
Abstract
Nitrile hydratase (NHase; E.C. 4.2.1.84) has been purified and characterized using ammonium sulfate precipitation, ion exchange chromatography and gel filtration chromatography from the mutant 4D of Rhodococcus rhodochrous PA-34. The SDS-PAGE and MALDI-TOF analysis of the purified enzyme revealed that it is dimmer consisting of α- and β-subunits with a molecular mass of 25 and 30 kDa, respectively. The Km and Vmax values were 102 mM and 350.8 μmol/min/mg using 3-cyanopyridine as substrate. The purified NHase was stable in higher concentration of potassium ions and in acidic pH 5.5 as compared to NHase of the wild R. rhodochrous PA-34. The analysis of the N-terminal amino acid sequence of this enzyme revealed that this enzyme has 90 % homology with the high molecular weight nitrile hydratase of R. rhodochrous J1.
Similar content being viewed by others

Introduction
The nitrile metabolism in microorganisms mainly involves nitrilase, nitrile hydratase and amidase enzymes that convert nitriles to amides or acids (Asano et al. 1980; Bhalla et al. 1992; Yamada and Kobayashi 1996). This enzyme is one of the first enzymes used in industry for the production of a commodity chemical, i.e., Acrylamide (Raj et al. 2006, 2007; Prasad et al. 2007, 2009). Besides this a number of commercially important products such as nicotinamide, pyrazinamide, thiophenamide etc. have also been synthesized from the nitriles using this enzyme (Raj et al. 2006, 2007; Prasad et al. 2007, 2009; Kobayashi et al. 1992). Yet, there are very few organisms whose nitrile hydratase has shown promise for industrial application (Raj et al. 2006, 2007; Prasad et al. 2007; Kobayashi et al. 1992). The genetically modified organisms or mutants generated through chemical or physical mutagenesis having higher activity have not been either generated or perfected for the large scale applications in the conversion of nitriles to corresponding amides (Pratush et al. 2010).
Rhodococcus rhodochrous PA-34 has been reported as a potential organism that can convert acrylonitrile, butyronitrile and 3-cyanopyridine to corresponding amides at a high concentration (Raj et al. 2006, 2007; Prasad et al. 2007; Bhalla and Kumar 2005). A mutant 4D has been generated by chemical mutagenesis of R. rhodochrous PA-34 using MNNG that exhibited twofold increase in its nitrile hydratase activity for the conversion of 3-cyanopyridine to nicotinamide (Pratush et al. 2010). In this communication, we report the purification and characterization of nitrile hydratase of mutant 4D of R. rhodochrous PA-34.
Materials and methods
Chemicals
Nicotinamide and 3-cyanopyridine used in the present study were purchased from Sigma and Alfa Aesar, respectively. Other chemicals used in the present study were of analytical grade from various commercial sources.
Microorganism
The mutant 4D of R. rhodochrous PA-34 generated earlier by Pratush et al. (2010) at the Department of Biotechnology, Himachal Pradesh University, Shimla, India.
Culture, nitrile hydratase assay and estimation of nicotinamide
Mutant 4D cells were grown for the production of nitrile hydratase by following the procedure as detailed by Pratush et al. (2010). The NHase activity was assayed in 1 ml reaction containing 880 μl, 0.3 M Potassium phosphate buffer (pH 5.5), 20 μl (0.08 mg/ml) of enzyme and 100 μl of substrate 0.5 M 3-cyanopyridine following the method reported previously (Prasad et al. 2004).
Purification of nitrile hydratase of mutant strain
All steps of purification were performed at 4 °C and 0.3 M potassium phosphate buffer pH 5.5 at 4 °C was used.
-
1.
Homogenization of resting cells of R. rhodochrous PA-34 mutant 4D
The resting cells of mutant 4D were disrupted using bead beater (BSP make) having Zirconium beads (0.1 mm diameter) for 30 min in 10 disruption cycles at 4 °C. The resulting cell-free extract (CFE) was used as crude enzyme for subsequent NHase purification.
-
2.
Ammonium sulfate fractionation of CFE
The cell-free extract was subjected to various % saturation concentration of ammonium sulfate (0–80 %). The fraction exhibiting maximum activity of NHase was termed as ASF and was taken for further purification of NHase.
-
3.
Gel filtration of ASF
Ammonium sulfate fractionation was filtered through 0.45-μm filter and directly loaded on to pre-packed Sephacryl S-300 gel filtration column (16 mm diameter × 600 mm length) equilibrated with buffer. The gel filtration chromatography was performed using AKTA prime™ V2.00 at a flow rate 0.3 ml/min of elution buffer (0.3 M potassium phosphate buffer pH 5.5). NHase active fractions were subjected to SDS polyacrylamide gel electrophoresis (PAGE). The fractions exhibiting NHase activity were pooled. These pooled fractions were termed as GFF and used for further purification of NHase.
-
4.
DEAE-ion exchange chromatography of GFF
The pooled filtered fractions of gel filtration (GFF) applied on an DEAE to an ion-exchange chromatography column (16 mm diameter × 100 mm length) equilibrated with 0.3 M potassium phosphate buffer pH 5.5. The column was eluted with a linear gradient of NaCl from 0.1 to 0.7 M in 0.3 M potassium phosphate buffer. NHase activity and protein concentration were estimated in each fraction. The fractions exhibiting single band on native-PAGE were pooled and termed as DEAEF (i.e., purified NHase).
Characterization of purified NHase
Buffer, pH, temperature, substrate specificity and effect of metal ions and inhibitors on activity of purified NHase of mutant 4D
Various buffer systems (such as sodium phosphate and Tris–HCl and potassium phosphate buffer each 0.1 M pH 7.2) were used to select a suitable buffer to assay the activity of the purified NHase. The ionic strength of buffer (0.1–0.5 M potassium phosphate buffer) and pH (5.0, 5.5, 6.0, 6.5, 7.0, 7.5, 8.0, and 8.5) of selected buffer (potassium phosphate buffer) were optimized for NHase assay.
To determine temperature optimum of NHase of mutant 4D, the activity of this enzyme was determined at 10–80 °C. Thermal stability and substrate specificity profile of the purified mutant 4D NHase were tested after an interval of 20 min by subjecting purified NHase to 45, 55 and 65 °C for 8 h with different substrates, i.e., 2-cyanopyridine, 3-cyanopyridine, 4-cyanopyridine, butyronitrile, benzonitrile and acrylonitrile, respectively.
The effects of metal ions (AgNO3, CaCl2, CdCl2, CoCl2, CuCl2, FeCl2, HgCl2, MgCl2 and MnCl2) and inhibitors (ammonium persulfate (APS), DTT, EDTA, hydroxylamine, iodoacetic acid, l-ascorbic acid, phenyl hydrazine, phenylmethanesulphonylfluoride, semicarbazide, sodium azide and urea) on NHase activity of mutant were investigated by pre-incubating the enzyme at 1 mM concentration of metal ions/inhibitors for 30 min at 55 °C and then the NHase activity was assayed.
Determination of Km and Vmax of NHase
The Km and Vmax of purified NHase of mutant 4D was calculated by determining initial velocity (v) of NHase at various concentrations of 3-cyanopyridine.
Determination of molecular mass of NHase N-terminal amino acid sequencing and MALDI-TOF analysis of purified NHase of R. rhodochrous PA-34 mutant 4D and its analysis
SDS and native-PAGE were carried out to determine the purity, molecular mass of NHase and its subunits by the method of Laemmli (1970). The N-terminal amino acid sequence of purified NHase of mutant 4D was done at the Institute of Microbial Technology (IMTECH), Sector 39A, Chandigarh (India). Matrix assisted laser dissociated ionization-time of flight (MALDI-TOF) analysis of purified NHase was done at Jawaharlal Nehru University, New Delhi (India). Multiple protein sequence alignment was carried out using Clustal W program (Thompson et al. 1997; Chenna et al. 2003).
Results and discussion
Purification of mutant 4D NHase
The disruption of 2.0 g of resting cells of mutant 4D (containing 11,240 U of NHase activity) released 119 mg of protein and 1,000 U NHase activity in CFE. The purification of NHase of mutant 4D from the CFE involved ammonium sulfate fractionation (ASF), gel filtration chromatography (GFF) on Sephacryl S-300, DEAE-ion exchange chromatography (DEAEF). NHase protein was precipitated at 30–40 % saturation of ammonium sulfate (ASF), and it contained 90.7 mg protein with specific activity of 9.2 U/mg proteins (Table 1). Ammonium sulfate fractionation (30–40 % saturation cut) resulted in onefold purification of enzyme (AFS) with an yield of 83 % of NHase activity (Table 1). The NHases of R. rhodochrous PA-34 (Prasad et al. 2009), R. rhodochrous J1 (low and high molecular weight both) (Wieser et al. 1998; Nagasawa et al. 1991) and Rhodococcus sp. strain YH3-3 (Kato et al. 1999), Brevibacterium R312 (Nagasawa et al. 1986) and Pseudomonas chlororaphis B23 (Nagasawa et al. 1987) were precipitated in the 30–70 % and 40–55 % cut off ammonium sulfate, respectively.
The dialysed protein of ASF was subjected to gel permeation chromatography using S-300 column (Fig. 1a). This step resulted in 1.7-fold of purification with an yield of 82 % having specific NHase activity of 15.2 U/mg protein. The fraction of GFF reached in NHase proteins was further loaded to the DEAE-ion exchange column (Fig. 1b), which resulted in the threefold purification of enzyme with a yield of 62 % and specific activity of 45 U/mg protein (Table 1). Earlier, the NHase of R. rhodochrous J1, Agrobacterium tumefaciens strain d3, B. pallidus Dac521, P. chlororaphis B23 and Brevibacterium R312 were purified by employing gel permeation chromatography techniques (Wieser et al. 1998; Nagasawa et al. 1986, 1987; Bauer et al. 1998; Cramp and Cowan 1999).

a Protein and NHase activity profile during gel permeation chromatography. b Protein and NHase activity profile by DEAE ion exchange chromatography
Characterization of NHase of mutant 4D
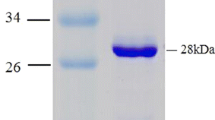
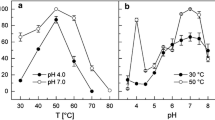
The purified NHase consisted of two polypeptides one comprising 25 kDa (α-subunit) and other was of 30.6 kDa (β-subunit) (Fig. 2a). It means that this enzyme is also constituted by two different polypeptides (α- and β-subunits) similar to the earlier reported NHases (Prasad et al. 2009; Wieser et al. 1998; Nagasawa et al. 1986). Native PAGE of purified NHase revealed a single band of 86 kDa (Fig. 2b). This indicated that the functional NHase might be constituted by one α- and two β-subunits (i.e., αβ2). The molecular mass of nitrile hydrates of different organisms varies from species to species, i.e., R. rhodochrous J1, R. rhodochrous sp. N774 and Cornybacterium sp. C5 have 59, 70 and 61 kDa, respectively (Kobayashi et al. 1991; Endo and Watanabe 1989; Yamamoto et al. 1992). The Km and Vmax values for the purified NHase of mutant 4D were 102 mM and 350.8 μmol/min/mg, respectively, using 3-cyanopyridine as substrate (Fig. 3), whereas Km and Vmax reported from other sources such as R. rhodochrous PA-34, L and H-NHase of R. rhodochrous J1 were 167 mM and 250 μmol/min/mg, 0.30 mM and 579 μmol/min/mg and 200 mM and 370 μmol/min/mg using 3-cyanopyridine as substrate, respectively (Prasad et al. 2009; Wieser et al. 1998). These results indicated that this enzyme had higher affinity for 3-cyanopyridine and had higher Vmax in comparison to its wild strain. Among the three types of buffers tested, maximum NHase activity (225 U) was rerecorded in 0.3 M potassium phosphate buffer (Fig. 4a). Below and above 0.3 M concentration of the buffer the activity of NHase drastically decreased (Fig. 4b). The mutant NHase was found to be stable in acidic conditions, i.e., pH 5.5 (Fig. 4c). These results are drastically different from the earlier reports of Banerjee et al. (2002). Moreover, this NHase exhibited higher concentration of potassium ions, i.e., 0.3 M (Fig. 4b), whereas other reported NHases showed higher activity at 0.1 M potassium phosphate buffer (Raj et al. 2006; Prasad et al. 2009). The optimum temperature for assay of NHase activity turns out to be 55 °C (Fig. 4d), whereas it is 40 °C for R. rhodochrous PA-34 and R. rhodochrous J1 (Prasad et al. 2009; Wieser et al. 1998). Most of the nitrile hydratases have exhibited maximum activity near ambient temperature between 20 and 35 °C (Banerjee et al. 2002). The thermophilic NHases of Bacillus RAPc8 and Pseudonocardia thermophila showed maximum activity at 60 °C (Yamaki et al. 1997; Pereira et al. 1998).

a Sodium dodecyl sulfate-polyacrylamide gel electrophoresis of NHase of mutant 4D at various steps of purification. SDS-protein molecular weight markers in kDa = kilo Dalton (lane1), cell-free protein extract (lane2), ammonium sulfate precipitation sample (lane3), gel filtration chromatography samples (lane4) and DEAE-ion exchange chromatography sample (lane5) SDS PAGE of purified NHase of mutant. b Native-PAGE analysis of purified NHase of mutant 4D, lane1 was loaded with following molecular mass standards: thyroglobulin (660 kDa), ferritin (440 kDa), catalase (232 kDa), lactate dehydrogenase (140 kDa) and albumin (66 kDa). Cell-free extract (lane2), ammonium sulfate fraction (lane3) and gel permeation column chromatography fraction (lane4) DEAE-ion exchange column chromatography fraction (lane5)

Lineweaver Burk plot of purified NHase of mutant

a Buffer system optimization for purified NHase of mutant. b Ionic strength optimization of buffer for purified NHase of mutant. c Buffer pH optimization for purified NHase of mutant. d Reaction temperature optimization for purified NHase of mutant. e Thermostability profile of purified NHase of mutant at 45, 55 and 65 °C. f Effect of different substrates on purified NHase of mutant
The NHase of mutant 4D showed its maximum activity at 55 °C and it remained stable up to 5 h at this temperature, whereas the enzyme completely lost its activity at 65 °C (Fig. 4e). However, a thermophilic NHase from B. pallidus Dac 521 had a half-life of 0.85 h at 50 °C. The NHase from mutant 4D was much more stable than earlier reported NHase at higher temperature (Banerjee et al. 2002). NHase of mutant 4D showed highest activity with 3-cyanopyridine (Fig. 4f). The purified NHase of mutant 4D, showed almost complete loss of activity in the presence of metal ions. The NHase activity was inhibited up to 65.5–73.7 % by MgCl2 and hydroxyl amine, respectively. An inhibition of 30.5 % in NHase activity was observed in the presence of PMSF in reaction (Table 2). The loss of activity of NHase of mutant 4D in the presence of metal ions indicated the involvement of Cys residue at its active site, which might have got complexed with metal ions leading to inactivation of the enzyme.
The N-terminal sequence of β- and α-subunits of mutant 4D NHase showed a significant (94 %) change in α-subunit when compared with the N-terminal sequence of α-subunit of wild R. rhodochrous PA-34, whereas the N-terminal sequence of β-subunit of mutant showed 90 % homology with the β-subunit of wild (Table 3). MALDI-TOF analysis revealed that purified NHase of mutant 4D had 90 % homology with the high molecular weight NHase of R. rhodochrous J1 (Kobayashi et al. 1991). The N-terminal and MALDI-TOF analysis of mutant 4D NHase protein revealed a significant change in the amino acid sequence of α- and β-subunits of wild strain and it exhibited high homology with H-NHase reported earlier from R. rhodochrous J1 (Kobayashi et al. 1991). These studies suggest that the wild strain R. rhodochrous PA-34 produces the L-NHase (low molecular weight nitrile hydratase), whereas the R. rhodochrous PA-34 mutant 4D produces H-NHase generated through chemical mutagenesis using MNNG. The chemical mutagenesis has switched off the expression of L-NHase gene and switch on the expression of H-NHase gene. However, further studies on cloning and sequencing of NHase operon of the wild and mutant 4D strains of R. rhodochrous PA-34 are needed further to explore this aspect at molecular level.
Conclusion
The NHase of R. rhodochrous PA-34 of mutant 4D produces high molecular nitrile hydratase with better thermal, ionic and acidic pH stabilities as compared to earlier reported nitrile hydratases including the NHase of wild strain.
References
Asano Y, Tani Y, Yamada H (1980) A new enzyme ‘nitrile hydratase’ which degrades acetonitrile in combination with amidase. Agri Biol Chem 44:2251–2252
Banerjee A, Sharma R, Banerjee UC (2002) The nitrile-degrading enzymes: current status and future prospects. Appl Microbiol Biotechnol 60:33–44
Bauer R, Knackmuss HJ, Stolz A (1998) Enantioselective hydration of 2-arylpropionitriles by a nitrile hydratase from Agrobacterium tumefaciens strain d 3. Appl Microbiol Biotechnol 49:89–95
Bhalla TC, Kumar H (2005) Nocardia globerula NHB-2: a versatile nitrile-degrading organism. Can J Microbiol 51:705–708
Bhalla TC, Miura M, Wakamoto A, Ohba Y, Furuhashi K (1992) Asymmetric hydrolysis of α-aminonitriles to optically active amino acids by a nitrilase of Rhodococcus rhodochrous PA-34. Appl Microbiol Biotechnol 37:184–190
Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, Thompson JD (2003) Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res 31:3497–3500
Cramp RA, Cowan DA (1999) Molecular characterisation of a novel thermophilic nitrile hydratase. Biochem Biophys Acta (BBA) Protein Struct Mol Enzymol 1431:249–260
Endo T, Watanabe I (1989) Nitrile hydratase of Rhodococcus sp. N-774 purification and amino acid sequences. FEBS Lett 243:61–64
Kato Y, Tsuda T, Asano Y (1999) Nitrile hydratase involved in aldoxime metabolism from Rhodococcus sp. strain YH3-3 purification and characterization. Eur J Biochem 263:662–670
Kobayashi M, Nishiyama M, Nagasawa T, Horinouchi S, Beppu T, Yamada H (1991) Cloning, nucleotide sequence and expression in Escherichia coli of two cobalt-containing nitrile hydratase genes from Rhodococcus rhodochrous J1. Biochem Biophys Acta 1129:23–33
Kobayashi M, Nagasawa T, Yamada H (1992) Enzymatic synthesis of acrylamide: a success story not yet over. Trends Biotechnol 10:402–408
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685
Nagasawa T, Ryuno K, Yamada H (1986) Nitrile hydratase of Brevibacterium R312-purification and characterization. Biochem Biophys Res Comm 139:1304–1312
Nagasawa T, Nanba H, Ryuno K, Takeuchi K, Yamada H (1987) Nitrile hydratase of Pseudomonas chlororaphis B23-purification and characterization. Eur J Biochem 162:691–698
Nagasawa T, Takechi K, Nardidei V, Mihara Y, Yamada H (1991) Optimum culture conditions for the production of cobalt-containing nitrile hydratase by Rhodococcus rhodochrous J1. Appl Microbiol Biotechnol 34:783–788
Pereira RA, Graham D, Rainey FA, Cowan DA (1998) A novel thermostable nitrile hydratase. Extremophiles 2:347–357
Prasad S, Raj J, Bhalla TC (2004) Optimization of culture conditions for hyper production of nitrile hydratase of Rhodococcus rhodochrous PA-34. Indian J Microbiol 44:251–256
Prasad S, Raj J, Bhalla TC (2007) Bench scale conversion of 3-cyanopyridine to nicotinamide using resting cells of Rhodococcus rhodochrous PA-34. Indian J Microbiol 47:34–41
Prasad S, Raj J, Bhalla TC (2009) Purification of a hyperactive nitrile hydratase from resting cells of Rhodococcus rhodochrous PA-34. Indian J Microbiol 49:237–242
Pratush A, Seth A, Bhalla TC (2010) Generation of mutant of Rhodococcus rhodochrous PA-34 through chemical mutagenesis for hyperproduction of nitrile hydratase. Acta Microbiol Immun Hungarica 57(2):135–146
Raj J, Prasad S, Bhalla TC (2006) Rhodococcus rhodochrous PA-34 a potential catalyst for acrylamide synthesis. Process Biochem 41(6):1359–1363
Raj J, Seth A, Prasad S, Bhalla TC (2007) Bioconversion of butyronitrile to butyramide using whole cells of Rhodococcus rhodochrous PA-34. Appl Microbiol Biotechnol 74:535–539
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997) The ClustalX windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Wieser M, Takeuchi K, Wada Y, Yamada H, Nagasawa T (1998) Low-molecular weight nitrile hydratase from Rhodococcus rhodochrous J1: purification, substrate specificity and comparison with the analogous high-molecular-mass enzyme. FEMS Microbiol Lett 169:17–22
Yamada H, Kobayashi M (1996) Nitrile hydratase and its application to industrial production of acrylamide. Biosci Biotechnol Biochem 60:1391–1400
Yamaki T, Oikawa T, Ito K, Nakamura T (1997) Cloning and sequencing of a nitrile hydratase gene from Pseudonocardia thermophila JCM 3095. J Ferment Bioeng 83:474–477
Yamamoto K, Fujimatsu I, Komatsu KI (1992) Purification and characterization of the nitrilase from Alcaligenes faecalis ATCC 8750 responsible for enantioselective hydrolysis of mandelonitrile. J Ferment Bioeng 73:425–430
Acknowledgments
The research work was supported by a research grant from University Grants Commission, Government of India, New Delhi, India. The authors also wish to acknowledge the internet facilities provided by Sub-Distributed Information centre, Department of Biotechnology, Himachal Pradesh University, Shimla-5, India.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pratush, A., Seth, A. & Bhalla, T.C. Purification and characterization of nitrile hydratase of mutant 4D of Rhodococcus rhodochrous PA-34. 3 Biotech 3, 165–171 (2013). https://doi.org/10.1007/s13205-012-0081-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13205-012-0081-5