
Abstract
Alumina supported Pt, Sn and PtSn catalysts were prepared by impregnation and tested in the gas phase hydrogenation of acetic acid, and characterized by N2-physisorption, XRD, H2-TPR, H2-pulse chemisorption, H2-TPD, DRIFTS (diffuse reflectance infrared Fourier transform spectroscopy), XPS, electron microscopy techniques (TEM, HRTEM and EDX). Results show that Sn addition to Pt/Al2O3 catalyst can improve the conversion of acetic acid and suppress the production of by-products, as well as enhance the selectivity towards ethanol. The optimization of activity and selectivity can be achieved by changing reaction temperature and pressure. Characterizations indicated that the better performance of PtSn/Al2O3 catalyst is due to the formation of PtSn alloy and Pt-SnOx species.
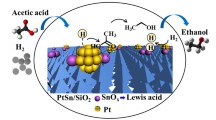
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
As one of the most important oxygen-containing organic materials, ethanol has a variety of applications, such as pharmaceutical and perfume preparation, as solvent and antiseptic, and as additives to fuel motor vehicles. Most ethanol in the world is produced through fermentation of agricultural products, such as corn and sugar cane, and only a small fraction is made synthetically. The chemosynthesised ethanol is mainly manufactured from ethylene hydration [1]. This process is conducted by reacting ethylene with steam at 573 K, 6.0–7.0 MPa and only 5 % of the ethylene is converted into ethanol at each pass. However, the food crisis and the oil depletion make the aforementioned routes unattractive for the large scale production of ethanol. More recently, much attention has been paid to the study of conversion of coal-based starting materials such as methanol or synthesis gas to ethanol. They include homologation and carbonylation processes and the direct conversion of synthesis gas to ethanol [2, 3]. Unfortunately, all the synthetic methods are not so satisfactory till now.
From the point of industrial view, a desirable synthesis method should be direct and robust and applicable on a large scale, and catalytic hydrogenation of acetic acid would meet these demands if suitable catalyst is available. On one hand, acetic acid is currently manufactured by three alternative processes: acetaldehyde oxidation, n-butane oxidation and methanol carbonylation, and the homogeneous methanol carbonylation technology developed by Monsanto exhibits high selectivity (based on methanol 99 % and carbon monoxide 85 %) and can be operated under mild conditions (453–493 K, 3.0–4.0 MPa) [4]. On the other hand, theoretically, acetic acid can be reduced to ethanol according to the following stoichiometry,
without the hazards of pollutants.
Furthermore, the study of acetic acid hydrogenation has attracted much attention during the past decades. Pestman et al. [5–7] have accomplished a series of experiments to explore the reactions of carboxylic acids on oxides and the products they obtained from acetic acid are acetone and aldehyde. Studies conducted by Rachmady and Vannice indicated that Pt supported on oxides is a promising catalyst for acetic acid hydrogenation and a product distribution of 50 % ethanol, 30 % ethyl acetate and 20 % ethane can be acquired on TiO2 supported Pt catalyst [8, 9].
The addition of a second metal to noble metal catalysts can modify their catalytic behavior, such as in the case of bimetallic PtSn catalysts. Sn added to platinum-based catalysts has proven to be beneficial to the activity maintenance and/or can modify the selectivity of catalysts in many processes, such as selective oxidation of CO [10], soot oxidation [11], catalytic reduction of NO [12], paraffins dehydrogenation [13–17], and hydrogenation of α, β-unsaturated aldehydes [18, 19]. The research of Coloma et al. [20, 21] revealed that the presence of Sn can greatly enhance the catalytic activity and improve the selectivity towards C=O bond hydrogenation in the gas phase hydrogenation of crotonaldehyde, and the reports of Miguel et al. [18] and Serrano-Ruiz et al. [19] had drew similar conclusions.
To our knowledge, the effect of Sn addition in alumina-supported PtSn catalyst on acetic acid hydrogenation has not been investigated yet. In this study, alumina-supported bimetallic PtSn catalysts and monometallic Pt, Sn catalysts were prepared, characterized and tested in the gas phase hydrogenation of acetic acid.
2 Experimental
2.1 Catalyst Preparation
Three bimetallic PtSn/Al2O3 catalysts with different Pt/Sn ratio (1/3, 2/2 and 3/1) and constant total loading (2.0 wt%) were prepared by co-impregnation. After being ground and sieved to desired size (0.3–0.45 mm), the Al2O3 support was calcined at 823 K for 12 h to remove organic impurities before impregnated with a aqueous solution of tin salt (SnCl2·4H2O) and platinum salt (H2PtCl6·6H2O). After aged for 12 h at room temperature, the catalysts were dried at 293 K over night and then calcined in air at 823 K for 12 h. For the purpose of comparison, two monometallic catalysts with a nominal loading of 1.0 wt% were also prepared. They were subjected to the same procedure other than that the impregnation solution only contains SnCl2·4H2O or H2PtCl6·6H2O. The catalysts were denoted as aPtbSn/A, where b and a represent ten times the nominal weight percentage of Sn and Pt, respectively, and A represents alumina. For instance, PtSn/Al2O3 catalyst with a Pt/Sn ratio of 1/3 was labeled as 5Pt15Sn/A.
2.2 Catalyst Characterization
The BET (Brunauer Emmett Teller) surface area, average pore diameter, and pore size distribution of the catalysts and the support were determined by means of nitrogen physisorption using a Micromeritics ASAP 2020M automated system. All samples were degassed at 500 K, 1.3 Pa for 6 h before N2 adsorption.
X-ray diffraction (XRD) powder patterns of the catalysts were recorded with a Rigaku D/max 2550 diffractometer using nickel filtered Cu Kα radiation (λ = 1.54056 Å) at room temperature. The X-ray tube was operated at 40 kV, 100 mA and scanned from 10° to 80°.
The morphology and particle size of the catalysts were observed using a JEOL-JEM 2100 transmission electron microscope operated at 100 kV. High resolution TEM combined with energy-dispersive X-ray microanalysis (EDX) was performed on a Tecnai G2 F30 S-TWIN electron microscope operating at 300 kV. The catalysts were ground to 300 mesh and dispersed in absolute ethanol by ultrasonic for 30 min, then a drop of the prepared solution was added to a carbon-coated copper grid. The catalysts were exposed to an infrared red lamp for 30 min to remove the ethanol before scanning.
Hydrogen temperature-programmed reduction (H2-TPR) was performed on Micromeritics AutoChem II 2920. Generally, 0.20 g sample was placed in a quartz U-tube reactor and calcined in situ at 773 K for 60 min under Ar (99.99 %) atmosphere and then cooled down to room temperature. The reductive mixture (10 % H2 in Ar) was fed to the reactor at a flow rate of 50 mL min−1. The TPR experiments were carried out from room temperature to 1,073 K with a ramp of 10 K min−1 and the hydrogen consumption was monitored by a thermal conductivity detector (TCD) after the base line was stable.
H2-pulse chemisorption experiments were performed to analyze the Pt dispersion of the catalysts in the same system as adopted in TPR. The samples were reduced under H2 (99.99 %, 30 ml min−1) at 623 K for 2 h, then purged in He (99.99 %, 30 ml min−1) at 673 K for 1.5 h and cooled down to 303 K in flowing He (30 ml min−1). Calibrated pulses of H2 were injected into the system repeatedly until no further H2 consumption was noted, and the time between pulses was 4 min. H2-TPD experiments were performed after H2 pulse chemisorption in the same system and the spectra was recorded from 303 to 1073 K.
DRIFTS (diffuse reflectance infrared Fourier transform spectroscopy) of acetic acid were carried out to determine the nature of the adsorbed species on the catalysts in a spectrometer (Nicolet 6700, Thermo, USA) which had been modified to allow in situ catalyst pretreatment. The infrared cell was equipped with temperature controlled parts and ZnSe window. The catalysts were reduced in situ for 2 h under atmospheric pressure by hydrogen at 623 K (30 ml min−1, 5 K min−1) and purged with pure nitrogen (99.99 %) for 1 h at 623 K. Acetic acid was introduced into the system by passing a N2 flow through a glass vessel containing liquid acetic acid (99.95 %) at room temperature under a pressure of 0.1 MPa. After the catalyst was saturated with acetic acid, the system was flushed with N2 for 1 h. Infrared spectra of adsorbed species were collected at atmospheric pressure and room temperature at a resolution of 4 cm−1. Each spectrum after adsorption was referenced to that of the freshly pretreated sample just prior to introduction of the adsorbate.
Photoelectron spectra were recorded over calcined and reduced samples using an ESCALAB 250Xi (Thermo Fisher, America) photoelectron spectrometer coupled with a Al Kα X-ray operated at 10 mA and 12 kV. The energy regions of the photoelectrons of interest were repeatedly scanned to get good signal-to-noise ratios. The intensity of the peaks were quantified by determining the integral of each peak after subtracting an S-shaped background and fitting the experimental peaks to Lorentzian/Gaussian lines (80 % L/20 % G). The C1 s line was taken as an internal standard at 285.0 eV.
2.3 Catalyst Evaluation
The activity and product selectivity of the catalysts were tested for the gas phase hydrogenation of acetic acid in a fixed bed reactor (Φ14 × 2 × 650 mm) under different conditions. Catalysts (2.5 g, about 4 ml) were reduced in situ in flowing hydrogen (0.1 MPa, 100 ml min−1) according to the following procedure: 2 K min−1 from ambient to 533 K and kept at this temperature for 1.5 h then 1 K min−1 to 623 K and held at 623 K for 2 h. The pressure of the system was controlled by regulating the reducing valve and the back-pressure valve after the catalyst bed was cooled down to the selected reaction temperature under H2 flow. Acetic acid was pumped and transported by H2 into the system and preheated to maintain gas phase before entering the reactor. The effluent was monitored every 12 h so as to get a steady-state performance after passing through the condenser and the liquid–vapor separator. Components of the tail gas were determined on-line and the products in the liquid phase were detected off-line by GC Agilent 7890A. In one sampling loop, CH4, C2H6, H2 and H2O were analyzed with a thermal conductivity detector (TCD) furnished with a molecular sieve 5A packed column (6 FT × 1/8 IN × 2.1 mm) and a Hayesep Q packed column (8 FT × 1/8 IN). In the other loop, C1-C6 oxy-compounds were analyzed with a flame ionization detector (FID) coupled with an HP-PLOT/Q capillary column (30 m × 0.53 mm × 40 μm) and an HP-INNOWAX (30 m × 0.53 mm × 1 μm) capillary column. Particularly, the acetic acid left in the liquid was determined by titration.
3 Results and Discussion
3.1 N2-Physisorption
The BET surface area (SBET), pore volume (Vp), and average pore diameter (Dp) of the catalysts and Al2O3 support are listed in Table 1. For Al2O3 support, the surface area is mainly formed by the micro/meso-pores. The introduction of metals could result in blockage of part of the small pores, which can be ascribed to the loss of surface areas and pore volumes, as well as the increase of average pore diameters. However, the differences among the catalysts are slight because of a low metal loading, which is similar with the report of Ayastuy et al. [10]. The N2 adsorption–desorption isotherms (Fig. 1) reveal that the catalysts and support all show the properties of type H4 hysteresis loop, which means that the pores are narrow slit shaped and the pore structure of the support has been changed little by metal loading [22].

N2 adsorption–desorption isotherms of the catalysts and the support
3.2 XRD
Figure 2 shows the XRD patterns of the catalysts and the support. The characteristic peaks of alumina located at 2θ = 38, 46 and 67° are very distinct and no other peak is detectable [23]. The absence of Pt, Sn and Pt–Sn alloy phases in our samples may be ascribed to the fact that the Pt and Sn are highly dispersed on the alumina support and/or that the size of the crystallite is too small (as revealed by TEM and H2-pulse chemisorption) to be detected by XRD characterization.

XRD patterns of the catalysts
3.3 H2-TPR
H2-TPR profiles of the monometallic catalysts (10Sn/A, 10Pt/A) and the bimetallic catalysts (5Pt15Sn/A, 10Pt10Sn/A and 15Pt5Sn/A) are shown in Fig. 3. The reduction peak of 10Sn/A is broad and H2 consumption is observed over a large temperature range with a maximum at about 750 K, while monometallic 10Pt/A catalyst shows a reduction peak at about 510 K. Bimetallic catalysts present similar reduction behavior with 10Pt/A. A broadening of reduction peaks and a shift of peak temperature towards higher zones is also observed along with the increase of Sn/Pt ratio. These findings are similar to the reports of Hoang et al. [24]. A plausible explanation is that Sn species are in the vicinity of Pt and the coreduction of Pt and Sn occurred in bimetallic PtSn catalysts, which probably lead to the formation of Pt–SnOx species or PtSn alloy [31].

H2-TPR profiles of the catalysts
3.4 H2-Pulse Chemisorption and H2-TPD
The Pt dispersion of the catalysts was calculated from H2-pulse chemisorption experiments at room temperature and displayed in Table 2, based on the assumption that H/Pt = 1. Our preliminary tests of H2-pulse chemisorption indicated that 10Sn/A and the Al2O3 support do not chemisorb H2. It is obviously that the introduction of Sn to monometallic Pt catalyst can lead to a decreased Pt dispersion, and the higher is the Sn/Pt ratio, the smaller is the Pt dispersion. This phenomenon is also suggested by others [21, 25] and can be attributed to the formation of Pt–SnOx species or PtSn alloy.
The characteristics of chemisorbed hydrogen on the catalysts were studied by H2-TPD. The H2-TPD profiles obtained after H2-pulse chemisorption at room temperature of the monometallic Pt and bimetallic PtSn catalysts are shown in Fig. 4. All the catalysts show two desorption peaks. The desorption peaks at low temperature region (about 380 K) are prominent while the peaks at high temperature zone (near 800 K) are so weak that they almost cannot be observed. Generally, low temperature desorption peaks are assigned to hydrogen desorbed from metallic Pt [26, 27], while high temperature desorption peaks are assigned to spillover hydrogen, strong chemisorbed hydrogen and hydrogen in subsurface layers of the platinum [22].

H2-TPD profiles of the catalysts
3.5 DRIFTS
Infrared spectra of species resulting from acetic acid adsorption on catalysts were obtained after the introduction of acetic acid to the reduced catalysts for 0.5 h, followed by purging the reactor cell for 1.5 h with high purity N2 (99.99 %) at atmospheric pressure and room temperature. Figure 5 show infrared spectra after adsorption of acetic acid on the monometallic and bimetallic catalysts. Four different surface species can be identified—acyl, acetate, adsorbed acetic acid and CO, the detailed assignment of bands can be found in literature [9]. All catalysts show almost the same spectra except that the band at 2,054 cm−1 assigned to adsorbed CO on 10Pt/A disappeared after the introduction of Sn, which indicates that the presence of Sn can modify the property of Pt and deter the decarbonylation of acetic acid [28]. This phenomenon can be accounted for the dramatical differences of activity and product distribution between monometallic Pt and bimetallic PtSn catalysts.

DRIFTS of the catalysts after the adsorption of acetic acid
3.6 XPS
XPS experiments were performed to determinate the chemical state of Pt, Sn in the fresh and reduced catalysts. It is hard to discriminate metal oxidation states by XPS and the data revealed by different authors contradict. Pt 4d lines were analyzed instead of Pt 4f because the energy region of Pt 4f was overlapped by the intense Al 2p peak. From Pt 4d5/2 spectrum of the calcined catalysts, only one peak locating at 316.1–317.2 eV was found, which can be assigned to Pt4+ or Pt2+ species [19, 29]. After reduction, the peak shifted to the region of 313.3–313.8 eV and can be ascribed to Pt0 [19]. Figures 6 and 7 show the Sn 3d5/2 spectrum of calcined and reduced catalysts. All the calcined catalysts show one sharp peak (486.6–487.1 eV) while the spectrum of the reduced catalysts can be resolved into 2 peaks (485.2–485.8 eV and 486.2–487.0 eV, respectively). The component at lower BE corresponds to zero-valent Sn or alloyed Sn, and that located at higher region can be ascribed to oxidic Sn [29–31, 32].

The 3d5/2 spectrum of the fresh catalysts

The 3d5/2 spectrum of the reduced catalysts
The quantitative results of XPS are shown in Table 3. Compare the atom ratio of fresh and reduced Pt, Sn monometallic catalysts, one can find out that H2 reduction can prompt the aggregation of Pt and Sn to the surface of the catalysts. As to the Pt-Sn bimetallic catalysts, H2 reduction lowered the atom ratio of Pt/Al while increased that of Sn/Al, which means that the addition of Sn can restrict the reduction of Pt and that is in agreement with the result of H2-TPR. Similar observations were made by different authors on supported Pt-Sn catalysts [18, 33, 34]. One plausible reason is that H2 reduction can lead to the formation of PtSn alloy or that Sn can be transferred to Pt surfaces and formed Pt-SnOx species.
3.7 Electron Microscopy Techniques
The morphology and size distribution of the active metal particles were further investigated by TEM and the typical images of 10Pt/A, 10Pt10Sn/A and 10Sn/A are shown in Fig. 8. At first sight, all the samples are consisted of agglomerated large particles of alumina and small round metal granules, whereas the boundary of metal particles on 10Sn/A is less clear than that of the samples containing Pt and the particle size is larger. Result from statistics (calculated from the formula: average particle size = ∑nid 3i /∑nid 2i ) also demonstrated the average particle size decreased from 5 nm for 10Sn/A to 3.3 nm for 10Pt10Sn/A and 1.9 nm for 10Pt/A. The difference between 10Pt/A and 10Pt10Sn/A probably can be explained in terms of alloy formation or that Pt is covered by SnOx species in its vicinity.

TEM images of a 10Pt/A, b 10Pt10Sn/A and c 10Sn/A
To investigate the nature of metallic phases on the bimetallic catalysts, 10Pt10Sn/A was further characterized by HRTEM and EDX. Figure 9 depicts representative lattice fringe images of PtSn particles and their corresponding EDX analysis of 10Pt10Sn/A after reduction. The particles showed low contrast with respect to the alumina background and were very hard to detect. The image of one region (region I) of the catalyst shows the lattice spacings of 0.203 and 0.215 nm and the EDX analysis confirms the presence of both Pt and Sn. Thus, this structure can be attributed to a PtSn alloy by comparison with the lattice parameters of the (110) and (102) reflection planes of the PtSn alloy according to PDF card #25-0614. Another region (region II) of the catalyst exhibits a lattice spacing of 0.192 nm and can be assigned to Pt because the lattice parameter of 220 plane of Pt is 0.196 nm (PDF card #04-0802). However, EDX shows a bimetallic composition though the signals of Sn are very weak. Furthermore, XPS result from Table 3 demonstrates that 54 % of the Sn on the reduced 10Pt10Sn/A catalyst was in the state of oxidic species. Thus, the Pt on region II is probably in the vicinity of oxidic Sn species and formed Pt-SnOx species. Combining the results of TPR, XPS, HR-TEM and EDX, it can be noted that both PtSn alloy and Pt-SnOx are formed on the surface of reduced PtSn catalyst.

Catalyst 10Pt10Sn/A: a, b Representative HRTEM micrographs and c, d corresponding EDX patterns
3.8 Acetic Acid Hydrogenation
A number of reactions [14] related to acetic acid can occur on supported Pt catalysts as listed in Eqs. (1–6).
Apart from ethanol and ethyl acetate (originated from the esterification of ethanol and acetic acid), there are also a small amount of CH4, C2H6 and acetaldehyde in the products. The conversion of acetic acid and the selectivity of products were calculated by formula (7) and (8):
where n(CH3COOH)in and n(CH3COOH)out are the flow rate of acetic acid at inlet and outlet, and ni and νi represent the flow rate of product i and the stoichiometry of product i, respectively.
3.9 Performance of the Catalysts
The performances of all the catalyst, both monometallic and bimetallic, were tested at 548 K, 2 MPa, LHSV = 0.6 h−1, H2/HOAC = 10, and the results are presented in Table 4. The activity in terms of acetic acid conversion follows the trend of 10Pt10Sn/A > 15Pt5Sn/A > 5Pt15Sn/A > 10Pt/A > 10Sn. The monometallic catalyst of 10Sn/A and 10Pt/A show distinct performance. The acetic acid conversion of 10Sn/A is only 2 % and the main product is acetone (79 %) while 10Pt/A can achieve 10 % conversion and a large portion of CH4 (22 %) emerged, which is originated from acetic acid decarbonylation. Selectivity of aldehyde shows little variation because it is a primary product of acetic acid reduction and it can be hydrogenated to ethanol quickly. Compare monometallic Pt catalyst with bimetallic PtSn catalysts, one can find out that the addition of Sn can exert 2 remarkable impacts. For one thing, the conversion of acetic acid increased dramatically; for the other thing, the production of CH4 was suppressed sharply. The observed promotional effect of Sn addition could be related to the formation of PtSn alloy and the existence of oxidized tin on platinum particles (Pt–SnOx). One proposal is that the oxidized metal species act as electrophilic or Lewis acid sites for the adsorption and activation of the C=O bond and enhance the activity of hydrogenation of C=O [34]. Another explanation is that the formed PtSn alloy particles can change the electronic properties of Pt and decrease the size of the surface Pt ensembles, thus inhibiting the cleavage of C–C and C–O bond that lead to the production of CH4 and C2H6. Previous studies of acetic acid hydrogenation mainly focused on the reaction mechanism or kinetic behavior, and the data about acetic acid conversion and product selectivity are not much [5–8]. The ethane selectivity of 10Pt10Sn/A is much lower than that of Pt/TiO2 and other oxide supported Pt catalysts [8]. In another study, 40 % ethanol selectivity and about 10 % acetic acid conversion was achieved on a PtFe/SiO2 catalyst [35]. Thus, 10Pt10Sn/A is a promising catalyst for the hydrogenation of acetic acid to ethanol as revealed in Table 4 and the effect of temperature and pressure was studied to optimize the product selectivity.
3.10 The Effect of Temperature on Acetic Acid Conversion and Product Selectivity
The impact of reaction temperature on the activity and selectivity of 10Pt10Sn/A was studied from 468 to 548 K and displayed in Fig. 10. The original hydrogenation product aldehyde varies little in the selected temperature range and CH4 and C2H6 were only founded at 548 K, so only the selectivity of aldehyde, ethanol and ethyl acetate were presented here.

The effect of temperature on a acetic acid conversion and b product selectivity over 10Pt10Sn/A
It is shown that the conversion of acetic acid increased dramatically from 16 to 68 % over the temperature range studied due to the promotion of reaction rate caused by the rise of temperature. The selectivity of ethanol dropped markedly as the continued increase of temperature and the trend of selectivity of ethyl acetate is against that of ethanol, which may be caused by the difference of apparent activation energies between hydrogenation and esterification [8].
3.11 The Effect of Pressure on Acetic Acid Conversion and Product Selectivity
The influence of pressure has been investigated over the range of 0.1–4.0 MPa when other operation conditions were fixed (T = 528 K, H2/CH3COOH = 10, LHSV = 0.6 h−1) and the results are presented in Fig. 11. The conversion of acetic acid rose with the increase of pressure, which results from the enhanced density of active acetate surface species and the improved collision probability of reactants and the active sites on catalyst surface. At the same time, the selectivity of ethanol increased while that of ethyl acetate decreased, and that can be explained from the perspective of thermodynamics. The reduction of acetic acid to ethanol and the esterification of acetic acid and ethanol to ethyl acetate are two consecutive reactions. The first one is a molecules reduced reaction while the molecule number of the second one is constant. Thus, the increase of pressure is beneficial to the production of ethanol.

The effect of pressure on a acetic acid conversion and b product selectivity over 10Pt10Sn/A
4 Conclusions
Pt/Al2O3, Sn/Al2O3 and PtSn/Al2O3 were prepared for the gas phase hydrogenation of acetic acid. The addition of Sn can enhance the catalytic activity and the selectivity towards ethanol and ethyl acetate, as well as restrain the production of CH4 and C2H6. Operation conditions such as temperature and pressure can also affect the activity and selectivity of PtSn catalyst. Results from XRD and TEM shows that metal particles are highly dispersed on alumina surface and the textural property of the support has been changed little. The shift of reduction temperature of PtSn catalyst in H2-TPR indicated that Sn addition may cause co-reduction of Pt and Sn, thus lead to the formation of PtSn species. H2-pulse chemisorption and acetic acid DRIFTS revealed that the introduction of Sn lowered the dispersion of Pt and modify the property of Pt. At the same time, XPS demonstrated that H2 reduction is beneficial to the enrichment of Pt, Sn on the surface of monometallic catalysts while the introduction of Sn in PtSn catalyst is detrimental to the transportation of Pt to the catalyst surface, which can be ascribed to the presence of PtSn alloy or Pt-SnOx species. HRTEM and EDX revealed the presence of PtSn alloy and Pt-SnOx species after the reduction of 10Pt10Sn/A. Thus, the better performance of PtSn catalysts can be attributed to the formation of PtSn alloy and Pt-SnOx particles.
References
Maki Y, Sato K, Isobe A, Iwasa N, Fujita S, Shimokawabe M, Takezawa N (1998) Appl Catal A 170:269–275
Liu YY, Murata M, Inaba M, Takahara I, Okabe K (2011) Catal Today 164:308–314
Mei DH, Rousseau R, Kathmann SM, Glezakou VA (2010) J Catal 271:325–342
Yoneda N, Kusano S, Yasui M, Pujado P, Wilcher S (2001) Appl Catal A 221:253–265
Pestman R, Koster RM, Boellaard E, van der Kraan AM, Ponec V (1998) J Catal 174:142–152
Pestman R, van Duijne A, Pieterse JAZ, Ponec V (1995) J Mol Catal A Chem 103:175–180
Pestman R, Koster RM, Pieterse JAZ, Ponec V (1997) J Catal 168:255–264
Rachmady W, Vannice MA (2000) J Catal 192:322–334
Rachmady W, Vannice MA (2002) J Catal 207:317–330
Ayastuy JL, González-Marcos MP, Gutiérrez-Ortiz MA (2011) Catal Commun 12:895–900
Corro G, Fierro JLG, Romero FB (2006) Catal Commun 7:867–874
Corro G, Fierro JLG, Montiel R, Banuelos F (2005) J Mol Catal A 228:275–282
Siri GJ, Ramallo-Lópezc JM, Casella ML, Fierrod JLG, Requejo FG, Ferretti OA (2005) Appl Catal A 278:239–249
Bocanegra SA, de Miguel SR, Borbath I, Margitfalvi JL, Scelza OA (2009) J Mol Catal A 301:52–60
de Miguel SR, Bocanegra SA, Julieta Vilella IM, Guerrero-Ruiz A, Scelza OA (2007) Catal Lett 119:5–15
Shashikala V, Jung H, Shin CH, Koh HL, Jung KD (2013) Catal Lett 143:651–656
Bocanegra SA, De Miguel SR, Castro AA, Scelza OA (2004) Catal Lett 96:129–131
de Miguel SR, Román-Marínez MC, Jablonski EL, Fierro JLG, Cazorla-Amorós D, Scelza OA (1999) J Catal 184:514–525
Serrano-Ruiz JC, Huber GW, Sánchez-Castillo MA, Dumesic JA, Rodríguez-Reinoso F, Sepúlveda-Escribano A (2006) J Catal 241:378–388
Coloma F, Sepfilveda-Escribano A, Fierro JLG, Rodriguez-Reinoso F (1996) Appl Catal A 136:231–248
Coloma F, Sepfilveda-Escribano A, Fierro JLG, Rodriguez-Reinoso F (1996) Appl Catal A 148:63–80
He SB, Sun CL, Bai ZW, Dai XH, Wang B (2009) Appl Catal A 356:88–98
Pisduangdaw S, Panpranot J, Methastidsook C, Chaisuk C, Faungnawakij K, Praserthdam P, Mekasuwandumrong O (2009) Appl Catal A 370:1–6
Hoang DL, Farrage SA-F, Radnik J, Pohl M-M, Schneider M, Lieske H, Martin A (2007) Appl Catal A 333:67–77
Llorca J, de la Piscina PR, Fierro J-LG, Sales J, Horns N (1997) J Mol Catal A 118:101–111
Bariås OA, Holmen A, Blekkan EA (1996) J Catal 158:1–12
Yu CL, Ge QJ, Xu HY, Li WZ (2006) Appl Catal A 315:58–67
Margitfalvi JL, Tompos A, Kolosova I, Valyon J (1998) J Catal 174:246–249
Corro G, Fierro J-LG, Montiel R, Castillo S, Moran M (2003) Appl Catal B 46:307–317
Vu BK, Song MB, Ahn IY, Suh Y-W, Suh DJ, Kim W-I, Koh H-L, Choi YG, Shin EW (2011) Appl Catal A 400:25–33
Homs N, Llorca J, Riera M, Jolis J, Fierro J-LG, Sales J, de la Piscina PR (2003) J Mol Catal A 200:251–259
Vilella IMJ, de Miguel SR, de Lecea CS-M, Linares-Solano Á, Scelza OA (2005) Appl Catal A 281:247–258
Neri G, Milone C, Galvagno S, Pijpers APJ, Schwank J (2002) Appl Catal A 227:105–115
Homs N, Llorca J, de la Piscina PR, Rodríguez-Reinoso F, Sepúlveda-Escribano A, Silvestre-Albero J (2001) Phys Chem Chem Phys 3:1782–1788
Rachmady W, Vannice MA (2002) J Catal 209:87–98
Acknowledgments
This work is financially supported by the National Science and Technology Supporting Plan (2006BAE02B02).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Zhang, K., Zhang, H., Ma, H. et al. Effect of Sn Addition in Gas Phase Hydrogenation of Acetic Acid on Alumina Supported PtSn Catalysts. Catal Lett 144, 691–701 (2014). https://doi.org/10.1007/s10562-014-1210-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10562-014-1210-z