
Abstract
A series of Ni–Co bimetallic catalysts supported on γ-Al2O3 with different Ni/Co mass ratio were prepared by incipient wetness impregnation method for glycerol hydrogenolysis. The catalyst with a Ni/Co mass ratio of 1:3 (denoted as Ni1Co3) exhibited the highest conversion. The performance was compared with that of catalysts promoted by Ce. Moreover, the addition of Ce showed a remarkable promoting effect on the catalytic performance when the cerium content was 2.5 wt%. The physicochemical properties of the supported Ni–Co catalysts were characterized by N2 physisorption, XRD, H2-TPR, NH3-TPD, XPS and TEM. H2-TPR profiles revealed that the coexistence of Ni and Co components on support changed the respective reduction behavior of Ni or Co alone, showing the synergistic effect between Ni and Co species. Compared with the TPR profiles of Ni1Co3, it was clearly observed that the reduction peak of nickel oxide and/or cobalt oxide shifted down to the lower temperature zone gradually with the addition of Ce. It was most probable that the addition of Ce favored the formation of the strong interaction between metal species and ceria. The TEM images showed that the addition of Ce component could improve the dispersion of Ni–Co species on support and inhibited the agglomeration of metal particles during the reaction process, which might be responsible for the enhanced stability.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
In recent years, increased consumption of fossil fuels and serious pollution problem make people pay much attention to the transformation of biomass and biomass-derived carbohydrates to fuels and value-added chemicals in a sustainable manner [1, 2]. The dramatic increase of biodiesel production by the transesterification of vegetable oils and animal fats enriched the yield of glycerol and consequently reduced its market price [3, 4]. Therefore, conversion of glycerol into value-added products can not only solve the glycerol surplus problem but also increase the profits of biodiesel manufacturing. Extensive investigations have been performed to convert glycerol to hydrogen and commodity chemicals. A promising route is catalytic hydrogenolysis of so-derived bio-glycerol to produce 1,2-propanediol (1,2-PDO) and ethylene glycol (EG), which are widely used as raw materials for polyester resins, food additives, cosmetics, pharmaceuticals and functional fluids. The hydrogenolysis of glycerol to glycols has been intensively studied for several years, and great progress has been made both in catalyst preparation and characterization [5, 6]. Different metal catalysts have been developed, such as noble metal catalysts (Ru, Rh, Pt, Pd, etc.) [7–10] as well as non-noble metal catalysts (Ni, Co, Cu, etc.) [11–14].
Among the catalysts used for glycerol hydrogenolysis, Ni-based catalysts are interesting materials since nickel is inexpensive and has the capability to break the C–C bond efficiently. Ueda [15] prepared a Ni/γ-Al2O3 catalyst promoted by a small amount of Pt which showed high selectivity of ethylene glycol from hydrogenolysis of glycerol. The performance was attributed to promotion of the retro-aldol reaction of glyceraldehydes and dissociation of the C–C bond by the Pt atoms on the Ni-rich Pt–Ni alloy surface. Meanwhile, Raksaphort et al. [16] studied the products distribution of glycerol hydrogenolysis over supported Co catalyst as well as the effect of support type and varied reaction conditions. The results disclosed that the acidity of catalyst played a more important role than the specific surface and pore volume. Among all the utilized catalysts, Co/γ-Al2O3 showed the highest acetol yield and glycerol conversion.
In general, bimetallic catalysts frequently exhibit better catalytic performances than their monometallic counterparts and have, therefore, attracted a great deal of attention from both academic and industrial fields [17]. The use of a second metallic component has proven to be a suitable method to improve the desired properties of catalysts in a variety of different applications. Yuan et al. [18] prepared a RuFe/CNT catalyst through a co-impregnation method for selective conversion of glycerol aqueous solution. It was indicated that higher performance of the RuFe/CNT catalyst was attributed to the synergistic effects of the formation of Ru–Fe alloys and the interactions between the RuFe bimetallic nanoparticles and iron oxides on CNT surfaces. On the other hand, promoters including alkali metal oxides and rare-earth metal oxides were usually introduced into the catalysts to improve the catalytic performance. Yu et al. [19, 20] prepared a type of Ni/AC catalyst and investigated the promoting effect of cerium on glycerol hydrogenolysis. The glycerol conversion could reach 90.4 % at 200 °C under 5 MPa of H2. Ceria was regarded as promoter that changed the reductive behavior of catalysts and accelerated the hydrogenation of acetol.
In the present work, a series of Ni–Co bimetallic catalysts supported on γ-Al2O3 were prepared to catalyze glycerol aqueous solution to produce glycols. The influence of Ni/Co mass ratio was investigated and then Ce was added to study the promoting effect. Techniques of catalyst characterization including XRD, TPR, XPS and TEM were carried out to discuss the structure–activity relationship of the Ni–Co/γ-Al2O3 bimetallic catalysts.
Experimental
Catalyst preparation
Ni–Co bimetallic catalysts were prepared by means of incipient wetness impregnation method. First, the purchased γ-Al2O3 (Sinopharm Chemical Reagent) was ground and sieved through 40–60 mesh. Desired amounts of Ni(NO3)2·6H2O and Co(NO3)2·6H2O were dissolved in deionized water and then impregnated with required amount of γ-Al2O3. After thorough stirring, the mixture was dried at 110 °C overnight in an oven and calcined at 500 °C for 4 h in air. Catalysts of 20.0 wt% Ni/γ-Al2O3, 16.7 wt% Ni–3.3 wt% Co/γ-Al2O3, 15.0 wt% Ni–5.0 wt% Co/γ-Al2O3, 10.0 wt% Ni–10.0 wt% Co/γ-Al2O3, 5.0 wt% Ni–15.0 wt% Co/γ-Al2O3, 3.3 wt% Ni–16.7 wt% Co/γ-Al2O3, 20.0 wt% Co/γ-Al2O3 were labeled as Ni1Co0, Ni5Co1, Ni3Co1, Ni1Co1, Ni1Co3, Ni1Co5, Ni0Co1, respectively.
In a similar procedure, Ce-modified Ni–Co bimetallic catalysts were prepared by adding desired amount of cerium nitrate into the aqueous solution containing nickel and cobalt before impregnation. 0, 2.5, 5.0, 7.5 and 10.0 wt% of ceria addition to Ni1Co3 were denoted as Ni1Co3, 2.5Ce–Ni1Co3, 5.0Ce–Ni1Co3, 7.5Ce–Ni1Co3 and 10.0Ce–Ni1Co3, respectively. Before use, the samples were reduced in a flowing stream of hydrogen at a steady rate of 50 ml/min in a tube furnace at 500 °C for 4 h.
Catalyst characterization
The specific surface area, average pore diameter and pore size distribution of the prepared catalysts were determined on a Micromeritics ASAP 2020 M instrument at liquid nitrogen temperature. Prior to N2 adsorption, all samples were degassed at 200 °C, 1.3 Pa for 6 h. Specific surface areas were determined by the multi-point Brunauer Emmet Teller (BET) method. Total pore volume and sizes were evaluated using the standard Barrett Joyner Halenda (BJH) treatment.
The X-ray diffraction (XRD) patterns of the prepared catalysts were carried out on a Rigaku D/max-2550 diffractometer using nickel-filtered Cu–Kα radiation (λ = 1.54056 Å) at room temperature. The X-ray tube was operated at 40 kV, 100 mA and scanned from 10° to 80°.
Hydrogen temperature-programmed reduction (H2-TPR) measurements were carried out on a Micromeritics AutoChem II 2920 instrument. Prior to the H2-TPR measurements, 0.20 g sample was placed in a quartz U-tube reactor, pretreated in argon stream at 300 °C for 60 min and then cooled to room temperature. Subsequently, a H2-Ar mixture (10 % H2 by volume) was fed to the reactor at a flow rate of 50 mL min−1 and the temperature was increased linearly from ambient to 800 °C at a ramp of 10 K min−1. The hydrogen consumption was continuously monitored by a thermal conductivity detector after the base line was stable.
Transmission electron microscope (TEM) images were taken on a JEM-2100 apparatus operated at 200 kV. The catalysts were ground through a 300 mesh, dispersed in ethanol by ultrasonic for 30 min and then a drop of the prepared solution was added to a carbon-coated copper grid. The samples were exposed to an infrared red lamp for 30 min to remove the ethanol before scanning.
XPS analysis was performed on reduced samples using a Perkin-Elmer PHI 5000C ESCA system, equipped with a monochromatic AlKα X-ray source, to determine the surface electronic states. The binding energy was calibrated by taking the C1s peak (284.6 eV) as a reference with the application of electron shower gun to reduce charging effect of the samples.
Activity testing
The hydrogenolysis reactions of glycerol aqueous solution were carried out in a 250-ml stainless steel autoclave equipped with stirrer, heater and sample port. Before reaction, the autoclave was purged with nitrogen to remove air for five times. After the reactor was heated to the required temperature, the desired hydrogen pressure was fed into and maintained during the reaction. The rotation rate of the stirrer was set constant at 500 rpm throughout the reaction. In a typical experiment, 2.0 g reduced catalyst and 100 ml of 20 wt% glycerol aqueous solution were loaded into the reactor. In order to investigate the influence of the reaction conditions, one factor was changed while the others were kept constant. After reaction, the reactor was cooled down to room temperature. The gas products were collected in a gas bag and analyzed by gas chromatograph (GC-900C, Shanghai) equipped with a 3-m TDX-01 stainless column and a thermal conductivity detector. Liquid products were filtrated to remove the solid catalyst powder and charged into a 10-ml glass vial and analyzed by a gas chromatograph (GC-900C, Shanghai) equipped with a flame ionization detector. A HP-INNOWAX column (30 m × 0.32 mm × 0.25 µm) was equipped to separate all components.
All products detected in the liquid were identified by a gas chromatography and mass spectrometry system (GC–MS, Agilent 6890) and quantified via an internal standard method. n-Butanol was used as the internal standard and DMF was used as the solvent during preparation of samples for analysis. Conversion of the glycerol was calculated as the mass ratio of the consumed glycerol in the reaction to the initial added glycerol. The selectivity was calculated based on the amount of products detected to be formed per the amount of glycerol actually reacted. For example, if 1 mol of glycerol was converted into 1 mol of ethylene glycol and 1 mol of methane, the selectivity of ethylene glycol and methane was calculated as 66.7 and 33.3 %, respectively. In this paper, the error of reproduction was measured by carbon balance, which was defined as (the total amount of glycerol, 1,2-PDO, EG, 1-O, 2-PO, C1 product)/(the amount of added glycerol). All the carbon balance in this work can reach more than 95 %.
Results and discussion
Physicochemical properties
As summarized in Table 1, the textural properties of γ-Al2O3 support, Ni–Co/γ-Al2O3 bimetallic catalysts and Ce-modified Ni1Co3 catalysts were listed. For γ-Al2O3 support, the surface area was mainly formed by the micro/meso pores and the impregnation of metals could result in blockage of part of the small pores which can account for the loss of surface areas and pore volumes along with the decrease of average pore diameters. The BET surface areas of the γ-Al2O3 was 235.2 m2 g−1, whereas 20 wt% of metal loading made the values decline to ~170 m2 g−1. It was also shown that the Ni/Co mass ratio had negligible impact on the physicochemical properties of all the catalysts. Moreover, a gradual decrease of surface areas as well as pore volume and diameters can be observed because of the introduction of cerium. This change in turn would have a significant influence on the catalytic performance (see Table 2).
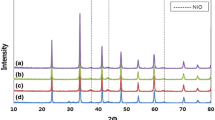
Figure 1 shows the XRD patterns of the Ni–Co bimetallic catalysts with different Ni/Co mass ratio. The diffraction peaks of alumina located at 2θ = 37.6°, 45.9° and 67.0° were distinct. Ni1Co0, Ni5Co1 and Ni3Co1 indicated peaks of NiO and/or NiAl2O4 at 37.0°, NiO at 43.3°, 62.9° and 75.4°. As cobalt concentration increased and nickel loading declined, new diffraction peaks at 19.0°, 31.2°, 36.8°, 44.8°, 55.7°, 59.4°, 65.2° and 77.7° appeared for Ni1Co1, Ni1Co3, Ni1Co5 and Ni0Co1 catalysts, representing peaks of Co3O4. At the same time, those peaks of NiO and/or NiAl2O4 disappeared gradually. Moreover, the peaks at 31.2°, 36.7°, 59.2° and 65.2° were also identified as CoAl2O4 specie.

XRD spectrum of catalysts with different Ni/Co ratio. a Ni0Co1; b Ni1Co5; c Ni1Co3; d Ni1Co1; e Ni3Co1; f Ni5Co1; g Ni1Co0
Figure 2 presents the patterns of Ni1Co3 catalysts promoted by different cerium amount from 0 to 10.0 wt%. According to the results of XRD, diffraction peaks referring to CeO2 were not visible until cerium loading was greater than 5.0 wt%. Peaks of CeO2 were not observed for samples with low Ce dosage, which illustrated that cerium species were uniformly dispersed in support. And CeO2 interacted with metal particles to form composites so that cerium-containing particles lay behind the detection limit of XRD. When the ceria loading was more that >5.0 wt%, diffraction peaks of 28.6°, 33.1°, 47.5° and 56.3° standing for CeO2 appeared. Moreover, the intensity of the CeO2 diffraction peaks increased with the increasing ceria content. The results indicated that the CeO2 species had an intimate contact with nickel and/or cobalt as well as the support when the content was low. The strong interaction between metal and support restrained the growth of CeO2 crystal but the excessive loading of ceria decreased the density of active sites, which would make the reaction rate decline.

XRD spectrum of catalysts with different Ce contents. a Ni1Co3; b 2.5Ce–Ni1Co3; c 5.0Ce–Ni1Co3; d 7.5Ce–Ni1Co3; e 10.0Ce–Ni1Co3
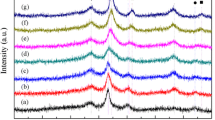
Figure 3 shows H2-TPR results of all the Ni–Co/γ-Al2O3 bimetallic catalysts. There existed two reduction peaks over Ni1Co0 catalyst: the first located at about 340 °C belonging to the reduction of well-dispersed NiO species, and the higher peak centered at about 700 °C was attributed to the reduction of bulk NiO species with strong interaction with the γ-Al2O3. However, the complete reduction of Ni species required reduction temperature of more than 850 °C as reported in literature [21], which was assigned to the reduction of NiAl2O4. However, cobalt catalyst (Ni0Co1) owned three reduction peaks: the hydrogen consumption at 360 °C was attributed to reduction of Co3O4 to CoO, with subsequent reduction of CoO to Co at 550 °C. And the peak at 670 °C probably accounted for the reduction of cobalt aluminate specie [22]. As the Co dosage increased over Ni–Co bimetallic catalysts, both of the two peaks of NiO species shifted downward into lower temperature area and the former peak disappeared gradually. At the same time, all the mentioned peaks of Co oxide species also moved down to lower temperature zone along with the increasing nickel content. This behavior was well documented in the literature [23] and was attributed to the strong interaction between nickel and cobalt species during the process of catalyst reduction. Moreover, this also can be explained by the fact that the strong interaction was caused probably by the formation of Ni–Co nanoparticles or alloy entities proposed by Yuan et al. [18]. The existence of Ni–Co species may bring about some influences on the absorption of C–O bond and thus affected the products selectivity.

H2-TPR profiles of Ni–Co bimetallic series catalysts
It is known that cerium oxide has an oxygen storage property because of high oxygen mobility in its lattice [24, 25]. Thus it is prone to interact with metal active components thereby affecting the reduction behavior of metal oxides and promoting the activity of the catalyst [26]. In addition, the results from Fig. 4 indicated that the former peak assigned to the reduction of NiO and/or Co3O4 shifted from 350 to 320 °C gradually. This behavior demonstrated that the incorporation of ceria decreased the reduction temperature of metal oxides species and caused a strong interaction with the active components and support. Yu reported that cerium addition to Ni/AC catalyst would decrease the reduction temperature and facilitate the hydrogenation of acetol [19]. Ye et al. [27] also found that the introduction of cerium to the Ni/Al2O3 catalysts slightly lowered the reduction temperature of nickel oxides but considerably enhanced the H2-chemisorption amount. These results can elucidate the increased glycerol conversion. Our experimental results also indicated that ceria was favorable for the generation of 1,2-PDO and the suppression of EG and methane selectivity.

H2-TPR profiles of Ni1Co3 catalysts modified by cerium
The NH3-TPD measurements show the amount of acid sites normalized by surface area, which represents the surface density of acid sites on the catalysts [28]. Figure 5 indicated NH3-TPD analysis to estimate the acidity of catalysts. Compared with γ-Al2O3 support, the intensity of strong acid sites (over 350 °C) was enhanced remarkably by Ni and Co loading. However, in the Ce-promoted samples, the peaks corresponding to strong acid sites shifted to lower values, indicating that the strong acid sites were more available for enhancing the dehydration of glycerol. This phenomenon elucidated the increased 1,2-PDO selectivity which is illustrated in Table 3.

NH3-TPD profiles of Ce-promoted Ni–Co catalysts. a Ni1Co3; b 2.5Ce–Ni1Co3; c 5.0Ce–Ni1Co3; d 7.5Ce–Ni1Co3; e 10.0Ce–Ni1Co3
XPS analysis can be helpful to understand the nature of electronic properties of bimetallic catalysts. Binding energy (BE) values of Ni 2p for reduced catalysts of Ni1Co0, Ni0Co1, Ni1Co3 and 2.5Ce–Ni1Co3 were summarized in Fig. 6a. Generally, the binding energy of Ni 2p3/2 in pure Ni is about 852.7 eV and in NiAl2O4 is about 855.8 eV. As shown in Fig. 6, there were two characteristic peaks standing for Ni 2p3/2 of pure Ni and NiAl2O4, respectively. And the BE of Ni 2p3/2 (corresponding to pure Ni) of Ni1Co0, Ni1Co3 and 2.5Ce–Ni1Co3 were lower than 852.7 eV significantly, which indicated that the Ni species had strong metal-support interaction with γ-Al2O3 support. The BE of Ni 2p3/2 in the 2.5Ce–Ni1Co3 catalyst was 852.1 eV for pure Ni and 856.9 eV for NiAl2O4, confirming that the addition of Ce led to strong interaction with Ni species and influenced the formation of NiAl2O4 [29]. Combined with the TPR profiles from Fig. 4, the reduction temperature of main peaks for Ni1Co3 catalyst was reduced remarkably due to the addition of Ce. This suggested that the Ni species having strong interactions with CeO2 in 2.5Ce–Ni1Co3 catalyst were more easily reduced. As for the BE of Co 2p in Ni–Co bimetallic catalysts, similar behaviors can also be observed, as shown in Fig. 6b.

a Ni 2p spectra of the Ni1Co0, Ni1Co3, 2.5 %Ce–Ni1Co3 catalysts and b Co 2p spectra of the Ni0Co1, Ni1Co3, 2.5 %Ce–Ni1Co3 catalysts
The function of ceria for improving the catalytic performance has mainly been attributed to the electron effect of Ce-ions as reported in literatures [30]. It was confirmed that the presence of Ce+4/Ce+3 species in the catalyst is beneficial for adjusting the electron density of metallic particles and thus improving the reaction activity. And our experimental results indicated that the catalyst exhibited an improved catalytic performance as compared with unmodified catalysts. The TEM images of Ni1Co3 and 2.5Ce–Ni1Co3 catalysts after reduction at 500 °C in hydrogen atmosphere for 4 h as well as after reaction are shown in Fig. 7. The average size of Ni and/or Co particles was decreased from 19.9 nm to 17.7 m, when 2.5 wt% of Ce was introduced. A smaller metal particle is believed that able to provide more active sites for the adsorption of H2, leading to a higher hydrogenolysis activity.

TEM images of fresh catalysts (a Ni1Co3, b 2.5Ce–Ni1Co3) and spent catalysts (c Ni1Co3, d 2.5Ce–Ni1Co3)
The particle size of Ni1Co3 catalyst after the reaction increased remarkably to around 23.0 nm, indicating that a significant agglomeration of metal particles occurred during the reaction. However, there were slight differences in the TEM images (17.7–18.6 nm) between the fresh catalyst and the one after reaction in the case of 2.5Ce–Ni1Co3 catalyst. Therefore, the catalysts modified by Ce were superior to the Ni1Co3 catalyst because the addition of Ce could enhance the interaction between Ni and/or Co particles and support. Agglomeration of nickel particles in the commercial catalyst was already described as a result of former investigations [31]. Combining with the experimental results, the agglomeration of metal particles was responsible for the catalyst deactivation which could be suppressed by enhancing the interaction between metal and support. This improvement of dispersion and stability for 2.5Ce–Ni1Co3 might account for the increase in glycerol conversion described in the following activity tests.
Catalytic performance of Ni–Co catalysts and Ce promoted Ni1Co catalysts
Conversion and selectivity data relative to the hydrogenolysis of glycerol in the presence of different Ni–Co bimetallic catalysts are shown in Table 2. Among the tested catalysts, the best conversion was achieved over Ni1Co3 catalyst (63.5 %). In contrast, the monometallic catalysts (Ni1Co0 and Ni0Co1) exhibited the lowest activity for glycerol conversion. Among the bimetallic catalysts tested in this work, the Ni/Co weight ratio had a significant influence on the glycerol conversion and products distribution. As shown in the experimental data, it seemed that the monometallic catalyst favored the C–O bond breakage leading to the formation of 1,2-PDO instead of EG. However, the bimetallic catalysts with different Ni/Co mass ratio were inclined to catalyze the C–C bond cleavage to facilitate the generation of EG as well as the gaseous degradation products such as CH4 and CO2. Moreover, the highest EG selectivity was obtained over Ni1Co3 catalyst, which might stem from the synergistic effect between the two types of metal species and/or the strong metal-support interaction (SMSI) revealed by the above results of TPR and XPS spectrum.
Table 3 presents the catalytic performance of Ni1Co3 catalysts modified by different Ce dosage. When cerium was added into Ni1Co3 catalyst, the glycerol conversion increased significantly from 63.5 to 71.3 %, with 68.5 % selectivity of 1,2-PDO and 12.6 % selectivity of EG. But as for higher concentration of cerium-doped catalysts (5.0, 7.5 and 10.0 % mass percent), the catalysts exhibited decreased activity and almost unchanged products distribution. The low activity may be attributed to the formation of CeO2 over the support which occupied the active sites and blocked the catalyst pores [32]. Moreover, the significant increase in the selectivity of 1,2-PDO was observed from all the Ce promoted catalysts. This result implied that the addition of Ce brought about not only enhancement of activity but also the increase in the formation of 1,2-PDO. It was also reported [19] that the cerium in the Ni–Ce/AC catalyst could accelerate the hydrogenation of the intermediate of acetol, which can be hydrogenated into 1,2-PDO easily.
To examine the effect of temperature on the glycerol conversion and selectivity of all the products, reactions were carried out at 200–240 °C at a hydrogen pressure of 6.0 MPa in the presence of 2.5Ce–Ni1Co3 catalyst. Figure 8 shows the corresponding results. With the increase of temperature from 200 to 240 °C, there was a constant increase in the glycerol conversion from 34.1 to 99.8 %. However, the selectivity of 1,2-PDO decreased gradually with the increase in temperature. This indicated that at a hydrogen pressure of 6.0 MPa, excessive hydrogenolysis resulted in the conversion of 1,2-PDO into lower alcohols like 1-PO and 2-PO. Moreover, from our initial screening studies, it was necessary to operate at higher pressures to prevent degradation of products.

Effect of reaction temperature on glycerol hydrogenolysis over 2.5Ce–Ni1Co3 catalyst. Reaction conditions: 6.0 MPa H2 pressure, 100 ml 20 wt% glycerol aqueous solution, 2.0 g catalyst, 10 h
To further investigate the effect of hydrogen pressure on the overall reaction, reactions were carried out at 2.0–10.0 MPa at a constant temperature of 220 °C as shown in Fig. 9. As expected, the conversion of the glycerol increased with the increase of hydrogen pressure from 2.0 to 10.0 MPa, while 1,2-PDO and EG selectivity increased slightly during this pressure range.

Effect of H2 pressure on glycerol hydrogenolysis over 2.5Ce–Ni1Co3 catalyst. Reaction conditions: 220 °C, 100 ml 20 wt% glycerol aqueous solution, 2.0 g catalyst, 10 h
The effect of reaction time on hydrogenolysis of glycerol over 2.5Ce–Ni1Co3 catalyst is shown in Fig. 10. At the end of initial 4 h, glycerol was swiftly converted and conversion was 38.5 %. When the reaction time was prolonged to 20 h, glycerol conversion reached 92.1 % with a gradual selectivity decrease both for 1,2-PDO and EG.

Effect of reaction time on glycerol hydrogenolysis over 2.5Ce–Ni1Co3 catalyst. Reaction conditions: 6.0 MPa H2 pressure, 220 °C, 100 ml 20 wt% glycerol aqueous solution, 2.0 g catalyst
In fact, there may be a competition of cleavage between the C–O bonds and C–C bonds. Many reports proposed and confirmed that glycerol hydrogenolysis was conducted by dehydration/hydrogenation mechanism [33–35] as shown in Fig. 11. From Route A, glycerol was first dehydrated to acetol and then the acetol was hydrogenated into 1,2-PDO. The catalysts used for glycerol hydrogenolysis were usually considered as bifunctional catalysts with the capacity of both dehydration and hydrogenation. It is known that the effect of support is related to the fact that support materials can influence the reaction routes (dehydration route or retro-aldolization) in the presence of metal catalyst. Herein, the γ-Al2O3 with acid sites favored the dehydration of glycerol to acetol. And the addition of Ce could promote the hydrogenation of acetol and formation of 1,2-PDO. However, the cleavage of C–C bond cannot be avoided in the presence of metallic Ni. As shown in Route B, a hypothetic summary of EG production on Ni–Co catalyst can be described as following: dehydrogenation of glycerol formed glyceraldehydes on metal surfaces, which then underwent retro-aldolization to form glycolaldehyde and formaldehyde. Consequently, the glycolaldehyde was hydrogenated into EG on metal sites. At the same time, the formaldehyde was converted into C1 products like methane and carbon dioxide.

Proposed reaction pathway of hydrogenolysis of glycerol to 1,2-PDO and EG over Ni–Co bimetallic catalysts
Conclusion
It was shown in this study that supported Ni–Co/γ-Al2O3 bimetallic catalysts were efficient catalysts for the hydrogenolysis of glycerol. The best catalytic performance was acquired over the Ni1Co3 catalyst and improved by the addition of Ce. Among all the catalysts tested, 2.5Ce–Ni1Co3 exhibited the highest glycerol conversion and the total glycol selectivity reached 81.1 %. XRD, TPR and XPS measurements indicated that the addition of Ce changed the reduction behavior of catalysts, enhancing the strong metal-support interaction between Ni–Co species and support. The TEM micrographs revealed that the smaller particle size and the inhibited agglomeration was attributed to the Ce addition.
References
Besson M, Gallezot P, Pinel C (2013) Conversion of biomass into chemicals over metal catalysts. Chem Rev 114(3):1827–1870
Serrano-Ruiz JC, Luque R, Sepulveda-Escribano A (2011) Transformations of biomass-derived platform molecules: from high added-value chemicals to fuels via aqueous-phase processing. Chem Soc Rev 40(11):5266–5281
Zhou CH, Zhao H, Tong DS, Wu LM, Yu WH (2013) Recent advances in catalytic conversion of glycerol. Catal Rev 55(4):369–453
Katryniok B, Paul S, Dumeignil F (2013) Recent developments in the field of catalytic dehydration of glycerol to acrolein. ACS Catal 3(8):1819–1834
Mane RB, Rode CV (2012) Simultaneous glycerol dehydration and in situ hydrogenolysis over Cu–Al oxide under an inert atmosphere. Green Chem 14(10):2780–2789
Nakagawa Y, Tomishige K (2011) Heterogeneous catalysis of the glycerol hydrogenolysis. Catal Sci Technol 1(2):179–190
Feng J, Xiong W, Xu B, Jiang W, Wang J, Chen H (2014) Basic oxide-supported Ru catalysts for liquid phase glycerol hydrogenolysis in an additive-free system. Catal Commun 46:98–102
Deng C, Duan X, Zhou J, Chen D, Zhou X, Yuan W (2014) Size effects of Pt–Re bimetallic catalysts for glycerol hydrogenolysis. Catal Today 234:208–214
Musolino MG, Scarpino LA, Mauriello F, Pietropaolo R (2011) Glycerol hydrogenolysis promoted by supported palladium catalysts. ChemSusChem 4(8):1143–1150
Nakagawa Y, Shinmi Y, Koso S, Tomishige K (2010) Direct hydrogenolysis of glycerol into 1,3-propanediol over rhenium-modified iridium catalyst. J Catal 272(2):191–194
Sharma RV, Kumar P, Dalai AK (2014) Selective hydrogenolysis of glycerol to propylene glycol by using Cu:Zn:Cr: Zr mixed metal oxides catalyst. Appl Catal A 477:147–156
Li T, Fu C, Qi J, Pan J, Chen S, Lin J (2013) Effect of zinc incorporation manner on a Cu–ZnO/Al2O3 glycerol hydrogenation catalyst. React Kinet Mech Catal 109(1):117–131
Huang J, Chen J (2012) Comparison of Ni2P/SiO2 and Ni/SiO2 for hydrogenolysis of glycerol: a consideration of factors influencing catalyst activity and product selectivity. Chin J Catal 33(4–6):790–796
Hoşgün HL, Yıldız M, Gerçel HF (2012) Hydrogenolysis of aqueous glycerol over raney nickel catalyst: comparison of pure and biodiesel by-product. Ind Eng Chem Res 51(10):3863–3869
Ueda N, Nakagawa Y, Tomishige K (2010) Conversion of glycerol to ethylene glycol over Pt-modified Ni catalyst. Chem Lett 39(5):506–507
Raksaphort S, Pengpanich S, Hunsom M (2014) Products distribution of glycerol hydrogenolysis over supported co catalysts in a liquid phase. Kinet Catal 55(4):434–445
Felicissimo MP, Martyanov ON, Risse T, Freund H-J (2007) Characterization of a Pd–Fe bimetallic model catalyst. Surf Sci 601(10):2105–2116
Li B, Wang J, Yuan Y, Ariga H, Takakusagi S, Asakura K (2011) Carbon nanotube-supported RuFe bimetallic nanoparticles as efficient and robust catalysts for aqueous-phase selective hydrogenolysis of glycerol to glycols. ACS Catal 1(11):1521–1528
Yu W, Zhao J, Ma H, Miao H, Song Q, Xu J (2010) Aqueous hydrogenolysis of glycerol over Ni–Ce/AC catalyst: promoting effect of Ce on catalytic performance. Appl Catal A 383(1–2):73–78
Yu W, Xu J, Ma H, Chen C, Zhao J, Miao H et al (2010) A remarkable enhancement of catalytic activity for KBH4 treating the carbothermal reduced Ni/AC catalyst in glycerol hydrogenolysis. Catal Commun 11(5):493–497
Hou Z, Yokota O, Tanaka T, Yashima T (2003) Characterization of Ca-promoted Ni/α-Al2O3 catalyst for CH4 reforming with CO2. Appl Catal A 253(2):381–387
Luo N, Ouyang K, Cao F, Xiao T (2010) Hydrogen generation from liquid reforming of glycerin over Ni–Co bimetallic catalyst. Biomass Bioenerg 34(4):489–495
Mauriello F, Ariga H, Musolino MG, Pietropaolo R, Takakusagi S, Asakura K (2015) Exploring the catalytic properties of supported palladium catalysts in the transfer hydrogenolysis of glycerol. Appl Catal B 166–167:121–131
Gorte RJ, Zhao S (2005) Studies of the water-gas-shift reaction with ceria-supported precious metals. Catal Today 104(1):18–24
Cheng Z, Wu Q, Li J, Zhu Q (1996) Effects of promoters and preparation procedures on reforming of methane with carbon dioxide over Ni/Al2O3 catalyst. Catal Today 30(1–3):147–155
Wang W, Stagg-Williams SM, Noronha FB, Mattos LV, Passos FB (2004) Partial oxidation and combined reforming of methane on Ce-promoted catalysts. Catal Today 98(4):553–563
Ye L, Duan X, Lin H, Yuan Y (2012) Improved performance of magnetically recoverable Ce-promoted Ni/Al2O3 catalysts for aqueous-phase hydrogenolysis of sorbitol to glycols. Catal Today 183(1):65–71
El Doukkali M, Iriondo A, Cambra JF, Gandarias I, Jalowiecki-Duhamel L, Dumeignil F, Arias PL (2014) Deactivation study of the Pt and/or Ni-based γ-Al2O3 catalysts used in the aqueous phase reforming of glycerol for H2 production. Appl Catal A 472:80–91
Cai X, Dong X, Lin W (2008) Effect of CeO2 on the catalytic performance of Ni/Al2O3 for autothermal reforming of methane. J Nat Gas Chem 17(1):98–102
Manfro RL, da Costa AF, Ribeiro NFP, Souza MMVM (2011) Hydrogen production by aqueous-phase reforming of glycerol over nickel catalysts supported on CeO2. Fuel Process Technol 92(3):330–335
Schimpf S, Louis C, Claus P (2007) Ni/SiO2 catalysts prepared with ethylenediamine nickel precursors: influence of the pretreatment on the catalytic properties in glucose hydrogenation. Appl Catal A 318:45–53
Zhang K, Zhang H, Ma H, Ying W, Fang D (2014) Effect of Sn addition in gas phase hydrogenation of acetic acid on alumina supported PtSn catalysts. Catal Lett 144(4):691–701
Miyazawa T, Kusunoki Y, Kunimori K, Tomishige K (2006) Glycerol conversion in the aqueous solution under hydrogen over Ru/C+ an ion-exchange resin and its reaction mechanism. J Catal 240(2):213–221
Feng J, Wang J, Zhou Y, Fu H, Chen H, Li X (2007) Effect of base additives on the selective hydrogenolysis of glycerol over Ru/TiO2 catalyst. Chem Lett 36(10):1274–1275
Montassier C, Dumas JM, Granger P, Barbier J (1995) Deactivation of supported copper based catalysts during polyol conversion in aqueous phase. Appl Catal A 121(2):231–244
Acknowledgments
This work was supported by the National Natural Science Foundation of China (21373083) and the Open Project of State Key Laboratory of Chemical Engineering (SKL-ChE-12C03).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Jiang, T., Kong, D., Xu, K. et al. Hydrogenolysis of glycerol aqueous solution to glycols over Ni–Co bimetallic catalyst: effect of ceria promoting. Appl Petrochem Res 6, 135–144 (2016). https://doi.org/10.1007/s13203-015-0128-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13203-015-0128-8