
Abstract
Purpose
To analyze the complete genome sequence of the Brevibacterium frigoritolerans ZB201705, a Brevibacterium strain was isolated from the maize rhizosphere in drought- and salt-stressed soil, and the activity of the strain under simulated drought and high salt conditions was assessed.
Methods
We used a combination of the PacBio RS and Illumina sequencing platforms to obtain the complete genome sequence of B. frigoritolerans ZB201705.
Results
The genome consists of 5,475,560 bp in a linear chromosome with no gaps, 4,391 protein-coding sequences, 39 ribosomal RNAs, and 81 transfer RNAs. The genome analysis revealed many putative gene clusters involved in defense mechanisms. In addition, an activity analysis of the strain under high-salt and simulated drought conditions helped clarify its potential tolerance to these abiotic stresses.
Conclusions
Our data revealed the complete genome sequence of the new isolated strain, and showed that it produces many proteins involved in drought and salt stress responses, suggesting that B. frigoritolerans ZB201705 may be a potential factor to increase crop yield under abiotic stresses. The information provided here on the genome of B. frigoritolerans ZB201705 provides valuable insight into rhizobacteria-mediated plant salt and drought tolerance and rhizobacteria-based solutions for agriculture under abiotic stress.
Similar content being viewed by others


Introduction
The genus Brevibacterium was suggested by Breed (1953) based on Brevibacterium linens as the type species. Thereafter, many species of Brevibacterium were isolated and studied. The genus includes at least 49 recognized species according to (www.bacterio.net/brevibacterium.html) (Choi et al. 2018). Brevibacterium has been isolated from various environments such as food (e.g., fermented seafood [Choi et al. 2013] and cheese [Leclercq-Perlat et al. 2000]), humans (e.g., clinical specimens [Wauters et al. 2001] and the human body [Wauters et al. 2003]), and saline environments (e.g., saline soil [Tang et al. 2008], beach sediment [Lee 2006], marine environments [Lee 2008], salt-lake sediment [Guan et al. 2010]), and other environment such as soil of ginseng field (Jung et al. 2018).
Some Brevibacterium, as plant growth-promoting bacteria (PGPB), can improve plant growth under different environmental conditions. B. frigoritolerans strain Imbl 2.1 has potential for bioremediation of phorate both in liquid cultures and agricultural soils (Jariyal et al. 2015). Recent study indicated that Brevibacterium linens RS16 can confer salt tolerance to Oryza sativa (Chatterjee et al. 2018).
Maize is one of the most important foods, feed, and industrial crops worldwide, and is highly sensitive to drought and salt (Morari et al. 2015). To identify beneficial rhizobacteria that promote maize growth, we isolated a Brevibacterium strain, B. frigoritolerans ZB201705, from the maize rhizosphere in drought- and salt-stressed soil and analyzed its complete genome sequence. In addition, we assessed the activity of the strain under high-salt and simulated drought conditions. The information provided here will provide valuable insight into PGPB for agriculture under abiotic stress.
Materials and methods
Sample collection and bacteria isolation
Brevibacterium frigoritolerans ZB201705 was isolated from drought-stressed saline rhizosphere soil of maize in Bayan Nur, Inner Mongolia Autonomous Region, China (40° 13′–42° 28′, E 105° 12′–109° 53′). Soil samples were collected at a depth of 5–10 cm in the maize rhizosphere. Five points were chosen according to the “S” form five-spot sampling method (Lu et al. 2013). Then, 0.1 kg of soil was collected at each point and pooled into one composite soil sample.
To isolate bacteria, soil samples were placed in paper bags and stored at 4 °C. Next, 100 mL of sterile water and 0.01 kg of soil were transferred to a Waring blender. The sample was homogenized for 1 min and the supernatant was collected. The supernatant was centrifuged for 10 min at 5000×g and re-dissolved in 0.5 mL of sterile water. Samples diluted to 10−3, 10−4, and 10−5 were streaked onto soil extract agar plates containing 5 g/L of beef extract, 10 g/L of peptone, 5 g/L of NaCl, 100 g/L of mannitol, 15 g/L of agar, and 1 L of distilled water (pH 7.2). After 2 days of incubation at 30 °C, colonies of different morphologies and sizes were frozen in 15% glycerol and stored at −80 °C.
Identification of sequence similarity
To identify the colony strains, the bacteria were first identified by Gram staining. Then, a similarity search with the 16S rRNA gene nucleotide sequence (accession number MH490935) was performed using the online program EzBioCloud (Yoon et al. 2017) and two housekeeping genes (recA and atpD) were used to identify the strains. Finally, the average nucleotide identity (ANI) was calculated using JSpeciesWS (Richter et al. 2016) to identify the interspecific relationships among strains.
Genomic DNA preparation, genome sequencing, and assembly
For genomic DNA isolation, strains were inoculated into 50 mL of liquid medium and cultivated overnight at 30 °C in a shaker at 150 rpm. Genomic DNA was then extracted using a Rapid Bacterial Genomic DNA Isolation Kit (Sangon Biotech Shanghai Co., Ltd., Shanghai, China).
Whole-genome sequencing was performed using a combination of the PacBio RS and Illumina sequencing platforms. The Illumina PE library and PacBio Library (8–10 kb) were each constructed. First, 52 Mb of continuous long read (CLR) PacBio sequences were sequenced on the PacBio RS platform using an SMRT cell with a sequence N50 length of 12,035 bp, of which 47.2 Mb of sequences had lengths ≥ 5 kb. Then, a 400 bp Illumina sequencing library was constructed, and 2.9 Gb of paired-end sequences were sequenced on the Illumina HiSeq 2000 platform. The Illumina data were used to evaluate the complexity of the genome and were assembled using Velvet ver. 1.2.10 genome assembler with a k-mer length of 99 (Zerbino and Birney 2008).
The complete genome sequence was assembled using both the PacBio and Illumina reads. The assembly was initially produced using an in-house assembly solution, in which a de Bruijn-based assembly algorithm and CLR correction algorithm were integrated. The final circularization step was completed manually using Circos ver. 0.64 software (http://circos.ca/) (Krzywinski et al. 2009).
Genome annotation
Prodigal software (Hyatt et al. 2010) was used to predict bacterial genes. Then, the protein sequences of the predicted genes were compared with the Clusters of Orthologous Groups (COG) protein database (http://www.ncbi.nlm.nih.gov/COG/) (Tatusov et al. 2003), and the corresponding COG annotation results were obtained. BLAST (blastx/blastp 2.2.24+) was used to compare the predicted genes with the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/genes.html). Specific pathways involving the corresponding genes were obtained according to the KEGG Orthology number in the comparison results. Finally, the BLAST results were analyzed using Gene Ontology annotations (http://www.geneontology.org/) with Blast2go.
Identification of B. frigoritolerans ZB201705 abiotic stress responses
For the anti-abiotic stress susceptibility tests (e.g., salt and drought), different NaCl concentrations (1, 3, and 5 M) and d-sorbitol concentrations (0, 1, 2, and 3 M) were tested on LB agar plates. For the salt and drought stress challenges, cultures were multiplied at 30 °C to a density of about 5 × 107 cells/mL. The cell concentrations were determined by plating appropriate dilutions onto agar containing 1, 3, and 5 M NaCl or 1, 2, or 3 M d-sorbitol for at least 12 h at 30 °C.
Statistical analyses
The obtained data were subjected to an analysis of variance using the general linear model software Agres and Agdata, and means were compared using the least significant difference test at a probability level ≤ 0.05.
Results
Organism information and classification
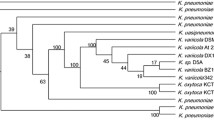
According to the Gram stain results, the cells were Gram-positive bacteria (Fig. S1). Based on the similarity of the 16S rRNA gene sequence determined using EzBioCloud, twelve strains with the greatest similarity were selected. The results (Table 1) showed 100% identity with B. frigoritolerans FJAT-2396, 98% with Bacillus psychrosaccharolyticus ATCC 23296, and 97% with Bacillus manliponensis JCM 15802. Nevertheless, analyses based on 16S rRNA gene sequencing alone are insufficient for species classification within this group. Therefore, the identities of two housekeeping genes (recA and atpD) in B. frigoritolerans ZB201705 and 12 other type strains were determined, which revealed two identities > 90% (Table 1). To classify the strains further, the twelve most similar strains were selected and their whole genome sequences were analyzed with ANI using JSpeciesWS (Richter et al. 2016). The highest ANI value was 97.1% for B. frigoritolerans FJAT-2396, which exceeded the species cutoff threshold (95%) (Goris et al. 2007). The whole genome sequences of the two strains were compared using Circos ver. 0.22 (Krzywinski et al. 2009), which revealed numerous differences between the two strains (Fig. 1). A comparison with the available type strain genomes indicated that our strain should be considered a subspecies of B. frigoritolerans. Based on these organism information and classification, we named the strain B. frigoritolerans ZB201705.

Comparison of whole genome sequence from the two strains was performed using the software Circos V0.22
Genome sequencing results
To obtain a genome sequence with no gaps, we used a combination of the PacBio RS and Illumina sequencing platforms. The assembled results contained 350 contigs with lengths ≥ 200 bp, with an N50 length of 53,736 bp, and indicated that the B. frigoritolerans ZB201705 genome consists of one circular chromosome with a size of 5,475,560 bp (Fig. 2) and one plasmid sequence (Fig. S2), with no gaps. The guanine–cytosine content of the genome was 40.5%. In total, 5,266 genes were predicted, accounting for 79.73% of the genome. Moreover, 39 ribosomal RNAs and 81 transfer RNAs were detected. Of the total number of predicted genes (5,266), 4,391 (83.38%) were putative protein-coding genes and 72.52% were assigned a putative function (Table 2).

The circular chromosome of Brevibacterium frigoritolerans ZB201705
Based on the COG analysis (Tatusov et al. 2003), the identified proteins were classified into 25 functional categories (Fig. 3). Among these categories, many proteins were involved in salt and drought stress responses. For example, the coenzyme transport and metabolism category (H) contained a sodium/proline symporter (Jung et al. 2012), the signal transduction mechanism category (T) contained the signal transduction histidine-protein kinase/phosphatase DegS (Kunst and Rapoport 1995), and the transcription category (K) contained the transcriptional regulatory protein DegU (Kunst and Rapoport 1995) and cold-shock protein CspB, which improves maize grain yield under water-limited conditions (Castiglioni et al. 2008). A previous study indicated that stress-induced reactive oxygen species accumulation is counteracted by enzymatic antioxidant systems comprised of a variety of scavengers, including superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX) (Wang et al. 2017). According to the COG functional categories of the genome, there were several related genes. For example, the inorganic ion transport transmembrane category (P) contained three SOD- and three CAT-related genes, and the secondary metabolite biosynthesis, transport, and catabolism category (O) contained one GPX-related gene. These results suggest that B. frigoritolerans ZB201705 has an important role in environmental pathogen defense and abiotic stress responses.

COG functional classifications of Brevibacterium frigoritolerans ZB201705 coding sequences
Activity of the strain under high salt and simulated drought conditions
We identified several environmental abiotic stress-related genes based on our COG analysis. With the aim of detecting its resistance to high salt and drought conditions, we cultured B. frigoritolerans ZB201705 on LB media with different concentrations of NaCl and d-sorbitol. The strain can tolerate up to 3 M d-sorbitol (~ 55%, w/v) and 5 M NaCl (~ 29%, w/v) with survival rates of 3.5% and 12.6%, respectively (Fig. 4).

Identification of the activity of Brevibacterium frigoritolerans ZB201705 under salt and drought stress
Nucleotide sequence accession numbers
The complete genomic sequence and 16S rRNA gene sequence of B. frigoritolerans ZB201705 have been deposited in the GenBank database under Accessions CP030063 and MH490935.
Discussion
Brevibacterium has been isolated from numerous environments, including saline environments such as saline soil, beach sediment, marine environments, and salt-lake sediment (Lee 2006; Lee 2008; Tang et al. 2008; Guan et al. 2010). However, few studies have referred specifically to B. frigoritolerans. One strain of B. frigoritolerans, Imbl 2.1, has shown potential for the bioremediation of phorate (Jariyal et al. 2015). Another strain, SMA23, may improve growth and nutrient uptake in wheat (Meena and Saharan 2017). As for plant growth-promoting bacteria (PGPB), free-living plant growth-promoting bacteria form symbiotic relationships with specific plants or bacterial endophytes that colonize some or part of the interior tissues of plants (Basu et al. 2017). Therefore, PGPB are generally used as inoculants for the biostimulation, biocontrol, and biofertilization of plants, and may improve plant growth under various environmental stress conditions (Numan et al. 2018).
The present study shows that B. frigoritolerans ZB201705 produces a large number of proteins involved in salt and drought stress responses, including DegS, DegU, CspB, and SOD. Anti-abiotic stress susceptibility tests revealed that this strain can tolerate up to 3 M d-sorbitol (~ 55%, w/v) and 5 M NaCl (~ 29%, w/v). These results suggest that B. frigoritolerans ZB201705 is a vital bacterium with potential environmental abiotic stress resistance applications, including the promotion of salt and drought tolerance in maize. The information provided here will provide valuable insight into PGPB for agriculture under abiotic stress.
References
Basu S, Rabara R, Negi S (2017) Towards a better greener future - an alternative strategy using biofertilizers. I: Plant growth promoting bacteria. Plant Gene 12:43–49
Breed RS (1953) The families developed from Bacteriaceae Cohn with a description of the family Brevibacteriaceae. Riass. Commun. VI Congr. Int Microbiol Roma 1:10–15
Castiglioni P, Warner D, Bensen RJ, Anstrom DC, Harrison J, Stoecker M, Abad M, Kumar G et al (2008) Bacterial RNA chaperones confer abiotic stress tolerance in plants and improved grain yield in maize under water-limited conditions. Plant Physiol 147:446–455
Chatterjee P, Samaddar S, Niinemets Ü, Sa TM (2018) Brevibacterium linens RS16 confers salt tolerance to Oryza sativa genotypes by regulating antioxidant defense and H+ ATPase activity. Microbiol Res 215:89–101
Choi EJ, Lee SH, Jung JY, Jeon CO (2013) Brevibacterium jeotgali sp. nov., isolated from jeotgal, a traditional Korean fermented seafood. Int J Syst Evol Microbiol 63:3430–3436
Choi KD, Siddiqi MZ, Liu Q, Muhammad Shafi S, Durrani Y, Lee SY, Kang MS, Im WT (2018) Brevibacterium hankyongi sp. nov., isolated from compost. Int J Syst Evol Microbiol 68:2783–2788
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM (2007) DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91
Guan TW, Zhao K, Xiao J, Liu Y, Xia ZF, Zhang XP, Zhang LL (2010) Brevibacterium salitolerans sp. nov., an actinobacterium isolated from salt-lake sediment. Int J Syst Evol Microbiol 60:2991–2995
Hyatt D, Chen GL, LoCascio PF, Land ML, Larimer FW, Hauser LJ (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119
Jariyal M, Gupta VK, Mandal K, Jindal V (2015) Brevibacterium frigoritolerans as a novel organism for the bioremediation of phorate. Bull Environ Contam Toxicol 95:680–686
Jung H, Hilger D, Raba M (2012) The Na+/L-proline transporter PutP. Front Biosci 17:745–759
Jung MS, Quan XT, Siddiqi MZ, Liu Q, Kim SY, Wee JH, Im WT (2018) Brevibacterium anseongense sp. nov., isolated from soil of ginseng field. J Microbiol 56:1–7
Krzywinski M, Schein J, Birol Ì, Connors J, Gascoyne R, Horsman D, Jones SJ, Marra MA (2009) Circos: an information aesthetic for comparative genomics. Genome Res 19:1639–1645
Kunst F, Rapoport G (1995) Salt stress is an environmental signal affecting degradative enzyme synthesis in Bacillus subtilis. J Bacteriol 177:2403–2407
Leclercq-Perlat MN, Oumer A, Bergere JL, Spinnler HE, Corrieu G (2000) Behavior of Brevibacterium linens and Debaryomyces hansenii as ripening flora in controlled production of smear soft cheese from reconstituted milk: growth and substrate consumption. J Dairy Sci 83:1665–1673
Lee SD (2006) Brevibacterium samyangense sp. nov., an actinomycete isolated from a beach sediment. Int J Syst Evol Microbiol 56:1889–1892
Lee SD (2008) Brevibacterium marinum sp. nov., isolated from seawater. Int. J Syst Evol Micro 58:500–504
Lu S, Quan W, Wang SM, Liu HL, Tan Y, Zeng GP, Zhang X (2013) Correlation of soil microbes and soil micro-environment under long-term safflower (Carthamus tinctorius L.) plantation in China. J Environ Biol 34:471–479
Meena TN, Saharan BS (2017) Plant growth promoting traits shown by bacteria Brevibacterium frigrotolerans SMA23 isolated from Aloe vera rhizosphere. Agr Sci Digest 37:226–231
Morari F, Meggio F, Lunardon A, Scudiero E, Forestan C, Farinati S, Varotto S (2015) Time course of biochemical, physiological, and molecular responses to field-mimicked conditions of drought, salinity, and recovery in two maize lines. Front Plant Sci 6:314
Numan M, Bashir S, Khan Y, Mumtaz R, Shinwari ZK, Khan AL, Khan A, AL-Harrasi A (2018) Plant growth promoting bacteria as an alternative strategy for salt tolerance in plants: a review. Microbiol Res 209:21–32
Richter M, Rosselló-Móra R, Oliver Glöckner F, Peplies J (2016) JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32:929–931
Tang SK, Wang Y, Schumann P, Stackebrandt E, Lou K, Jiang CL, Xu LH, Li WJ (2008) Brevibacterium album sp. nov., a novel actinobacterium isolated from a saline soil in China. Int. J Syst Evol Micro 58:574–577
Tatusov RL, Fedorova ND, Jackson JD, Jacobs AR, Kiryutin B, Koonin EV, Krylov DM, Mazumder R et al (2003) The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41
Wang Y, Wu YP, Wang YY, Xu H, Mei XQ, Yu DY, Wang YB, Li WF (2017) Antioxidant properties of probiotic bacteria. Nutrients 9:521
Wauters G, Charlier J, Janssens M, Delmée M (2001) Brevibacterium paucivorans sp. nov., from human clinical specimens. Int J Syst Evol Microbiol 51:1703–1707
Wauters G, Avesani V, Laffineur K, Charlier J, Janssens M, Bosterhaut BV, Delmée M (2003) Brevibacterium lutescens sp. nov., from human and environmental samples. Int. J Syst Evol Micro 53:1321–1325
Yoon SH, Ha SM, Kwon S, Lim J, Kim Y, Seo H, Chun J (2017) Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int J Syst Evol Microbiol 67:1613–1617
Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18:821–829
Funding
This work was supported by the Beijing Academy of Agriculture and Forestry Sciences (Grant number KJCX20170404, KJCX20170203 and QNJJ201724) and the Beijing Association for Science and Technology.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Research involving human participants and/or animals
This article does not contain any studies with human participants or animals performed by any of these authors.
Informed consent
Informed consent is not required in this work.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 323 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Zhang, C., Li, X., Yin, L. et al. Analysis of the complete genome sequence of Brevibacterium frigoritolerans ZB201705 isolated from drought- and salt-stressed rhizosphere soil of maize. Ann Microbiol 69, 1489–1496 (2019). https://doi.org/10.1007/s13213-019-01532-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13213-019-01532-0