (5) are widely distributed in Ascomycota. Several orders (e.g. Asterinales, Erysiphales, Meliolales, Myriangiales) contain almost exclusively plant-associated species with very similar patterns of infection, while major plant pathogens are also found in Botryosphaeriales, Diaporthales, Hypocreales, Magnaporthales, Pleosporales, Venturiales and others. Numerous strict plant pathogens are located in basidiomycetous orders belonging to Ustilaginomycotina and Pucciniomycotina.
Most fungi that are able to grow at 37 °C have also been encountered in human infection (4) (red bars in Fig. 1). Infection of mammal hosts requires tolerance of body temperature at or close to 37 °C. Only a small number of fungi are thermophilic without having any apparent invasive ability, e.g. species of Myceliophthora, Byssochlamys or Thermoascus (the group is too small to be displayed in Fig. 1). The main order comprising species considered as pathogens [i.e. causing transmissible disease; (de Hoog et al. 2018)] is Onygenales. The order contains agents of systemic disease (i.e. exhibiting a specialised invasive phase in tissue) in the Ajellomycetaceae and Onygenaceae, in addition to the dermatophytes classified in Arthrodermataceae. Members of Pneumocystidales are pulmonary colonisers. As only a very small number of species has been described in this order so far, numerous additional mammal host-specific taxa are likely to exist showing parallel evolution with their hosts (Guillot et al. 2001). Members of the small order Malasseziales are lipophilic fungi associated with mammals as superficial commensals, but can be involved in infection, e.g. in patients receiving lipid-rich parenteral nutrition (Baker et al. 2016). In total, about 100 species may have some kind of advantage of the use of a mammal host, via infection or colonisation. All remaining fungi (approximately 550 species) listed in the Atlas of Clinical Fungi are categorised here as being opportunists, i.e. having infectious ability but showing no sign of specialisation for the mammal host and having an environmental primary habitat (de Hoog et al. 2015).
Opportunistic fungi are found scattered all over the fungal tree (Fig. 1), distributed over 21 orders (15.0% of all discerned orders): three out of 18 orders of lower fungi (16.7%), three out of 42 orders of Basidiomycota (7.1%) and 15 out of 80 orders of Ascomycota (18.8%) contain potentially infectious fungi. In absolute numbers the Eurotiales (60), Hypocreales (64), Onygenales (78), and Pleosporales (64) contain large numbers of clinically relevant species. Relatively, compared to the number of currently known species in the order, Chaetothyriales (6.6% opportunists), Onygenales (19.0%) in Ascomycota, Trichosporonales (7.8%) in Basidiomycota, and Mucorales (10.5%) in Mucoromycotina show the largest infectious potential.
An association of clinical relevance (4) with surface colonising life-style (1) is found in five orders, with osmotolerance/osmophily (2) in six orders, psychrotolerance/psychrophily (3) in five orders, and with preponderantly plant-inhabiting (mostly saprobe or opportunist) life styles (5) in four orders, and without clear association with any of the listed parameters in three orders; this includes the pulmonary and cutaneous colonisers of Pneumocystidales and Malasseziales. Statistically significant correlation was confirmed between medical relevance and general extremotolerance/osmophily (P = 0.0001), and between medical relevance and osmotolerance (P = 0.0007). There was also a significant correlation with psychrotolerance (P = 0.0043). However, there was no significant association between medical relevance and surface colonising lifestyle (P = 0.324).
Genomic signatures of extremotolerance and opportunistic pathogenicity
To search for possible genomic signatures of opportunism in fungi that are able to cause human infections, we performed the genomic comparison of 11 dothideomycetous and 9 eurotiomycetous species, selected here to represent black fungi, a group which harbours many examples of polyextremotolerant species that are also able to switch to opportunism. The investigated fungi cover a range of ecological strategies, colour coded on Fig. 2 (extremotolerant in blue, animal opportunists and plant pathogens in red and green, respectively). As explained in the Methods, we focused on the predicted proteins (primarily their copy numbers), known to be involved in virulence and stress tolerance and searched for common patterns in distantly related species with similar lifestyles.
Secretome
The fungal secretome was previously implicated in promoting pathogenicity (Ranganathan and Garg 2009). In this study we show that the numbers of proteases are significantly increased in the plant pathogens (Fig. 3c1). There are fewer proteases seen in the extremotolerant species, although this is mainly due to the protease-rich plant pathogens in the non-extremotolerant group, and the difference did not reach statistical significance. When comparing the most abundant individual enzyme families (Fig. 3c2), the plant pathogens have significantly more aspartic endopeptidases or pepsin family A1 proteases (Fig. 3c2, A01A) and serine endopeptidases or subtilisin family proteases (Fig. 3c2, S08A), while the extremotolerant species have significantly fewer serine exopeptidases (Fig. 3c2, S28).

Predicted secreted proteins. a Total secreted proteins. b Secreted carbohydrate-active enzymes (CAZymes) b1 and a heatmap of the CAZymes families with more than five representatives in the species studied (b2). c Secreted proteases (c1) and a heatmap of the MEROPS families with more than two representatives in the species studied (c2). T-tests were used to determine the significances of the differences between the abundance of the proteins for the following pairs of species groups: extremotolerant versus non-extremotolerant; plant and animal pathogens versus non-pathogens; Eurotiomycetes versus Dothideomycetes. Significant differences (P < 0.05) are indicated with an asterisk (*)
The differences in carbohydrate-active enzymes (CAZymes) abundance between plant pathogens and non-plant pathogenic species did not reach statistical significance. Nevertheless, three out of four plant pathogens analysed here have higher numbers of CAZymes than seen for 16 non-plant pathogenic species (except A. nidulans and H. werneckii; Fig. 3b1). Moreover, the plant pathogenic fungi are significantly enriched with members of the CAZymes auxiliary activity family (AA7), carbohydrate-binding module families (CBM13, CBM18, CBM35, CBM50), carbohydrate esterase family (CE5), glycoside hydrolase families (GH2, GH12, GH92), and glycosyl transferase family (GT34) (Fig. 3b2). The extremotolerant species have significantly more members of the CAZymes GH32 family, which comprises invertases and other enzymes that are involved in energy storage and recovery. Despite these patterns, no lifestyle-linked clustering of the species was observed based on their enzyme profiles (Fig. 3b2, c2).
Polyketide synthases
Polyketide synthases (PKSs) are responsible for the synthesis of secondary metabolite polyketides. Our phylogenetic analysis of ketosynthase domains characteristic of PKSs, produces four well-defined clusters that correspond to fatty-acid synthases (FAS), non-reducing (NR)-PKSs, highly reducing (HR)-PKSs, and partially reducing (PR)-PKSs (Fig. 4a). All of the major subgroups of NR-PKSs (groups I-VII) and HR-PKSs (lovastatins, fumonisins, T-toxins) were identified in the phylogenetic tree. The total number of PKSs analysed is significantly higher for the plant pathogens compared to the non-pathogenic fungi and animal opportunists (Fig. 4b), which is due to the significant enrichment of NR-PKSs and HR-PKSs (Fig. 4d, e). There is no difference in the PKS numbers between Dothideomycetes and Eurotiomycetes. Genes encoding 1,3,6,8-tetrahydroxynaphthalene (THN) synthases involved in the synthesis of melanin, as well as genes involved in the 3,4-dihydroxyphenylalanine (DOPA)-melanin synthesis pathway and pyomelanin synthesis (l-tyrosine degradation) pathway are present in the majority of the 22 analysed species (Supplemental Table S1).

Predicted polyketide synthases. a Phylogeny of the ketosynthase domains determined using the Natural Product Domain Seeker. b–e Abundance of the PKS subgroups in the studied fungal species. FAS fatty-acid synthases (c), PR-PKS partially reducing PKS, NR-PKS non-reducing PKS (d), HR-PKS fully reducing PKS (e). T-tests were used to test the significances of the differences between the abundances of these proteins for the following pairs of species groups: extremotolerant versus non-extremotolerant; plant and animal pathogens versus non-pathogens; Eurotiomycetes versus Dothideomycetes. Significant differences (P < 0.05) are indicated with an asterisk (*)
Non-ribosomal peptide synthetases
The here reconstructed phylogeny of the A domains from the non-ribosomal peptide synthetases (NRPSs) or NRPS-like proteins identified by antiSMASH software reveals two major groups: the NRPSs and the adenylating enzymes, such as long-chain fatty-acyl-CoA synthases (Fig. 5a). Where possible, the NRPS phylogenetic groups based on the A domains were named after the previously characterised homologues (Fig. 5a). Different A domains from single NRPS genes with several A domains mostly occur in the same clade, with the exceptions being diketopiperazine synthetases and Pes1 homologues, the A domains of which are placed in two or more well-separated clusters (Fig. 5a).

Predicted non-ribosomal peptide synthetases. a Phylogeny of the adenylation domains (A domains) determined using the antibiotics and secondary metabolite analysis shell. The dots mark the positions of the reference proteins from A. fumigatus and A. nidulans (purple) and C. heterostrophus (pink) used to infer the functions of the protein clusters (Cramer et al. 2006; von Döhren 2009; O’Hanlon et al. 2012; Condon et al. 2013; Bushley and Turgeon 2010). The names of the reference proteins are in round brackets, while the inferred products of the NRPSs are in square brackets. Note that different domains from the same proteins similar to the GliP and NPS4/Pes1 proteins are included in more than one cluster. b–e Abundance of the NRPS proteins in the studied fungal species, without the adenylating enzymes (b), and for the selected protein subfamilies of the adenylating enzymes (c), and intracellular (d) and extracellular (e) siderophores. T-tests were used to test the significances of the differences between the abundances of these proteins for the following pairs of species groups: extremotolerant versus non-extremotolerant; plant and animal pathogens versus non-pathogens; Eurotiomycetes versus Dothideomycetes. Significant differences (P < 0.05) are indicated with an asterisk (*)
The total number of NRPSs are significantly higher in the plant pathogens, both with the inclusion (data not shown) and the exclusion (Fig. 5b) of the large group of adenylating enzymes, although no lifestyle-associated patterns are observed for the adenylating enzymes alone (Fig. 5c). The numbers of NRPSs involved in the synthesis of extracellular (but not intracellular) siderophores are higher in most of the animal opportunists and plant pathogens compared to the non-pathogens, although the differences reached statistical significance only for the plant pathogens (Fig. 5e).
Selected fungal virulence factors
Several protein families that experienced significant expansion or contraction in various parts of the phylogenetic tree were investigated in more detail. When the expansion and contraction of these protein families are compared within and between the groups of species with similar lifestyles, there are no significant differences in most cases, although there are some exceptions, a selection of which is shown in Fig. 6. We show that the number of fungal TP-binding cassette (ABC) type multidrug resistance (MDR) efflux transporters is higher in the plant pathogens than in most other fungi, but the between-group difference is not significant. The number of siderophore-iron transporters does not differ between the fungal groups with different lifestyles (Fig. 6b). In 12 extremotolerant species the number of siderophore genes is higher than in five non-extremophile species, the exceptions being B. compniacensis, C. apollinis and B. dothidea. However, the between-group differences did not reach statistical significance. Tripeptidyl-peptidases (TPPs), secreted serine proteases capable of protein degradation at acidic pH differ significantly in number between Eurotiomycetes and Dothideomycetes (Fig. 6c) as well as in the animal opportunists versus plant pathogens. Finally, proteins similar to the effector Ecp6, a scavenger of chitin fragments, are enriched in the plant pathogens (Fig. 6d). No significant differences were observed between Dothideomycetes and Eurotiomycetes.

Abundance of the selected fungal virulence factors in the fungal species studied. a Multidrug resistance (MDR) efflux transporters. b Siderochrome–iron transporters. c Tripeptidyl peptidases. d Extracellular proteins 6. The protein groups were selected based on significant protein family expansion or contraction, as determined by analysis using CAFE 3. T-tests were used to test the significances of differences between the abundances of the proteins for the following pairs of species groups: extremotolerant versus non-extremotolerant; plant and animal pathogens versus non-pathogens; Eurotiomycetes versus Dothideomycetes. Significant differences (P < 0.05) are indicated with an asterisk (*)
Discussion
Extremotolerance and opportunism are linked at the level of fungal orders
Infection of mammal hosts is a rare ability in the fungal kingdom, as shown by our study. Of the 140 orders distinguished in this paper based on current taxonomy (Fig. 1), only 21 (15.0%) contain species that repeatedly show this infectious potential. Among these, most opportunistic orders and the main order containing true pathogens causing transmissible disease [Onygenales (de Hoog et al. 2018)] belong to Ascomycota (14 out of 80 orders, 17.3%). Of the much fewer opportunistic orders in Basidiomycota (3 out of 42, 7.1%) Malasseziales are generally regarded as asymptomatic cutaneous commensals/pulmonary colonisers and the same is true for the ascomycetous Pneumocystidales (Baker et al. 2016). Members of the remaining 18 orders are able to cause infection in specific circumstances but typically have an environmental habitat.
Our results show that opportunism in mammals is correlated with extremotolerance. Association of the 21 orders with all categories of medical relevance and general extremotolerance was found to be significant (P = 0.0001). However, while some large melanised and extremotolerant groups within the fungal kingdom are only rarely encountered in human infection, others (such as Chaetothyriales) have a particularly high number of opportunists. This apparent discrepancy can be explained by distinguishing between two modes of extremotolerance: ‘polyextremotolerance’, where tolerance to a variety of stress types is coupled with large adaptability and resulting in great potential for habitat shifts, and ‘monoextremotolerance’ describing a specialisation for a particular type of stress and with low potential for habitat shifts. While the first type of extremotolerance was significantly associated with opportunism in our analysis (as described above) the second (surface colonising lifestyle) was not. A similar observation was made by Prenafeta-Boldu et al. (2006), introducing dual ecology as a mechanism enhancing habitat shifts. Other factors besides stress tolerance may play a role, such as the ability to assimilate monoaromatic hydrocarbons, linked to the tendency of fungi to infect the central nervous system (Prenafeta-Boldu et al. 2006), but this was not tested here due to the limited available data.
In addition to almost complete association of thermotolerance with clinical occurrence, osmotolerance/osmophily was also identified as a major factor correlating with opportunism (P = 0.0007), confirming the connection proposed by de Hoog et al. (2005). Interestingly, despite the ability to grow at 37 °C being the main virulence factor in fungi (Robert and Casadevall 2009), opportunism also correlated to tolerance to low temperatures (P = 0.0043). In 97 orders no obvious extremotolerance of any kind is known; of these, only five (5.2%) contain species with an infectious potential. Two of these concern the lower fungi in Entomophthoromycotina, where opportunism has been hypothesised to be enhanced by intestinal occurrence in cold-blooded tropical animals (Gugnani and Okafor 1980). The large and species-rich classes of Pezizomycetes, Orbiliomycetes, Lecanoromycetes, and Leotiomycetes do not contain any clinical representatives.
An association between human opportunism and plant pathogenicity that has been put forward earlier on the basis of similar virulence factors (Kempf et al. 2002; Kirzinger et al. 2011; Melotto et al. 2014) is not supported by our results. Of the 26 fungal orders containing major plant pathogens, only three also contain a few or more clinical species. One of these is Pleosporales, an order showing a wide ecological diversity including extremotolerance (Ahmed et al. 2014). The Hypocreales contains species causing both human infections (only 64 out of 4479 described species) as well as major plant diseases, as an example of cross-kingdom pathogenicity (van Baarlen et al. 2007; Segorbe et al. 2017) or more accurately, cross-kingdom opportunism. Other such examples include species of Fusarium, where both plant and human infection is frequent and strains have an unusually wide host range (Al-Hatmi et al. 2016; Šišić et al. 2018). Similarly, Sharma et al. (2014) noted the plant-virulence of clinical strains of Exserohilum rostratum (Pleosporales), but this species is not host-specific, it has a wide host range on diverse grasses, and is also found in soil and on rotten materials (Ellis 1971). However, in orders where host-specific plant-pathogenicity is among the prevalent life styles (e.g. Myriangiales, Asterinales, Meliolales, Magnaporthales, Erysiphales, Taphrinales, Tilletiales, Exobasidiales, Ustilaginales, Microstromatales, Pucciniales), no human opportunistic species are encountered. In the genus Alternaria mammal opportunism is found almost exclusively in the few saprobic representatives (de Hoog and Horre 2002).
Few genomic signatures reflect the lifestyles of black fungi
If, as we suggest, fungal opportunism is a side effect of polyextremotolerance and not a specialised lifestyle, opportunists are expected to have few differences from their non-opportunistic counterparts. This is supported by the results of our analysis of genomic signatures in 11 dothideomycetous and 9 eurotiomycetous fungi. We focused on enzymes for secondary metabolite production, carbohydrate-active enzymes (CAZymes), and small secreted proteins and peptidases, proteins with documented or proposed roles as effectors in virulence.
Although proteases are believed to be important for animal pathogenesis (Monod et al. 2002), in our dataset they were not enriched in opportunistic species compared to strictly non-opportunistic species (Fig. 3c1). The same was true for individual protease families (Fig. 3c2) and also for CAZyme families. In contrast, CAZyme family GH32 (invertases and other enzymes involved in energy storage and recovery) was significantly enriched in extremotolerant species, possibly reflecting their energetically demanding life in extreme conditions (Oren 2011). Generally, however, no lifestyle-linked clustering of the species was observed based on their CAZyme profiles (Fig. 3b2, c2), confirming the observations of Krijger et al. (2014).
Secondary metabolites such as polyketides are important for interactions of fungi with other organisms (Fischbach and Walsh 2006), yet no enrichment in PKSs catalysing their synthesis was observed in human opportunists (Fig. 4). One of the PKS products, melanin, plays an important role in virulence and resistance to clinically used antifungal agents (van Baarlen et al. 2007; Feng et al. 2001; Nosanchuk et al. 2015; Schnitzler et al. 1999) [although these roles have been disputed by some authors (Song et al. 2017)] and protection against abiotic stress (Slepecky and Starmer 2009; Gostinčar et al. 2012; Kogej et al. 2007; Kejžar et al. 2013), but again we found no significant differences between the opportunistic and other species in numbers of enzymes involved in the PKS melanin synthesis pathway and also in the alternative DOPA-melanin synthesis pathway (Langfelder et al. 2003), and pyomelanin synthesis/l-tyrosine degradation pathway (Schmaler-Ripcke et al. 2009) (Supplemental Table S1). The genes known to be involved in the three different melanin synthesis pathways are present in the majority of the 22 species studied here (Supplemental Table S1).
Similarly to all above discussed genes, genes for non-ribosomal peptide synthetases (NRPSs) were not enriched in animal opportunists, although they are involved in the synthesis of important virulence determinants, such as iron-chelating siderophores (Bushley and Turgeon 2010; Silva et al. 2011), and toxins, such as enniatin, victorin, HC-toxin and AM-toxin (Walton 1996; Haese et al. 1993). Iron overload in a host is known to exacerbate many infectious diseases, such as cryptococcosis in people infected with HIV, and conversely, iron withholding is an important defence strategy for mammalian hosts (Jung et al. 2006). There were no lifestyle-associated differences in the number of siderophore–iron transporters, which are responsible for the uptake of siderophore–iron chelates (Philpott 2006) and linked to stress response and virulence in Cryptococcus neoformans (Singh et al. 2015; Jung et al. 2006), or the number of multidrug resistance (MDR) efflux transporters (Fig. 6a), which provide fungi with protection against antibiotics, plant defence compounds, and fungicides (de Waard et al. 2006). The differences in the number of secreted serine proteases [previously linked to virulence in A. fumigatus (Reichard et al. 2006)] between animal opportunists and plant pathogens can be attributed to their phylogenetic history, as the same difference was observed between Eurotiomycetes and Dothideomycetes—most here studied animal opportunists are Eurotiomycetes and most plant pathogens are Dothideomycetes (Fig. 6c).
While no genomic traits of analysed black fungi were linked to their opportunism, several traits were significantly associated with their plant pathogenicity. Proteases, believed to be involved in fungal signalling, nutrition, degradation of host tissues, and digestion of plant defence proteins (Ohm et al. 2012), were significantly enriched in the plant pathogens (Fig. 3c1). Some of the differences are in line with previous observations, for example in the case of A01 and S08A proteases (ten Have et al. 2004; Armijos Jaramillo et al. 2013), additionally validating our general approach. Similarly, certain CAZyme families (AA7, CBM13, CBM18, CBM35, CBM50, CE5, GH2, GH12, GH92, GT34; Fig. 3b2) were shown to be enriched in plant pathogens, likely linked to the role of enzymes from these families in breaking down the barrier of the plant cell-wall polysaccharides and using plant polysaccharides as a carbon source (Ohm et al. 2012; Lowe et al. 2015). Additionally, plant pathogens differed from other analysed species in the total number of PKSs, in having a significantly higher abundance of extracellular siderophore NRPSs (Fig. 5e) and in the enrichment of the effector Ecp6 (Fig. 6d). The latter is easily explained as the Ecp6 is a known scavenger of chitin fragments that are released by chitinases, thereby preventing recognition of the fungus by the host immune receptors for chitin (Sanchez-Vallet et al. 2013; de Jonge et al. 2010).
We can conclude that while our analysis successfully identified genomic traits linked to plant pathogenicity, we could find no such genomic signatures connected to the fungal opportunistic lifestyle—supporting our hypothesis that virulence traits of opportunistic fungi are exaptations, which can also be found in other, non-opportunistic species.
Implications of the overlap between adaptation to environmental stress and opportunism
The importance of stress tolerance acquired out-of-host in pathogenesis was first proposed for the well-known pathogen C. neoformans (Brown et al. 2007; van Burik and Magee 2001). This interpretation is even more plausible in the case of emerging opportunistic pathogens such as black yeasts, which cause substantially fewer infections than C. neoformans. If the adaptations that allow opportunistic fungi to survive within a human host are indeed exaptations (i.e., mechanisms that originally evolved for different purposes, such as tolerance to environmental stress) and that are only later found to be useful during an infection, there should be few (if any) traits that can be directly linked to the opportunistic potential of the fungal species. Indeed, unlike bacteriologists, mycologists have largely been unsuccessful in finding classical virulence factors, even in notorious human pathogens like Candida albicans and A. fumigatus (Casadevall and Pirofski 2014). Similarly, in this study we show that apart from growth at 37 °C there appear to be few other traits that can distinguish opportunistic human pathogens from their non-pathogenic relatives. No such traits were found through the comparative genomics investigations, while in contrast, plant pathogens were distinguished in several aspects. Taken together, these observations indicate that most generalistic polyextremotolerant fungi can be seen as potential opportunistic pathogens as long as they can grow at the temperatures in the mammalian body. Fortunately, this is a trait that (for now) most fungi lack (Robert and Casadevall 2009).
The observation that polyextremotolerance and opportunistic pathogenesis repeatedly share a common phylogenetic history supports the hypothesis that traits important for fungal pathogenicity are shaped by selection pressures outside of the host. This corresponds to the concept of “accidental virulence” as postulated by Casadevall and Pirofski (2007). However, care should be taken not to view the infection events from a pan-adaptationist perspective. On the one hand, it is unclear whether the opportunist causing the infection is able to escape from the host back into the environment—if it is not, any adaptations to host acquired during the infection are meaningless from an evolutionary perspective. On the other hand, if the opportunist is able to return to the environment, any newly acquired adaptations will be either beneficial, neutral or detrimental for its survival in the environment. Evidently, adaptations detrimental to environmental fitness would be selected against once outside the host. Adaptations with a neutral effect would likely be drowned in the much larger gene pool of environmental strains. Finally, for adaptations beneficial to both survival in the host and in the out-of-host environment it is difficult to envisage why they would need to arise in the small populations during rare and time-limited infection events rather than in much larger out-of-host populations. The speculation of Casadevall and Pirofski (2007) that a passage in animal hosts might increase the fitness of environmental microbes in their out-of-host environment therefore appears unlikely except for species for which animals are a major and regular habitat. An accidental infection is thus likely a (literal or evolutionary) dead end, since the persistence of hypothetical adaptations acquired during infection is improbable. For this reason the gradual evolution of true pathogens through a series of repeated opportunistic infections (in the absence of host-to-host transmission) would be expected to happen extremely rarely, if at all. Our hypothesis of polyextremotolerance (coupled with the ability to grow at 37 °C) as the background of opportunism explains why opportunistic species are relatively rare, and why evolution towards true pathogenicity hardly ever happens at all.
Nevertheless, many species of black fungi are now increasingly being recognised as a medical issue (Silveira and Nucci 2001; Chowdhary et al. 2015). Besides greater numbers of susceptible hosts and improved diagnostics, changes of our lifestyle have been proposed as another reason for this trend (Casadevall et al. 2011; Gostinčar et al. 2011, 2015). In addition to the well-known dry fraction of indoor environments, wet cells have recently revealed a gamut of opportunistic species. Both A. melanogenum and E. dermatitidis were found to be common in tap water, bathrooms, steam-baths and dishwashers (Novak Babič et al. 2016; Hamada and Abe 2010; Zalar et al. 2011). Changes that render indoor habitats inhospitable to a majority of microbes frequently make conditions favourable for other, more resilient and more adaptable species—polyextremotolerant fungi with greater potential to cause opportunistic human infections (Gostinčar et al. 2011, 2015). These are problematic especially if they are also enriched for thermotolerance, such as E. dermatitidis, Saprochaete clavata and Magnusiomyces capitatus in dishwashers (Zalar et al. 2011; Zupančič et al. 2016) or for the ability to metabolise phenols and hydrocarbons (Prenafeta-Boldu et al. 2006, 2012).
This study shows that complex phenotypes such as the ability to cause opportunistic infections of mammals do not evolve from scratch. The genomic toolkit that is the basis of selection is neither unlimited in quantity nor infinitely malleable. It can differ substantially between different taxonomic groups, resulting in repeated emergence of opportunistic pathogens in some groups, but not in others. Our results support the hypothesis that opportunistic fungi do not specialise for pathogenicity as such but that their invasive potential is tightly linked to their polyextremotolerant ecology and most likely uncoupled from their hosts. As a consequence, tackling the emerging problem of opportunistic fungi will require an epidemiological approach very different from the one applied to true pathogens.
References
Ahmed SA, van de Sande WWJ, Stevens DA, Fahal A, van Diepeningen AD, Menken SBJ, de Hoog GS (2014) Revision of agents of black-grain eumycetoma in the order Pleosporales. Persoonia 33:141–154. https://doi.org/10.3767/003158514x684744
Al-Hatmi AMS, Meis JF, de Hoog GS (2016) Fusarium: molecular diversity and intrinsic drug resistance. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1005464
Amselem J, Cuomo CA, van Kan JAL, Viaud M, Benito EP, Couloux A, Coutinho PM, de Vries RP, Dyer PS, Fillinger S, Fournier E, Gout L, Hahn M, Kohn L, Lapalu N, Plummer KM, Pradier JM, Quevillon E, Sharon A, Simon A, ten Have A, Tudzynski B, Tudzynski P, Wincker P, Andrew M, Anthouard V, Beever RE, Beffa R, Benoit I, Bouzid O, Brault B, Chen ZH, Choquer M, Collemare J, Cotton P, Danchin EG, Da Silva C, Gautier A, Giraud C, Giraud T, Gonzalez C, Grossetete S, Guldener U, Henrissat B, Howlett BJ, Kodira C, Kretschmer M, Lappartient A, Leroch M, Levis C, Mauceli E, Neuveglise C, Oeser B, Pearson M, Poulain J, Poussereau N, Quesneville H, Rascle C, Schumacher J, Segurens B, Sexton A, Silva E, Sirven C, Soanes DM, Talbot NJ, Templeton M, Yandava C, Yarden O, Zeng QD, Rollins JA, Lebrun MH, Dickman M (2011) Genomic analysis of the aecrotrophic fungal pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002230
Armijos Jaramillo VD, Vargas WA, Sukno SA, Thon MR (2013) Horizontal transfer of a subtilisin gene from plants into an ancestor of the plant pathogenic fungal genus Colletotrichum. PLoS ONE 8(3):e59078. https://doi.org/10.1371/journal.pone.0059078
Arnaud MB, Chibucos MC, Costanzo MC, Crabtree J, Inglis DO, Lotia A, Orvis J, Shah P, Skrzypek MS, Binkley G, Miyasato SR, Wortman JR, Sherlock G (2010) The Aspergillus Genome Database, a curated comparative genomics resource for gene, protein and sequence information for the Aspergillus research community. Nucleic Acids Res 38:420–427. https://doi.org/10.1093/Nar/Gkp751
Baker RM, Stegink RJ, Manaloor JJ, Schmitt BH, Stevens JC, Christenson JC (2016) Malassezia pneumonia: a rare complication of parenteral nutrition therapy. JPEN J Parenter Enteral Nutr 40(8):1194–1196. https://doi.org/10.1177/0148607115595224
Blin K, Medema MH, Kazempour D, Fischbach MA, Breitling R, Takano E, Weber T (2013) antiSMASH 2.0—a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res 41(1):204–212. https://doi.org/10.1093/nar/gkt449
Brown SM, Campbell LT, Lodge JK (2007) Cryptococcus neoformans, a fungus under stress. Curr Opin Microbiol 10(4):320–325. https://doi.org/10.1016/j.mib.2007.05.014
Bushley KE, Turgeon BG (2010) Phylogenomics reveals subfamilies of fungal nonribosomal peptide synthetases and their evolutionary relationships. BMC Evol Biol. https://doi.org/10.1186/1471-2148-10-26
Casadevall A (2007) Determinants of virulence in the pathogenic fungi. Fungal Biol Rev 21(4):130–132. https://doi.org/10.1016/j.fbr.2007.02.007
Casadevall A, Pirofski LA (2007) Accidental virulence, cryptic pathogenesis, Martians, lost hosts, and the pathogenicity of environmental microbes. Eukaryot Cell 6(12):2169–2174. https://doi.org/10.1128/Ec.00308-07
Casadevall A, Pirofski LA (2014) Microbiology: ditch the term pathogen. Nature 516(7530):165–166. https://doi.org/10.1038/516165a
Casadevall A, Fang FC, Pirofski LA (2011) Microbial virulence as an emergent property: consequences and opportunities. PLoS Pathog. https://doi.org/10.1371/journal.ppat.1002136
Cerqueira GC, Arnaud MB, Inglis DO, Skrzypek MS, Binkley G, Simison M, Miyasato SR, Binkley J, Orvis J, Shah P, Wymore F, Sherlock G, Wortman JR (2014) The Aspergillus Genome Database: multispecies curation and incorporation of RNA-Seq data to improve structural gene annotations. Nucleic Acids Res 42(D1):705–710. https://doi.org/10.1093/nar/gkt1029
Chen ZH, Martinez DA, Gujja S, Sykes SM, Zeng QD, Szaniszlo PJ, Wang Z, Cuomo CA (2014) Comparative genomic and transcriptomic analysis of Wangiella dermatitidis, a major cause of phaeohyphomycosis and a model black yeast human pathogen. G3 Genom Genet 4(4):561–578. https://doi.org/10.1534/g3.113.009241
Chowdhary A, Perfect J, de Hoog GS (2015) Black molds and melanized yeasts pathogenic to humans. Cold Spring Harb Perspect Med. https://doi.org/10.1101/cshperspect.a019570
Condon BJ, Leng YQ, Wu DL, Bushley KE, Ohm RA, Otillar R, Martin J, Schackwitz W, Grimwood J, MohdZainudin N, Xue CS, Wang R, Manning VA, Dhillon B, Tu ZJ, Steffenson BJ, Salamov A, Sun H, Lowry S, LaButti K, Han J, Copeland A, Lindquist E, Barry K, Schmutz J, Baker SE, Ciuffetti LM, Grigoriev IV, Zhong S, Turgeon BG (2013) Comparative genome structure, secondary metabolite, and effector coding capacity across Cochliobolus pathogens. PLoS Genet. https://doi.org/10.1371/journal.pgen.1003233
Cramer RA, Stajich JE, Yamanaka Y, Dietrich FS, Steinbach WJ, Perfect JR (2006) Phylogenomic analysis of non-ribosomal peptide synthetases in the genus Aspergillus. Gene 383:24–32. https://doi.org/10.1016/j.gene.2006.07.008
Darriba D, Taboada GL, Doallo R, Posada D (2011) ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27(8):1164–1165. https://doi.org/10.1093/bioinformatics/btr088
de Hoog GS, Horre R (2002) Molecular taxonomy of the Alternaria and Ulocladium species from humans and their identification in the routine laboratory. Mycoses 45(8):259–276. https://doi.org/10.1046/j.1439-0507.2002.00747.x
de Hoog GS, Zalar P, Gerrits van den Ende AHG, Gunde-Cimerman N (2005) Relation of halotolerance to human-pathogenicity in the fungal tree of life: an overview of ecology and evolution under stress. In: Gunde-Cimerman N, Oren A, Plemenitaš A (eds) Adaptation to life at high salt concentrations in Archaea, Bacteria, and Eukarya. Springer, Dordrecht, pp 373–395
de Hoog GS, Guarro J, Gené J, Figueras MJ (2015) Atlas of clinical fungi. The ultimate benchtool for diagnostics. https://doi.org/10.1111/j.1439-0507.1996.tb00148.x
de Hoog G, Ahmed S, Danesi P, Guillot J, Gräser Y (2018) Distribution of pathogens and outbreak fungi in the fungal kingdom. In: Seyedmousavi S, de Hoog G, Guillot J, Verweij PE (eds) Emerging and epizootic fungal infections in animals. Springer, Dordrecht, pp 3–16
de Jonge R, van Esse HP, Kombrink A, Shinya T, Desaki Y, Bours R, van der Krol S, Shibuya N, Joosten MHAJ, Thomma BPHJ (2010) Conserved fungal LysM effector Ecp6 prevents chitin-triggered immunity in plants. Science 329(5994):953–955. https://doi.org/10.1126/science.1190859
de Waard MA, Andrade AC, Hayashi K, Schoonbeek HJ, Stergiopoulos I, Zwiers LH (2006) Impact of fungal drug transporters on fungicide sensitivity, multidrug resistance and virulence. Pest Manag Sci 62(3):195–207. https://doi.org/10.1002/ps.1150
Elias PM (2007) The skin barrier as an innate immune element. Semin Immunopathol 29(1):3–14. https://doi.org/10.1007/s00281-007-0060-9
Ellis MB (1971) Dematiaceous Hyphomycetes. CMI, Kew
Feng B, Wang X, Hauser M, Kaufmann S, Jentsch S, Haase G, Becker JM, Szaniszlo PJ (2001) Molecular cloning and characterization of WdPKS1, a gene involved in dihydroxynaphthalene melanin biosynthesis and virulence in Wangiella (Exophiala) dermatitidis. Infect Immun 69(3):1781–1794
Fischbach MA, Walsh CT (2006) Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem Rev 106(8):3468–3496. https://doi.org/10.1021/cr0503097
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHugh W, Ma LJ, Smirnov S, Purcell S, Rehman B, Elkins T, Engels R, Wang S, Nielsen CB, Butler J, Endrizzi M, Qui D, Ianakiev P, Bell-Pedersen D, Nelson MA, Werner-Washburne M, Selitrennikoff CP, Kinsey JA, Braun EL, Zelter A, Schulte U, Kothe GO, Jedd G, Mewes W, Staben C, Marcotte E, Greenberg D, Roy A, Foley K, Naylor J, Stange-Thomann N, Barrett R, Gnerre S, Kamal M, Kamvysselis M, Mauceli E, Bielke C, Rudd S, Frishman D, Krystofova S, Rasmussen C, Metzenberg RL, Perkins DD, Kroken S, Cogoni C, Macino G, Catcheside D, Li W, Pratt RJ, Osmani SA, DeSouza CP, Glass L, Orbach MJ, Berglund JA, Voelker R, Yarden O, Plamann M, Seiler S, Dunlap J, Radford A, Aramayo R, Natvig DO, Alex LA, Mannhaupt G, Ebbole DJ, Freitag M, Paulsen I, Sachs MS, Lander ES, Nusbaum C, Birren B (2003) The genome sequence of the filamentous fungus Neurospora crassa. Nature 422(6934):859–868
Goodwin SB, Ben M’Barek S, Dhillon B, Wittenberg AHJ, Crane CF, Hane JK, Foster AJ, Van der Lee TAJ, Grimwood J, Aerts A, Antoniw J, Bailey A, Bluhm B, Bowler J, Bristow J, van der Burgt A, Canto-Canche B, Churchill ACL, Conde-Ferraez L, Cools HJ, Coutinho PM, Csukai M, Dehal P, De Wit P, Donzelli B, van de Geest HC, Van Ham RCHJ, Hammond-Kosack KE, Henrissat B, Kilian A, Kobayashi AK, Koopmann E, Kourmpetis Y, Kuzniar A, Lindquist E, Lombard V, Maliepaard C, Martins N, Mehrabi R, Nap JPH, Ponomarenko A, Rudd JJ, Salamov A, Schmutz J, Schouten HJ, Shapiro H, Stergiopoulos I, Torriani SFF, Tu H, de Vries RP, Waalwijk C, Ware SB, Wiebenga A, Zwiers LH, Oliver RP, Grigoriev IV, Kema GHJ (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. https://doi.org/10.1371/journal.pgen.1002070
Gostinčar C, Grube M, de Hoog GS, Zalar P, Gunde-Cimerman N (2010) Extremotolerance in fungi: evolution on the edge. FEMS Microbiol Ecol 71(1):2–11
Gostinčar C, Grube M, Gunde-Cimerman N (2011) Evolution of fungal pathogens in domestic environments? Fungal Biol 115(10):1008–1018. https://doi.org/10.1016/j.funbio.2011.03.004
Gostinčar C, Muggia L, Grube M (2012) Polyextremotolerant black fungi: oligotrophism, adaptive potential, and a link to lichen symbioses. Front Microbiol 3:390. https://doi.org/10.3389/fmicb.2012.00390
Gostinčar C, Ohm RA, Kogej T, Sonjak S, Turk M, Zajc J, Zalar P, Grube M, Sun H, Han J, Sharma A, Chiniquy J, Ngan CY, Lipzen A, Barry K, Grigoriev IV, Gunde-Cimerman N (2014) Genome sequencing of four Aureobasidium pullulans varieties: biotechnological potential, stress tolerance, and description of new species. BMC Genomics 15(1):549. https://doi.org/10.1186/1471-2164-15-549
Gostinčar C, Gunde-Cimerman N, Grube M (2015) Polyextremotolerance as the fungal answer to changing environments. In: Bakermans C (ed) Microbial evolution under extreme conditions. Life in extreme environments, vol 2. de Gruyter, Berlin, pp 185–208. https://doi.org/10.1515/9783110340716.185
Grigoriev IV, Nikitin R, Haridas S, Kuo A, Ohm R, Otillar R, Riley R, Salamov A, Zhao X, Korzeniewski F, Smirnova T, Nordberg H, Dubchak I, Shabalov I (2014) MycoCosm portal: gearing up for 1000 fungal genomes. Nucleic Acids Res 42(1):699–704. https://doi.org/10.1093/nar/gkt1183
Gugnani HC, Okafor JI (1980) Mycotic flora of the intestine and other internal organs of certain reptiles and amphibians with special reference to characterization of Basidiobolus isolates. Mykosen 23(5):260–268
Guillot J, Demanche C, Hugot JP, Berthelemy M, Wakefield E, Dei-Cas E, Chermette R (2001) Parallel phylogenies of Pneumocystis species and their mammalian hosts. J Eukaryot Microbiol 48(1):113–115
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59(3):307–321. https://doi.org/10.1093/sysbio/syq010
Haese A, Schubert M, Herrmann M, Zocher R (1993) Molecular characterization of the enniatin synthetase gene encoding a multifunctional enzyme catalyzing N-Methyldepsipeptide formation in Fusarium scirpi. Mol Microbiol 7(6):905–914. https://doi.org/10.1111/j.1365-2958.1993.tb01181.x
Hall TA (1999) BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp Ser 41:95–98
Hamad M (2008) Antifungal immunotherapy and immunomodulation: a double-hitter approach to deal with invasive fungal infections. Scand J Immunol 67(6):533–543. https://doi.org/10.1111/j.1365-3083.2008.02101.x
Hamada N, Abe N (2010) Comparison of fungi found in bathrooms and sinks. Biocontrol Sci 15(2):51–56
Han MV, Thomas GWC, Lugo-Martinez J, Hahn MW (2013) Estimating gene gain and loss rates in the presence of error in genome assembly and annotation using CAFE 3. Mol Biol Evol 30(8):1987–1997. https://doi.org/10.1093/molbev/mst100
Jung WH, Sham A, White R, Kronstad JW (2006) Iron regulation of the major virulence factors in the AIDS-associated pathogen Cryptococcus neoformans. PLoS Biol 4(12):2282–2295. https://doi.org/10.1371/journal.pbio.0040410
Karanyi Z, Holb I, Hornok L, Pocsi I, Miskei M (2013) FSRD: fungal stress response database. Database. https://doi.org/10.1093/database/bat037
Katoh K, Toh H (2008) Recent developments in the MAFFT multiple sequence alignment program. Brief Bioinform 9(4):286–298
Keeling KB, Pavur RJ (2011) Statistical accuracy of spreadsheet software. Am Stat 65(4):265–273. https://doi.org/10.1198/tas.2011.09076
Kejžar A, Gobec S, Plemenitaš A, Lenassi M (2013) Melanin is crucial for growth of the black yeast Hortaea werneckii in its natural hypersaline environment. Fungal Biol 117(5):368–379. https://doi.org/10.1016/j.funbio.2013.03.006
Kempf VAJ, Hitziger N, Riess T, Autenrieth IB (2002) Do plant and human pathogens have a common pathogenicity strategy? Trends Microbiol 10(6):269–275. https://doi.org/10.1016/S0966-842x(02)02372-7
Kirzinger MWB, Nadarasah G, Stavrinides J (2011) Insights into cross-kingdom plant pathogenic Bacteria. Genes 2(4):980–997. https://doi.org/10.3390/genes2040980
Kis-Papo T, Weig AR, Riley R, Persoh D, Salamov A, Sun H, Lipzen A, Wasser SP, Rambold G, Grigoriev IV, Nevo E (2014) Genomic adaptations of the halophilic Dead Sea filamentous fungus Eurotium rubrum. Nat Commun. https://doi.org/10.1038/ncomms4745
Kogej T, Stein M, Volkmann M, Gorbushina AA, Galinski EA, Gunde-Cimerman N (2007) Osmotic adaptation of the halophilic fungus Hortaea werneckii: role of osmolytes and melanization. Microbiology 153(Pt 12):4261–4273
Krijger JJ, Thon MR, Deising HB, Wirsel SGR (2014) Compositions of fungal secretomes indicate a greater impact of phylogenetic history than lifestyle adaptation. BMC Genomics. https://doi.org/10.1186/1471-2164-15-722
Krogh A, Larsson B, von Heijne G, Sonnhammer ELL (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol Biol 305(3):567–580. https://doi.org/10.1006/jmbi.2000.4315
Kumamoto CA (2008) Niche-specific gene expression during C. albicans infection. Curr Opin Microbiol 11(4):325–330. https://doi.org/10.1016/j.mib.2008.05.008
Langfelder K, Streibel M, Jahn B, Haase G, Brakhage AA (2003) Biosynthesis of fungal melanins and their importance for human pathogenic fungi. Fungal Genet Biol 38(2):143–158. https://doi.org/10.1016/S1087-1845(02)00526-1
Lenassi M, Gostinčar C, Jackman S, Turk M, Sadowski I, Nislow C, Jones S, Birol I, Gunde-Cimerman N, Plemenitaš A (2013) Whole genome duplication and enrichment of metal cation transporters revealed by de novo genome sequencing of extremely halotolerant black yeast Hortaea werneckii. PLoS ONE 8(8):e71328. https://doi.org/10.1371/journal.pone.0071328
Lian X, de Hoog GS (2010) Indoor wet cells harbour melanized agents of cutaneous infection. Med Mycol 48(4):622–628. https://doi.org/10.3109/13693780903405774
Liu L, Zhang Z, Shao CL, Wang JL, Bai H, Wang CY (2015) Bioinformatical analysis of the sequences, structures and functions of fungal polyketide synthase product template domains. Sci Rep. https://doi.org/10.1038/srep10463
Lowe RGT, McCorkelle O, Bleackley M, Collins C, Faou P, Mathivanan S, Anderson M (2015) Extracellular peptidases of the cereal pathogen Fusarium graminearum. Front Plant Sci. https://doi.org/10.3389/fpls.2015.00962
Lu T, Yao B, Zhang C (2012) DFVF: database of fungal virulence factors. Database. https://doi.org/10.1093/database/bas032
Melotto M, Panchal S, Roy D (2014) Plant innate immunity against human bacterial pathogens. Front Microbiol. https://doi.org/10.3389/fmicb.2014.00411
Miller M, Pfeiffer W, Schwartz T (2010) Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In: Proceedings of the Gateway Computing Environments Workshop (GCE), vol 1 (1), pp 1–8
Monod M, Capoccia S, Lechenne B, Zaugg C, Holdom M, Jousson O (2002) Secreted proteases from pathogenic fungi. Int J Med Microbiol 292(5–6):405–419. https://doi.org/10.1078/1438-4221-00223
Nosanchuk JD, Stark RE, Casadevall A (2015) Fungal melanin: what do we know about structure? Front Microbiol. https://doi.org/10.3389/fmicb.2015.01463
Novak Babič M, Zalar P, Ženko B, Džeroski S, Gunde-Cimerman N (2016) Yeasts and yeast-like fungi in tap water and groundwater, and their transmission to household appliances. Fungal Ecol 20:30–39. https://doi.org/10.1016/j.funeco.2015.10.001
O’Hanlon KA, Gallagher L, Schrettl M, Jochl C, Kavanagh K, Larsen TO, Doyle S (2012) Nonribosomal peptide synthetase genes pesL and pes1 are essential for fumigaclavine C production in Aspergillus fumigatus. Appl Environ Microbiol 78(9):3166–3176. https://doi.org/10.1128/Aem.07249-11
Ohm RA, Feau N, Henrissat B, Schoch CL, Horwitz BA, Barry KW, Condon BJ, Copeland AC, Dhillon B, Glaser F, Hesse CN, Kosti I, Labutti K, Lindquist EA, Lucas S, Salamov AA, Bradshaw RE, Ciuffetti L, Hamelin RC, Kema GH, Lawrence C, Scott JA, Spatafora JW, Turgeon BG, de Wit PJ, Zhong S, Goodwin SB, Grigoriev IV (2012) Diverse lifestyles and strategies of plant pathogenesis encoded in the genomes of eighteen Dothideomycetes fungi. PLoS Pathog 8(12):e1003037. https://doi.org/10.1371/journal.ppat.1003037
Oren A (2011) Thermodynamic limits to microbial life at high salt concentrations. Environ Microbiol 13(8):1908–1923. https://doi.org/10.1111/j.1462-2920.2010.02365.x
Park BH, Karpinets TV, Syed MH, Leuze MR, Uberbacher EC (2010) CAZymes Analysis Toolkit (CAT): web service for searching and analyzing carbohydrate-active enzymes in a newly sequenced organism using CAZy database. Glycobiology 20(12):1574–1584. https://doi.org/10.1093/glycob/cwq106
Pel HJ, de Winde JH, Archer DB, Dyer PS, Hofmann G, Schaap PJ, Turner G, de Vries RP, Albang R, Albermann K, Andersen MR, Bendtsen JD, Benen JA, van den Berg M, Breestraat S, Caddick MX, Contreras R, Cornell M, Coutinho PM, Danchin EG, Debets AJ, Dekker P, van Dijck PW, van Dijk A, Dijkhuizen L, Driessen AJ, d’Enfert C, Geysens S, Goosen C, Groot GS, de Groot PW, Guillemette T, Henrissat B, Herweijer M, van den Hombergh JP, van den Hondel CA, van der Heijden RT, van der Kaaij RM, Klis FM, Kools HJ, Kubicek CP, van Kuyk PA, Lauber J, Lu X, van der Maarel MJ, Meulenberg R, Menke H, Mortimer MA, Nielsen J, Oliver SG, Olsthoorn M, Pal K, van Peij NN, Ram AF, Rinas U, Roubos JA, Sagt CM, Schmoll M, Sun J, Ussery D, Varga J, Vervecken W, van de Vondervoort PJ, Wedler H, Wosten HA, Zeng AP, van Ooyen AJ, Visser J, Stam H (2007) Genome sequencing and analysis of the versatile cell factory Aspergillus niger CBS 513.88. Nat Biotechnol 25(2):221–231
Petersen TN, Brunak S, von Heijne G, Nielsen H (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8(10):785–786. https://doi.org/10.1038/nmeth.1701
Philpott CC (2006) Iron uptake in fungi: a system for every source. BBA Mol Cell Res 1763(7):636–645. https://doi.org/10.1016/j.bbamcr.2006.05.008
Prenafeta-Boldu FX, Summerbell R, de Hoog GS (2006) Fungi growing on aromatic hydrocarbons: biotechnology’s unexpected encounter with biohazard? FEMS Microbiol Rev 30(1):109–130. https://doi.org/10.1111/j.1574-6976.2005.00007.x
Prenafeta-Boldu FX, Guivernau M, Gallastegui G, Vinas M, de Hoog GS, Elias A (2012) Fungal/bacterial interactions during the biodegradation of TEX hydrocarbons (toluene, ethylbenzene and p-xylene) in gas biofilters operated under xerophilic conditions. FEMS Microbiol Ecol 80(3):722–734. https://doi.org/10.1111/j.1574-6941.2012.01344.x
Punta M, Coggill PC, Eberhardt RY, Mistry J, Tate J, Boursnell C, Pang N, Forslund K, Ceric G, Clements J, Heger A, Holm L, Sonnhammer ELL, Eddy SR, Bateman A, Finn RD (2012) The Pfam protein families database. Nucleic Acids Res 40:290–301. https://doi.org/10.1093/Nar/Gkr1065
R Development Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna
Ranganathan S, Garg G (2009) Secretome: clues into pathogen infection and clinical applications. Genome Med. https://doi.org/10.1186/gm113
Rawlings ND, Barrett AJ, Bateman A (2012) MEROPS: the database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res 40(D1):343–350. https://doi.org/10.1093/Nar/Gkr987
Reichard U, Lechenne B, Asif AR, Streit F, Grouzmann E, Jousson O, Monod M (2006) Sedolisins, a new class of secreted proteases from aspergillus fumigatus with endoprotease or tripeptidyl-peptidase activity at acidic pHs. Appl Environ Microbiol 72(3):1739–1748. https://doi.org/10.1128/Aem.72.3.1739-1748.2006
Robbertse B, Yoder RJ, Boyd A, Reeves J, Spatafora JW (2011) Hal: an automated pipeline for phylogenetic analyses of genomic data. PLoS Curr 3:RRN213. https://doi.org/10.1371/currents.rrn1213
Robert VA, Casadevall A (2009) Vertebrate endothermy restricts most fungi as potential pathogens. J Infect Dis 200(10):1623–1626. https://doi.org/10.1086/644642
Sanchez-Vallet A, Saleem-Batcha R, Kombrink A, Hansen G, Valkenburg DJ, Thomma BPHJ, Mesters JR (2013) Fungal effector Ecp6 outcompetes host immune receptor for chitin binding through intrachain LysM dimerization. Elife. https://doi.org/10.7554/elife.00790
Sanderson MJ (2003) r8s: inferring absolute rates of molecular evolution and divergence times in the absence of a molecular clock. Bioinformatics 19(2):301–302. https://doi.org/10.1093/bioinformatics/19.2.301
Schmaler-Ripcke J, Sugareva V, Gebhardt P, Winkler R, Kniemeyer O, Heinekamp T, Brakhage AA (2009) Production of pyomelanin, a second type of melanin, via the tyrosine degradation pathway in Aspergillus fumigatus. Appl Environ Microbiol 75(2):493–503. https://doi.org/10.1128/Aem.02077-08
Schnitzler N, Peltroche-Llacsahuanga H, Bestier N, Zundorf J, Lutticken R, Haase G (1999) Effect of melanin and carotenoids of Exophiala (Wangiella) dermatitidis on phagocytosis, oxidative burst, and killing by human neutrophils. Infect Immun 67(1):94–101
Schoch CL, Crous PW, Groenewald JZ, Boehm EWA, Burgess TI, de Gruyter J, de Hoog GS, Dixon LJ, Grube M, Gueidan C, Harada Y, Hatakeyama S, Hirayama K, Hosoya T, Huhndorf SM, Hyde KD, Jones EBG, Kohlmeyer J, Kruys A, Li YM, Lucking R, Lumbsch HT, Marvanova L, Mbatchou JS, Mcvay AH, Miller AN, Mugambi GK, Muggia L, Nelsen MP, Nelson P, Owensby CA, Phillips AJL, Phongpaichit S, Pointing SB, Pujade-Renaud V, Raja HA, Plata ER, Robbertse B, Ruibal C, Sakayaroj J, Sano T, Selbmann L, Shearer CA, Shirouzu T, Slippers B, Suetrong S, Tanaka K, Volkmann-Kohlmeyer B, Wingfield MJ, Wood AR, Woudenberg JHC, Yonezawa H, Zhang Y, Spatafora JW (2009) A class-wide phylogenetic assessment of Dothideomycetes. Stud Mycol 64:1–15. https://doi.org/10.3114/sim.2009.64.01
Segorbe D, Di Pietro A, Perez-Nadales E, Turra D (2017) Three Fusarium oxysporum mitogen-activated protein kinases (MAPKs) have distinct and complementary roles in stress adaptation and cross-kingdom pathogenicity. Mol Plant Pathol 18(7):912–924. https://doi.org/10.1111/mpp.12446
Sharma K, Goss EM, Dickstein ER, Smith ME, Johnson JA, Southwick FS, van Bruggen AHC (2014) Exserohilum rostratum: characterization of a cross-kingdom pathogen of plants and humans. PLoS ONE. https://doi.org/10.1371/journal.pone.0108691
Silva MG, Schrank A, Bailao EFLC, Bailao AM, Borges CL, Staats CC, Parente JA, Pereira M, Salem-Izacc SM, Mendes-Giannini MJS, Oliveira RMZ, Silva LKRE, Nosanchuk JD, Vainstein MH, Soares CMD (2011) The homeostasis of iron, copper, and zinc in Paracoccidioides brasiliensis, Cryptococcus neoformans var. grubii, and Cryptococcus gattii: a comparative analysis. Front Microbiol. https://doi.org/10.3389/fmicb.2011.00049
Silveira F, Nucci M (2001) Emergence of black moulds in fungal disease: epidemiology and therapy. Curr Opin Infect Dis 14(6):679–684
Singh A, Rella A, Schwacke J, Vacchi-Suzzi C, Luberto C, Del Poeta M (2015) Transmembrane transporter expression regulated by the glucosylceramide pathway in Cryptococcus neoformans. BMC Res Notes 8(1):681. https://doi.org/10.1186/s13104-015-1613-y
Sinha S, Flibotte S, Neira M, Formby S, Plemenitas A, Cimerman NG, Lenassi M, Gostincar C, Stajich JE, Nislow C (2017) Insight into the recent genome duplication of the halophilic yeast Hortaea werneckii: combining an improved genome with gene expression and chromatin structure. G3 Genes Genom Genet 7(7):2015–2022. https://doi.org/10.1534/g3.117.040691
Šišić A, Baćanović-Šišić J, Al-Hatmi AMS, Karlovsky P, Ahmed SA, Maier W, de Hoog GS, Finckh MR (2018) The ‘forma specialis’ issue in Fusarium: a case study in Fusarium solani f. sp. pisi. Sci Rep. https://doi.org/10.1038/s41598-018-19779-z
Slepecky RA, Starmer WT (2009) Phenotypic plasticity in fungi: a review with observations on Aureobasidium pullulans. Mycologia 101(6):823–832. https://doi.org/10.3852/08-197
Song YG, Laureijssen-van de Sande WWJ, Moreno LF, van den Ende BG, Li RY, de Hoog S (2017) Comparative ecology of capsular Exophiala species causing disseminated infection in humans. Front Microbiol. https://doi.org/10.3389/fmicb.2017.02514
Talavera G, Castresana J (2007) Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56(4):564–577. https://doi.org/10.1080/10635150701472164
ten Have A, Dekkers E, Kay J, Phylip LH, van Kan JAL (2004) An aspartic proteinase gene family in the filamentous fungus Botrytis cinerea contains members with novel features. Microbiology 150:2475–2489. https://doi.org/10.1099/mic.0.27058-0
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acid Res 24:4876–4882
van Baarlen P, van Belkum A, Summerbell RC, Crous PW, Thomma BPHJ (2007) Molecular mechanisms of pathogenicity: how do pathogenic microorganisms develop cross-kingdom host jumps? FEMS Microbiol Rev 31(3):239–277. https://doi.org/10.1111/j.1574-6976.2007.0065.x
van Burik JAH, Magee PT (2001) Aspects of fungal pathogenesis in humans. Annu Rev Microbiol 55:743–772
Varga J, Rigo K, Kocsube S, Farkas B, Pal K (2003) Diversity of polyketide synthase gene sequences in Aspergillus species. Res Microbiol 154(8):593–600. https://doi.org/10.1016/S0923-2508(03)00169-4
von Döhren H (2009) A survey of nonribosomal peptide synthetase (NRPS) genes in Aspergillus nidulans. Fungal Genet Biol 46:S45–S52. https://doi.org/10.1016/j.fgb.2008.08.008
Walton JD (1996) Host-selective toxins: agents of compatibility. Plant Cell 8(10):1723–1733
Zalar P, Novak M, De Hoog GS, Gunde-Cimerman N (2011) Dishwashers—a man-made ecological niche accommodating human opportunistic fungal pathogens. Fungal Biol 115(10):997–1007. https://doi.org/10.1016/j.funbio.2011.04.007
Ziemert N, Podell S, Penn K, Badger JH, Allen E, Jensen PR (2012) The Natural Product Domain Seeker NaPDoS: a phylogeny based bioinformatic tool to classify secondary metabolite gene diversity. PLoS ONE. https://doi.org/10.1371/journal.pone.0034064
Zupančič J, Babič MN, Zalar P, Gunde-Cimerman N (2016) The black yeast Exophiala dermatitidis and other selected opportunistic human fungal pathogens spread from dishwashers to kitchens. PLoS ONE. https://doi.org/10.1371/journal.pone.0148166
Acknowledgements
The authors acknowledge the financial support from the state budget of the Slovenian Research Agency (Research Programmes P1-0170 and P1-0207, Infrastructural Centre Mycosmo, MRIC UL, Postdoctoral Project Z7-7436 to J. Zajc). The authors would like to thank Chris Berrie for language editing assistance.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gostinčar, C., Zajc, J., Lenassi, M. et al. Fungi between extremotolerance and opportunistic pathogenicity on humans. Fungal Diversity 93, 195–213 (2018). https://doi.org/10.1007/s13225-018-0414-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13225-018-0414-8