
Abstract
Bipolar moss species, extreme examples of large and disjunctive geographical ranges, belong to the most important components of the vegetation cover in Antarctica. Their appropriate taxonomic interpretations are essential for fine-scale biogeographical considerations but our knowledge on their phylogenetic affinities still remains limited. Here, we address the history of the Antarctic populations of Drepanocladus longifolius, a moss species widely considered a bipolar taxon. First, based on a worldwide sampling, we verify its assumed bipolar status. Then, we ask whether its current Antarctic populations have persisted throughout at least the last major glaciation in situ or, alternatively, are they a recent, postglacial element. Phylogenetic analyses together with investigation of morphological characters were used to infer relationships among accessions representing the whole distribution range of the taxon. Morphological and phylogenetic data strictly segregated specimens from the Southern and the Northern Hemisphere, with an exception of Australian populations that were included in the latter group. The Antarctic, sub-Antarctic and South American populations belong to a single clade and are treated as D. longifolius s. str., which consequently is not a bipolar taxon. All Northern Hemisphere populations and the ones from Australia represent D. capillifolius, a lately neglected taxon, which thus displays a true bipolar distribution pattern but not comprising the Antarctic and South American populations. These results provide a sound example of key importance of a detailed taxonomical and phylogenetic treatment as a basis for biogeographical studies. In D. longifolius s. str., a homogeneity in all non-coding nuclear and plastid regions was observed throughout the whole range. It contrasted with the presence of one missense mutation in the rps4 gene in all examined Antarctic populations, which correlated with their distinct ecological situation. While identity of non-coding sequences in D. longifolius populations indicates a postglacial recruitment of extant Antarctic populations from extra-regional locations rather than a long-term in situ survival, the mutation in the rps4 gene may be hypothesised to reflect an adaptive response of plants to the Antarctic environment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Evolutionary history of the Antarctic moss flora
Antarctic terrestrial and freshwater habitats are believed to have been almost entirely destroyed during glaciations in the Pliocene (5–2.6 Ma) and the Pleistocene (2.6 Ma–10 Ka), including the Last Glacial Maximum (LGM; around 22–17 Ka) (Convey et al. 2008, 2009). Nonetheless, it is likely that some species of lichens and mosses could have survived this climatic disaster in local refugia, for example, in the Mount Kyffin area in southern Victoria Land (Green et al. 2011). Extant terrestrial vegetation communities in Antarctica are mostly composed of cryptogamic species, especially bryophytes and lichens that are accompanied by significant numbers of species of microalgae and cyanobacteria (Ochyra et al. 2008a). The greatest diversity of bryophytes is described from the Antarctic Peninsula and Scotia Arc archipelagos, whereas in the coastal continental Antarctic, few species can survive the extreme conditions (Peat et al. 2007; Ochyra et al. 2008a; Convey 2013).
Colonisation of the Antarctic continent presents considerable challenges. This is due, among others, to the influence of oceanic fronts and strong circumpolar currents which encircle the Antarctica, increase its isolation and could have prevented many taxa from dispersing to this continent. Despite this, a recent (postglacial) origin for most of the bryophyte flora has been suggested (Peat et al. 2007; Ochyra et al. 2008a; Fraser et al. 2012; Kato et al. 2013; Biersma et al. 2018). On the other hand, abundance of plant fossils combined with a relatively high degree of endemism observed in some groups suggest that this continent may have played an important role in shaping biodiversity and plant distributions in the Southern Hemisphere. Indeed, some recent molecular studies indicated genetic isolation of Antarctic populations and supported possible long-term persistence within fragmented Antarctic habitats of such terrestrial organisms as springtails (Arthropoda: Collembola) (Stevens and Hogg 2003; McGaughran et al. 2010; McGaughran et al. 2011), midges (Arthropoda: Diptera) (Allegrucci et al. 2006), mites (Arthropoda: Acari) (Mortimer et al. 2011), green algae (De Wever et al. 2009) and, more recently, also bryophytes (Hills et al. 2010; Pisa et al. 2014; Biersma et al. 2017). Nevertheless, knowledge on the biogeographical history of the extant Antarctic terrestrial biota remains rudimentary and reconstructing a framework of colonisation and dispersal events within a phylogeographic context is much needed for various taxonomical and biogeographical elements.
To date, few possibly comprehensive molecular studies on Antarctic bryophytes are at hand to assist the discussion on their history throughout the glacial events of the Pleistocene. Available case studies suggest that the present-day Antarctic moss flora has been shaped by two dissimilar processes, namely post-LGM colonisation and range expansions from putative glacial refugia distributed in continental Antarctica (Hills et al. 2010; Kato et al. 2013; Pisa et al. 2014; Biersma et al. 2017, 2018). During the LGM, all low-altitude coastal regions in the maritime Antarctic are thought to have been covered by grounded ice and ice shelves, both probably extending away from the coastline (Convey et al. 2008). Based on current glaciological reconstruction, only two potential refugia were proposed in coastal areas of this region, at Alexander Island (for nematodes and microbes) and at Marguerite Bay (for arthropods) (Convey et al. 2009). In the coastal continental Antarctic regions, some biological and geological data support the existence of terrestrial and freshwater habitats throughout the Pleistocene and back to, at least, 1–2 Ma, if not to the Miocene glaciations of 3–5 Ma (Peat et al. 2007, and references therein). For example, Hills et al. (2010) and Pisa et al. (2014) supported Pleistocene persistence of a moss species Bryum argenteum Hedw. in Victoria Land refugia, suggesting that geothermal habitats in Antarctica might have played a key role in the long-term presence of moss species. Similarly, Biersma et al. (2017) investigated genetic variation among Antarctic populations of Polytrichum juniperinum Hedw., and have suggested their long-term presence within Antarctica. In addition to molecular studies, Burgess et al. (1994) recorded moss sub-fossils from lake sediments dated to at least 24,950 B.P. from the Larsemann Hills, providing an indication that some moss species could have survived the formation of the permanent ice sheets.
Bipolar plant distribution: a fascinating biogeographical phenomenon
The bipolar plant disjunctions belong to the most fascinating biogeographical phenomena. Bipolar disjunctive plants are distributed in the cool-temperate and polar regions of both the Northern and Southern Hemispheres with or without intermediate populations in tropical mountain areas, but absent from the tropical lowlands (Du Rietz 1940). The vast majority of bipolar disjuncts are clearly of Holarctic origin. There are only very few examples of mosses of Holantarctic origin and thus of the opposite way of dispersal, for instance Andreaea mutabilis Hook.f. & Wilson (Murray 1988), Bucklandiella lamprocarpa (Müll.Hal.) Bedn.-Ochyra & Ochyra (Ochyra et al. 1988; Bednarek-Ochyra and Ochyra 1998) and Polytrichum juniperinum (Biersma et al. 2017).
The mosses include many drought-tolerant taxa with lightweight spores and therefore candidates for long-distance dispersal (Van Zanten and Pócs 1981). Interestingly, bipolar species belong to the most widespread and common biogeographical elements among Antarctic mosses and constitute nearly half of the moss flora of Antarctica (Ochyra et al. 2008a). The problem of bipolar taxa of bryophytes has been discussed in many accounts (e.g. Schofield and Crum 1972; Schofield 1974; Schuster 1983; Ochyra and Zander 2002; Ochyra and Buck 2003; Ochyra et al. 2008b; Biersma et al. 2017; Lewis et al. 2017). It should be highlighted, however, that in many cases, the biogeographical status of species clearly depends on their taxonomic circumscriptions. Misidentification of species is one of the most problematic issues in resolving the biogeographical and evolutionary origins of bipolar taxa in Antarctica.
Here, we dissect this problem using an example of a plant species with controversial delimitation which heavily affects its biogeographical circumscription: the pleurocarpous moss species Drepanocladus longifolius (Mitt.) Paris.
Competing circumscriptions of Drepanocladus longifolius
While Drepanocladus longifolius has long been considered a Southern Hemisphere species, at present, it is usually treated as a bipolar species (Hedenäs 1998, 2003, 2008; Dierßen 2001; Hill et al. 2006; Ignatov et al. 2006; Hedenäs and Rosborg 2008). It results from a taxonomic decision assuming its conspecificity with D. capillifolius (Warnst.) Warnst., a species distributed in the Northern Hemisphere (Hedenäs 1997).
Drepanocladus longifolius is a distinct pleurocarpous species which is readily recognised by its lanceolate and gradually long-acuminate leaves with a strong, percurrent to long-excurrent costa (Ochyra et al. 2008a). As is the case with other mosses associated with aquatic or otherwise wet habitats, this species exhibits a remarkable phenotypic plasticity and consequently some distinct phenotypes have been recognised as species in their own right. Altogether, five additional species and one variety with stout and excurrent costae were described and placed in the genus Drepanocladus (Müll.Hal.) G.Roth in the narrow sense: D. abbreviatus Cardot & Broth., D. barbeyi (Renauld & Cardot) Paris, D. perplicatus (Dusén) G.Roth. and D. fluitans (Hedw.) Warnst. var. australe (Cardot) Broth. from South America, D. capillifolius from Europe, as well as D. crassicostatus Janssens from North America. Moreover, two species clearly belonging within that complex were described from sub-Antarctic Îles Crozet as Cratoneuron drepanocladioides Broth. and from Lake Titicaca in Peru as Sciaromium lacustre Herzog & P.W.Richards. The latter was a deepwater moss exhibiting a number of unique adaptations to such habitats and this prompted Ochyra (1986) to place it in a separate genus Richardsiopsis Ochyra. All these taxa were critically revised by Hedenäs (1997) who merged them with D. longifolius, except for D. perplicatus which was accepted as a separate species, with S. lacustre being conspecific with it. Thus, the Southern Hemisphere D. longifolius has been considered conspecific with the Northern Hemisphere D. capillifolius making it a bipolar species. The status of D. longifolius as a bipolar species has gained wide acceptance. It has been maintained that D. longifolius has a panholarctic distribution from the northern temperate to the southern arctic zones in the Northern Hemisphere and recurs in the Southern Hemisphere where it ranges throughout the South American Cordillera from Colombia to Tierra del Fuego and the Falkland Islands (Hedenäs 1997, 1998, 2003, 2008) and extends also to the northern maritime Antarctic and south-eastern Australia (Ochyra et al. 2008a).
In contrast, some authors claimed the specific distinctness of Drepanocladus longifolius and D. capillifolius on account of their morphology and anatomy, habits and geographical distribution. In his world taxonomic monograph of the D. aduncus (Hedw.) Warnst. group, Żarnowiec (2001) treated D. capillifolius as a distinct species with a pan-boreal-temperate range in the Holarctic and which differs in a set of morphological characters, including the shape of the alar cells from D. longifolius. This concept has also been supported by Ochyra and Matteri (2001) and Ochyra et al. (2008a).
The source of the discussed problem lies presumably in the great phenotypic variability among members of presented genus (Hedenäs 1998; Vanderpoorten et al. 2002a, 2002b). So far, published phylogenies have shown incongruence between molecular and morphological data sets for representatives of Drepanocladus s. str. (Hedenäs and Rosborg 2008; Hedenäs 2008, 2011). It has been unclear whether this variation is induced by environmental factors, or if there is a genetic component as well (Hedenäs 2008). It has been often explained by frequent homoplasy among morphological characters, possibly associated with particular habitat conditions (Żarnowiec 2001; Shaw et al. 2008; Stech et al. 2008; Hedenäs 2008). Despite this, the shape of the costa and alar cells has been reported as stable morphological diagnostic characters in this group (Żarnowiec 2001).
Drepanocladus longifolius—recent colonist or in situ survivor?
Contemporary Antarctic populations of D. longifolius are known exclusively from the maritime Antarctic (Ochyra et al. 2008a; Li et al. 2009). The recent origin of the Antarctic populations may be supported by occurrence of this species on sub-Antarctic islands and on neighbouring lands of South America and Australia. Moreover, sub-fossil findings in the Admiralty Bay area on King George Island, dated to 4950 B.P., also point to recent colonisation of this moss species (Birkenmajer et al. 1985).
On the other hand, aquatic forms of Drepanocladus longifolius from the Antarctic differ considerably from non-Antarctic populations. Antarctic forms of D. longifolius are apparently unique amongst other aquatic mosses in being submerged in water of lakes (Li et al. 2009). Moreover, it has never been reported on ground around the lakes, whereas other mosses are land forms which have become adapted to the aquatic environment. In contrast, plants from South American mainland and Tierra del Fuego usually thrive in swampy habitats and are seldom submerged in pools and streams. Being permanently submerged in deepwater results in admittedly different morphology. Aquatic mosses significantly differ from their terrestrial counterparts in size and shape of the leaves (Priddle 1979). Accordingly, Antarctic forms have straight, narrowly lanceolate and long-acuminate leaves, whereas the South American plants have predominantly strongly falcato-secund leaves (Ochyra et al. 2008a; Li et al. 2009).
Due to the morphological variability between the plants of Drepanocladus longifolius from the maritime Antarctic and South America, we are particularly interested in the origin, colonisation history and the degree to which Antarctic populations are genetically isolated from those in other Southern Hemisphere landmasses.
Goals of this study
With this study, we aim to contribute to an understanding of Antarctic biogeography. First, we assess the accurate taxonomic status of the austral and boreal populations of Drepanocladus species with excurrent costae to support or to refute the bipolar status of D. longifolius. After establishing the large-scale framework, we address phylogeographical relationships and genetic divergence of the populations of D. longifolius in the Southern Hemisphere with a special focus on possible origins of populations from the sub-Antarctic and Antarctic. More specifically, we address the following questions: whether the extant local populations of D. longifolius represent ancient floras which survived LGM in situ or, alternatively, have dispersed more recently in the postglacial period from disjunct populations located outside Antarctica (e.g. South America, sub-Antarctic islands, Australia). Finally, we briefly discuss our results in terms of the assembly processes of sub-Antarctic and Antarctic moss floras and of the challenges in accurate interpretation of bipolar taxa.
Material and methods
Plant material and taxon sampling
Plant sampling for our study was based on collections deposited in the bryophyte herbarium of the W. Szafer Institute of Botany, Polish Academy of Sciences (KRAM). This possibility allowed us to overcome the main problem of obtaining a meaningful geographical sampling, especially from the Southern Hemisphere and including the Antarctic region. We sampled from the whole distribution range of the species.
A total of 42 collections of the broadly defined Drepanocladus longifolius sensu Hedenäs (1997) were used in the phylogenetic analyses (Fig. 1; Table 1, Supplementary Table 1). In the case of the Northern Hemisphere, analysed plant material originated from North America (USA: Nevada, Colorado; Canada: Ontario, Yukon, Alberta), Asia (Russia: Siberia) and Europe [Poland; in addition, one specimen from Sweden deposited in the Swedish Museum of Natural History (SB52662) was verified taxonomically; DNA sequences of this specimen had been already available in the public database]. The Southern Hemisphere material included plants from all disjunct parts of the species distribution: South America, Australia, sub-Antarctic islands (South Georgia, Îles Crozet, Îles Kerguelen) and peri-Antarctic islands, including Signy Island (South Orkney Islands), King George Island and Livingston Island (South Shetland Islands), and Vega Island and James Ross Island (James Ross Island group off the Nordenskjöld Coast on the east coast of the Antarctic Peninsula). We also included two specimens which were identified by Hedenäs (1997) as D. perplicatus, accepted as a separate species with excurrent costae. This species was considered a South American endemic known from altimontane elevations in the Andes of Ecuador and Peru, the Serra do Itatiaia in south-eastern Brazil and Patagonia (Hedenäs 1997).

Geographical distribution of the studied accessions and detected genetic lineages corresponding to Drepanocladus longifolius (blue dots) and D. capillifolius (green triangles) according to present circumscription. In addition, populations of D. longifolius bearing a missense mutation in the rps4 gene are marked with blue stars
Our original sequences (185) were supplemented by 103 sequences previously published by Hedenäs and Rosborg (2008) and Vanderpoorten et al. (2003) to enlarge the taxon sampling (‘extended data set’ hereafter). Seventeen species in total were included in the analysis. Nine of them represented the genus Drepanocladus as well as supposedly D. latinervis Warnst. (species identification according to Hedenäs and Rosborg (2008) and marked here as Drepanocladus sp.). Based on the species circumscription of Vanderpoorten et al. (2002a, 2002b) and phylogenetic studies of Hedenäs and Rosborg (2008) showing the close relationship of Drepanocladus and Pseudocalliergon (Limpr.) Loeske, we additionally included five species which represent the latter genus. Finally, three species were designated as outgroup taxa, based on previous analyses (Vanderpoorten et al. 2002a, 2002b, 2003): Campylium stellatum (Hedw.) Lange & C.E.O.Jensen, Amblystegium serpens (Hedw.) Schimp. and Cratoneuropsis relaxa (Hook.f. & Wilson) M.Fleisch.
Geographical origin of the material studied, herbarium specimen and GenBank accession numbers of sequences are provided in Table 1 and in Supplementary Table 1.
Morphological analysis and taxonomic verification of specimens used in the genetic analysis
Identification of all specimens used for DNA isolation was critically verified taxonomically. The general morphology of all individual specimens was analysed using a stereomicroscope. More detailed observations on their anatomical structure were examined under a compound microscope.
DNA extraction, PCR amplification and sequencing
Dried plant material was ground using a TissueLyser II (Qiagen, Hilden, Germany) and whole genomic DNA was isolated using DNeasy Plant Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. Due to difficulties in amplification of regions from the degraded DNA inherent in herbarium samples, genomic DNA of all investigated samples was additionally purified and concentrated prior of PCR reaction using the Genomic DCC™-10 (Zymo Research Corporation, Irvine, California, USA). We targeted seven DNA regions. Five of them were previously used for phylogenetic inference in the group (Hedenäs and Rosborg, 2008). Additionally, we assessed utility of two further chloroplast DNA (cpDNA) regions previously unexplored in the genus (rps4 gene and atpH-atpI intergenic spacer). Thus, from the chloroplast genome, we sequenced intergenic spacers atpB-rbcL, atpH-atpI, trnL gene plus the adjacent trnL-trnF (GAA) spacer together as a single amplicon (trnL-F), transfer RNAGly (UCC) intron (trnG), intron of the rpL16 gene (rpL16) and 30S ribosomal protein S4 gene (rps4). The nuclear genome was represented by the ITS1-5.8S-ITS2 ribosomal DNA region (ITS). Primer names and sequences for all genomic regions utilised in this study are listed (Supplementary Table 2).
PCR amplification of the chloroplast regions was based on two protocols. A basic protocol used 25 μL reaction volumes containing 1× RedTaq PCR reaction buffer (Sigma-Aldrich, St. Louis, Missouri, USA), 0.2 mM dNTPs in equimolar ratio (Sigma-Aldrich, St. Louis, Missouri, USA), 0.08 mg/ml bovine serum albumin (New England BioLabs, Boston, Massachusetts, USA), 0.05 U/μL RedTaq DNA polymerase (Sigma-Aldrich, St. Louis, Missouri, USA), 0.2 μM each of the primers (Sigma-Aldrich, St. Louis, Missouri, USA) and 1 μL template DNA. The amplification conditions were as follows: 80 °C for 5 min (initial denaturation), followed by 35 cycles of 1 min at 95 °C (denaturation), 1 min at 50 °C (annealing) with a ramp of 0.3 °C/s, 4 min at 65 °C (extension), followed by 5 min at 65 °C (final extension). When this regime failed, a second protocol was used with 25-μL reaction volumes containing 1× AccuTaq LA buffer (Sigma-Aldrich, St. Louis, Missouri, USA), 0.4 mM dNTPs in equimolar ratio (Sigma-Aldrich, St. Louis, Missouri, USA), 0.2 U/μL AccuTaq LA DNA polymerase (Sigma-Aldrich, St. Louis, Missouri, USA), 0.32 μM each of the primers (Sigma-Aldrich, St. Louis, Missouri, USA) and 1 μL template DNA. The following PCR profile was used for this reaction: 96 °C for 30 s, 35 cycles of 1 min at 94 °C, 45 s at 48 °C and 10 min at 68 °C, followed by 30 min at 68 °C.
Amplification of nuclear ITS was carried out using the RedTaq reaction mix as described above and the following cycling protocol: 5 min at 94 °C, 35 cycles of 1 min at 94 °C, 1 min at 48 °C and 1 min (increased by 4 s per cycle) at 72 °C, followed by 5 min at 72 °C. ITS1 and ITS2 regions were amplified separately to increase PCR success. Amplification products were checked on 1% agarose gel and amplicons were cleaned using the DCC™-5 (Zymo Research Corporation, Irvine, California, USA). Sequencing was completed using BigDye Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher Scientific, San Jose, California, USA) together with BDX64 Sequencing Enhancement Buffer (Nimagen, Nijmegen, Netherlands), performed according to the manufacturer’s protocol for 32× dilution. Cycle sequencing reaction was: 3 min at 96 °C, followed by 30 cycles of 10 s at 96 °C, 5 s at 50 °C and 2 min at 60 °C. The products were sequenced in two directions using the amplification primers. All amplification and sequencing reactions were carried out using Mastercycler Nexus GSX1 and Nexus SX1e thermocyclers (Eppendorf, Hamburg, Germany). Sequencing products were purified using ethanol/EDTA precipitation and separated on ABI Prism 3130 automated DNA sequencer (Applied Biosystems, Foster City, California, USA). All newly generated sequences were deposited in the National Center for Biotechnology Information (NCBI) GenBank database.
Sequence data alignment
Sequences of each DNA region were aligned using CodonCode Aligner v.6.0.2 (CodonCode Corporation, Dedham, Massachusetts, USA) with Muscle algorithm set to default parameters. Alignments were checked by eye and trimmed manually using BioEdit v.7.2.5 (Hall 1999). All alignments can be found in Supplementary Files 1–5. In cases where several specimens representing one geographical region with identical sequences were present, we treated them as one (see Table 1 and Supplementary Table 1). For this purpose, identical consensus sequences were merged for certain genetic and geographical region, and subsequently were used in the analyses.
In the case of DNA regions previously used by Hedenäs and Rosborg (2008) and Vanderpoorten et al. (2003), our data were aligned together with these external sequences imported from the NCBI GenBank database.
For the remaining two newly explored cpDNA regions (atpH-atpI, rps4) for which no earlier data were available, pairwise distance values (p-distance) were calculated using MEGA7 v7.0.14 (Kumar et al. 2016) to assess sequence identities within our own data set.
Phylogenetic analysis—atpB-rbcL, rpL16, trnG, trnL-trnF and ITS
We employed Bayesian phylogenetic inference (BI) and maximum likelihood (ML) analyses to establish relationships among the studied populations including testing probable delimitation of southern and northern populations of Drepanocladus longifolius s.l. and assessing genetic diversity and affinities of the Antarctic populations.
BI and ML analyses were performed independently for the combined chloroplast sequence data set (cpDNA) and for the nuclear ITS. Assuming linked inheritance, all available cpDNA sequences were concatenated into a single data set (atbB-rbcL + rpL16 + trnG + trnL-trnF) with SeaView v.4.6.1. (Gouy et al. 2009). Samples that lacked sequence information for certain genetic regions were included but the missing nucleotide sites were marked with ‘?’, treated by the software as missing information.
RAxML v8.2.11 implemented in the Geneious v.10.1.3 (Biomatters, USA) was used to compute a maximum likelihood tree using the GTRGAMMA model, chosen because it includes the parameter G for rate heterogeneity among sites (Stamatakis et al. 2008; Stamatakis 2014). In RAxML, by default, G has 25 rate categories making the estimation of proportion of invariable sites (I) unnecessary because G mathematically accounts for I (Stamatakis 2006). ML branch support was estimated using 1000 rapid ML bootstrap pseudoreplicates.
Prior to Bayesian inference, appropriate evolutionary models were selected for the concatenated data set of four aligned chloroplast regions and for the nuclear ITS region using jmodeltest v2.1.6 (Darriba et al. 2012). To assess probable evolutionary models, likelihood scores were computed with the following settings: 11 substitution schemes, base frequencies on (+F), the proportion of invariable sites on (+I), rate variation on with 8 rate categories (+G, nCat = 8), ML optimised base tree, NNI search algorithm. Hence, each partition was assigned an appropriate site model. According to the Akaike information criterion (AIC), the TPM1uf+G and TPM1uf+I models were selected for cpDNA and ITS data, respectively.
BI analyses were run using MrBayes v3.2.2 (Ronquist et al. 2012). The cpDNA and ITS analyses were run for 1.0 × 107 generations. The first 25% of trees were discarded as burn-in, and from the remaining trees, 50% majority rule consensus trees were calculated. We used Tracer v1.6 to assess convergence, and to check if all parameters had effective sample sizes (ESS) greater than 200 (Rambaut et al. 2014). Maximum clade credibility trees were imported and visualised using FigTree v1.4.2 (http://tree.bio.ed.ac.uk/software/figtree/). When analysing tree topologies, clades were treated as distinct and well supported if they received Bayesian posterior probabilities (PP) ≥ 0.95, and maximum likelihood bootstrap percentages (BS) ≥ 80.
Estimates of evolutionary divergence between sequences—atpH-atpI, rps4
Because of the lack of sequence information in public databases for the atpH-atpI intergenic spacer and rps4 gene for the studied group, we analysed these separately. For this analysis, evolutionary divergence over sequence pairs between created groups was estimated using MEGA7 software. We defined seven geographical groups: Europe (Poland), Asia (Russia), North America (USA, Canada), South America (Ecuador, Peru, Bolivia, Chile, Argentina, Falkland Islands, including also Drepanocladus perplicatus from Brazil), sub-Antarctic islands (Îles Crozet, Îles Kerguelen, South Georgia), peri-Antarctic islands (King George Island, Livingstone Island) and Australia. The number of base differences per site between given groups was estimated separately for the atpH-atpI intergenic spacer and rps4 gene.
In addition, the coding rps4 nucleotide alignment was translated into protein alignment, using Geneious software. The open reading frame (ORF) has been chosen based on the longest ORF. To check correctness of the selected frame, we uploaded protein sequences into the NCBI blastp suite tool. Before the analysis, we successfully confirmed our protein sequence similarity to plastid 30S ribosomal protein S4 of Drepanocladus aduncus specimen with the accession number AY908241.
Data accessibility statement
DNA sequences (as listed in Table 1 and in Supplementary Table 1) have been deposited in GenBank.
Results
Morphological characters of the specimens used in the genetic analysis
Our observations performed on specimens spanning all major geographical regions of occurrence across the Northern and Southern Hemisphere confirmed the previously reported principal differences between Drepanocladus capillifolius and D. longifolius s. str. treated as two allopatric taxa (Ochyra and Matteri 2001; Żarnowiec 2001; Ochyra et al. 2008a) and can be summarised as follows: in D. longifolius, the alar cells form a small, inflated, ovate to ovate-triangular marginal auriculate group in the leaf angles, sharply separated from the adjacent basal cells and occupying about one third or less of the leaf insertion; in contrast, in D. capillifolius, they form a very distinct, inflated, transversely triangular group, reaching the costa or nearly so (Fig. 2, Supplementary Fig. 1a–d). The morphological delimitation of specimens was fully congruent with their phylogenetic delimitation (see below).

Micromorphological differences between Drepanocladus longifolius and D. capillifolius. Alar cells of D. longifolius a, b—from Lyall 47, Falkland Islands. Alar cells of D. capillifolius c―from Nelson 4262, USA, Wyoming (KRAM), d―from isolectotype of Hypnum capillifolium var. fallax Renauld, Canada, Quebec. Scale bar 100 μm
PCR amplification and sequencing success
Out of the 42 isolated DNA samples, amplification of the whole set of nuclear and chloroplast regions was successful in 16 specimens. In a further 19 isolates, we were able to obtain amplicons from large subsets of genomic regions. We were unable to produce amplicons in seven specimens, which were thus excluded from further analyses. In total, we obtained 185 individual sequences (Table 1, Supplementary Table 1).
Phylogenetic analysis and genetic divergence
All DNA regions used were informative for the phylogenetic analysis and delimitation of Drepanocladus species with excurrent costae. The combined data set of cpDNA sequences used for the phylogenetic analysis was 1967 bp long and included 21 sequences from the respective geographical areas from our analysis, and 22 sequences from the study of Hedenäs and Rosborg (2008) (Supplementary File 1). The ITS alignment was 712 bp long and included 15 sequences from the respective geographical areas from our analysis, 20 sequences from Hedenäs and Rosborg (2008) and one sequence from Vanderpoorten et al. (2003) (Supplementary File 2).
We did not use sequences of the following specimens: Pseudocalliergon brevifolium (Lindb.) Hedenäs from Svalbard (Hedenäs and Rosborg, 2008), due to alignment difficulties, and chloroplast sequences of D. longifolius from Chile (Vanderpoorten et al. 2003), due to low-quality sequences.
Comparison of alignment lengths, parsimony informative characters and mean pairwise distance values (p-distance) between all analysed sequences obtained from single ITS and cpDNA data set analysis are provided (Supplementary Table 3).
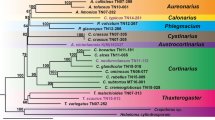
No topological conflicts were found between the BI and ML analyses, which yielded identical phylogenetic structure. Thus, we presented cpDNA and ITS trees with Bayesian branch support values and maximum likelihood bootstrap percentages (Fig. 3a, b).

Bayesian inference (BI) trees of Drepanocladus longifolius and related taxa reconstructed from a concatenated alignment of chloroplast markers atpB-rbcL, trnL-trnF, rpL 16, trnG, and b nuclear internal transcribed spacer (ITS1-5.8S-ITS2). Branch support values are Bayesian posterior probabilities (left side) and maximum likelihood bootstrap percentages (right side). Clades of D. longifolius s. str. and D. capillifolius are marked in blue and green, respectively (analogous to symbols in Fig. 1); sample names for specimens in these clades correspond with their respective geographical areas, and marked in Table 1, and in Supplementary Table 1. Specimens where complete data were available are in coloured font. Minor within-clade variation displayed D. longifolius s. str. and D. capillifolius is due to missing data in part of specimens
The BI and ML analyses of chloroplast and nuclear sequences strongly supported two major clades in Drepanocladus longifolius s.l., segregating specimens from the Southern Hemisphere (D. longifolius s. str.) and the Northern Hemisphere (D. capillifolius), with one notable exception (see below). Monophyly of D. longifolius and D. capillifolius clades has been supported in total by nine substitutions and one indel in both plastid and ITS data.
The cpDNA tree revealed four distinct and well-supported clades (Fig. 3a). The fully supported Drepanocladus longifolius s.s clade (PP = 1.0; BS = 98) consisted of genetically uniform specimens from South America, sub-Antarctic Islands and Antarctica. Moreover, representatives of D. perplicatus from South America, considered a separate species with an excurrent costa, had the same multilocus genotype and were contained in the D. longifolius clade. In contrast, previously published sequences treated as D. longifolius [two samples from Sweden: L.Hedenäs, M.Aronsson (S; B7692); L.Hedenäs (S; B52662)] were not contained within the D. longifolius clade but were identical to all other accessions from the Northern Hemisphere. All these samples formed a second well-supported clade, hereafter referred to as the D. capillifolius clade (PP = 0.98; BS = 95). Unexpectedly, this clade contained also samples from Australia, always described as D. longifolius in the literature (e.g. Streimann and Klanzenga 2002; Ochyra et al. 2008a). A third distinct, monophyletic group (PP = 1.00; BS = 97) was formed by accessions of D. sendtneri (Schimp. ex H.Müll.) Warnst., being sister to the D. longifolius clade. Representatives of D. arcticus (R.S.Williams) Hedenäs, D. polygamus (Schimp.) Hedenäs, D. latinervis Warnst. and an undetermined Drepanocladus sp. specimen from Hedenäs and Rosborg (2008) formed a fourth robust clade (PP = 1.00, BS = 100).
The nuclear ITS phylogeny showed mostly unresolved and weakly supported groups (Fig. 3b). Nevertheless, it also clearly divided Southern and Northern Hemisphere specimens into two major and well delimited clades, Drepanocladus longifolius (PP = 0.94; BS = 85) and D. capillifolius (PP = 0.97; BS = 80), respectively. In this respect, ITS data were fully congruent with the combined chloroplast DNA data set, except that Pseudocalliergon trifarium (F.Weber & D.Mohr) Broth. sample was nested within the D. capillifolius group. This specimen in the ITS analysis of Hedenäs and Rosborg (2008) also belongs to the D. capillifolius clade (Hedenäs and Rosborg, 2008 as D. longifolius). It should be noted that P. trifarium sample is separated from all analysed Drepanocladus species in cpDNA phylogeny also in Hedenäs and Rosborg (2008) but this discrepancy was not discussed by the authors. Unfortunately, we were unable to study this material, and thus, we treat the affinity of this specimen with caution. Besides the two above major clades obtained from ITS data, another with significant posterior probabilities grouped D. arcticus accessions (PP = 0.98; BS = 97), but unlike in the combined chloroplast data set analysis, the D. arcticus representatives grouped independently, rather than being sister to the D. latinervis and D. polygamus species.
In the case of atpH-atpI and rps4 sequences, genetic markers showed low p-distance values among defined geographical groups (0.00000–0.00402 for atpH-atpI; 0.00000–0.00826 for rps4), indicating their weak differentiation (Supplementary Table 4). Nonetheless, we observed again a clear delimitation of Drepanocladus longifolius s. str. and D. capillifolius specimens based on genetic distances. It is supported by two substitutions and one indel among the analysed plant material (Supplementary Files 3,4). In samples from the Antarctic islands (South Shetland Islands: King George Island and Livingstone Island), a single nucleotide substitution in rps4 gene was detected, differentiating the Antarctic populations from the remaining groups. Interestingly, translation of this nucleotide alignment reveals a missense mutation in the rps4 gene encoding protein S4 (Supplementary File 5).
Discussion
Relationships between austral and boreal populations: complicated fate of bipolar taxa
In the first step of the present study, we attempted to circumscribe the evolutionary relationships of the Antarctic populations of Drepanocladus longifolius to verify its presumed bipolar character and thus establish a basis for a further detailed phylogeographical analysis. While currently, a wide concept of D. longifolius as a bipolar taxon predominates in literature (Hedenäs 1997, 1998, 2003; Hedenäs and Rosborg 2008), some authors propose that populations from the Southern and Northern Hemispheres are accommodated by two distinct taxa, D. longifolius in a narrow sense and D. capillifolius, respectively (Dierßen 2001; Ochyra and Matteri 2001; Żarnowiec 2001; Ochyra et al. 2008a; Allen 2014; see also Introduction). Distinction between these two concepts is an essential prerequisite for perceiving the history of Antarctic and other austral parts of the range.
Our morphological and molecular data sets coherently supported distinction of austral and boreal populations as two divergent, allopatric species and showed that a wide bipolar distribution is not an appropriate framework for our case study. Even though the phylogenetic trees are not fully resolved and polytomies blur specific relationships among taxa, the two evolutionary groups form monophyletic and well-supported clades in our plastid and nuclear DNA data sets. Importantly, they are also coherently delimited morphologically based on diagnostic shape and size of the alar cells’ group. Hence, Drepanocladus longifolius, comprising the Antarctic populations, is limited to the Southern Hemisphere and thus is not a bipolar species. In fact, it is a south-cool-temperate amphi-Atlantic species, having the main centre of its occurrence in southern South America, from where it penetrates into the tropics along the Andean chain and extends to the northern maritime Antarctic and sub-Antarctic islands of South Georgia, Îles Crozet and Îles Kerguelen.
Northern Hemisphere populations belong to Drepanocladus capillifolius. Consequently, our data do not support the concept of Hedenäs (1997, 1998, 2003) and Hedenäs and Rosborg (2008) suggesting the conspecificity of D. longifolius and D. capillifolius making it a single, widely distributed bipolar species. Unexpectedly, however, the demonstrated affinity of the populations from south-eastern Australia to the otherwise northern taxon, gives D. capillifolius the status of a truly bipolar species. Thus, a bipolar taxon has indeed been identified but it does not comprise the Antarctic and South American populations.
Our case study provides a sound example of key importance of a detailed taxonomical and phylogenetic treatment as a basis for biogeographical (including phylogeographical) studies. This is especially true for large and disjunct distribution ranges exemplified at its extreme by bipolar patterns.
Antarctic populations in the context of the Southern Hemisphere phylogeography
In our comprehensive analysis based on sequences of seven DNA regions, all specimens of Drepanocladus longifolius from the maritime Antarctic populations formed a monophyletic and fully supported group that also contained plants from the sub-Antarctic islands and South America. Our analyses demonstrated a complete lack of variability in all non-coding plastid and nuclear DNA regions examined in this group. The genetic variation observed in D. longifolius was limited to a single nucleotide substitution in the rps4 gene found in the Antarctic populations from King George Island and Livingstone Island (see also next subchapter).
This general sequence similarity (with the exception of the rps4 single nucleotide substitution) among disjunct Drepanocladus longifolius populations supports the notion of its postglacial recruitment from extra-regional locations, rather than long-term persistence in situ in the Antarctic. Such a scenario is congruent with the biogeographical history of another hydrophytic Antarctic moss species, Pohlia wilsonii (Mitt.) Ochyra, for which immigration into Antarctic lakes via long-distance dispersal from southern South America was suggested (Kato et al. 2013 as Leptobryum wilsonii (Mitt.) Broth.). To date, most phylogenetic studies based on variable molecular markers support dispersal scenarios, rather than geographical vicariance, as the most probable explanation of disjunct ranges of bryophytes (Shaw et al. 2003; Vanderpoorten et al. 2008; Kato et al. 2013; Biersma et al. 2018; but see Hills et al. 2010; Pisa et al. 2014; Biersma et al. 2017).
Postglacial (re)colonisation of the Antarctic by Drepanocladus longifolius is further supported by several lines of evidence. The oldest known remains of D. longifolius in the area were reported by Ingólfsson et al. (1992) (as D. cf. aduncus) from sediments deposited at Cape Lachman, James Ross Island, and dated to 9525 ± 65 years BP. These low-altitude coastal regions in the maritime Antarctic are believed to have at most been ice-free during the last 10,000 years after the ice had retreated (Peat et al. 2007). It therefore supports, in congruence with our data, a dynamic post-LGM colonisation of this moss species rather than its persistence throughout the Pleistocene in the maritime Antarctic.
It is very likely that Drepanocladus longifolius or a closely related taxon occurred in the Antarctic much earlier but it was eradicated from this continent following past climatic deterioration. In the exceptionally well-preserved fossils from a small moraine-dammed basin near Mount Boreas in the McMurdo Dry Valleys sector of the Transantarctic Mountains in Victoria Land, the remains of a Drepanocladus with an excurrent costa were discovered (Lewis et al. 2008). They were determined as D. longifolius but because the alar region of the leaves is not clearly visible, in accordance with our results, it is impossible to state correctly whether the material represented D. longifolius s. str. or D. capillifolius. Taking into consideration the fact that Victoria Land was situated in the close proximity of south-east Australia and juxtaposed Tasmania (Griffiths 1971), one can speculate that this material may represent D. capillifolius rather than D. longifolius in its present circumscription. In any event, Lewis et al. (2008) have confirmed that Drepanocladus species with an excurrent costa inhabited the Antarctic continent before the onset of cooling that first brought a full polar climate to Antarctica at least 13.85 Ma, referred to as the middle-Miocene climatic transition (MMCT). It is assumed that these dry and freezing conditions have not subsequently been reversed. It is noteworthy that no species of Drepanocladus have been re-established in the continental Antarctic as this genus is currently absent from this area. This may suggest that Drepanocladus species are not able to withstand full glacial regimes, permanent ice cover and/or ice scour.
In the Antarctic lakes, Drepanocladus longifolius lives predominantly at greater depths of water, often under a thick ice cover which may persist on the lakes for long periods of time (Light and Heywood 1973). For example, in winter, the thickness of the ice covering the West Lake on King George Island is ca. 1–1.2 m, whereas D. longifolius was documented growing submerged in this lake at 5–6 m depth (Li et al. 2009). Interestingly, D. longifolius has never been found in lakes where ice scour could occur, as reported from Signy Island (South Orkney Islands) by Light and Heywood (1973). Priddle (1979) reported that specimens of two aquatic moss species, D. longifolius [as D. cf. aduncus] and Warnstorfia sarmentosa (Wahlenb.) Hedenäs, transported frozen for experiments, did not recover viability after thawing. On the other hand, La Farge et al. (2013) demonstrated in vivo regeneration of moss species that had been frozen under a glacier for approximately 400 years. Bryophytes are well known for their ability to survive cold conditions and thus it appears that such abilities may vary considerably among moss species.
Long-distance dispersal by wind is often invoked to explain biotic similarities between distant landmasses in the Southern Hemisphere (Muñoz et al. 2004 and references therein). Spores of bryophytes can be transported by air currents over short and even moderately long distances, provided that spores are very small (6–40 μm in diameter) (Van Zanten 1976). In this context, it is important to note that Drepanocladus longifolius is a dioicous species rarely producing sporophytes. As a consequence, the plants are mostly sterile (Hedenäs 1997; Żarnowiec 2001; Ochyra et al. 2008a). Taking this into account, airborne spore transport seems rather unlikely as a mechanism explaining effective (re)colonisation of Antarctic lakes by this species following Pleistocene glaciations. In this case, it seems more plausible that D. longifolius could have dispersed from source populations (continental refugia?) by migratory birds rather than by wind. Birds are known as dispersal vectors responsible for different types of disjunctions of many plant and lichen taxa (Du Rietz 1940). Lewis et al. (2014) provided first evidence for avian ectozoochory as an important factor in regional and global dispersal of bryophytes. They suggested that vegetative fragments of mosses are significant dispersal units for ectozoochory. In appropriate conditions (suitable habitat and period of the year), moss fragments could easily develop into mature plants, according to their totipotency capacities (Ward 1960; Sabovljević et al. 2003; Vujičić et al. 2012).
Although lack of genetic variability of populations suggests a post-LGM colonisation for Drepanocladus longifolius in the Antarctic, for the same reason, it is difficult to propose location of its refugia and reconstruct its dispersal route. North to south colonisation, most likely from the continental part of its South American range, seems the most plausible scenario. However, a hypothesis of local terrestrial refugia in the considerably more southern areas, such as southern South America and/or sub-Antarctic Islands could not be excluded. Contemporary molecular studies, as well as palaeoecological and palaeoenvironmental research, support the persistence of plants on sub-Antarctic islands through the most recent glacial periods (Van der Putten et al. 2004; Van der Putten et al. 2009; Van der Putten et al. 2010; Wagstaff et al. 2011; Bartish et al. 2012). Moreover, there is geological evidence that some terrestrial areas of the sub-Antarctic islands, e.g. Îles Crozet, the Falkland Islands and Macquarie Island, remained at least in part ice free during the LGM (Hall 2004). Nevertheless, patterns of glaciations of sub-Antarctic islands remain unclear. Cold tolerant taxa may have endured also glacial periods in multiple local refugia distributed in central and southern Patagonia west and east of the Andes and in the Tierra del Fuego archipelago (southern South America). Some of these survivors likely managed range expansion towards higher latitudes after glacial periods (Fraser et al. 2012 and references therein).
In summary, the historical biogeography of Drepanocladus longifolius seems to conform to a scenario which assumes that the majority of the Antarctic flora has originated fairly recently, with re-colonisation following the retreat of glaciers and ice sheets from their Pleistocene maxima (Peat et al. 2007 and references therein).
Phylogeographical signal or trace of genetic adaptation in the Antarctic populations?
The unique genetic polymorphism observed in our Drepanocladus longifolius data set was found in the only coding DNA region analysed. It constituted a missense single nucleotide substitution in a gene encoding plastid ribosomal protein S4, observed in the Antarctic populations from King George Island and Livingstone Island. While it may indicate a divergence of these populations due to isolation, this interpretation is challenged by a complete lack of variability observed in several non-coding regions examined. Interestingly, the detected single nucleotide polymorphism, causing alteration in amino-acid sequence of the gene, is correlated not only with geographical distribution but also with observed variation in ecological forms of the moss (deeply submerged aquatic in the Antarctic vs. shallow aquatic in other regions). Thus, it may alternatively be hypothesised that this genetic variation could have arisen as an adaptive response to the different light conditions. In land plants, protein S4 is involved in the assembly process of the 30S ribosomal subunit (Tahar et al. 1986; Harris et al. 1994; Yamaguchi et al. 2002). In general, the S4 protein is responsible for translational accuracy and is sensitive to environmental conditions (Harris et al. 1994; Manuell et al. 2007). It is well known that protein synthesis in the chloroplast is regulated in a light-dependent manner through effects on oxidation-reduction potential (Sharma et al. 2007). Deng et al. (2016) presented some significant positive correlation between the number of moss rps4 gene copies and chlorophyll a content, indicating a high accuracy in determining moss biomass.
In the maritime Antarctic, Drepanocladus longifolius is a typically hydrophytic moss species, growing permanently submerged in water of lakes and being a dominant element of the vegetation there. Notwithstanding the general light specificity of polar regions, due in particular to extended isolation from sunlight during the polar night, lakes additionally present a different photosynthetic environment from the land ecosystem. Priddle (1979) suggested that the different depth distribution of two aquatic mosses from Signy Island, D. longifolius (as D. cf. aduncus) and Warnstorfia sarmentosa were linked with their growth plasticity what could also be associated with their photosynthetic capacity. It should be noted that the low light levels in the deeper water of Antarctic lakes is not a limiting factor for vigorous growth of the aquatic mosses.
While the adaptive nature of the rps4 gene mutation observed in Drepanocladus longifolius remains hypothetical, it is suggested that single-site mutations could be a response to selective pressures (Tahar et al. 1986). Confronting this modification with a lack of non-coding (neutral) variability and palaeobotanical data, both indicating a recent (postglacial) establishment of extant lineages, may reflect an adaptive response of plants to the harsh Antarctic environment. This aspect requires additional, dedicated studies.
Conclusions
Complete lack of variability in all examined non-coding plastid and nuclear DNA regions among disjunct Drepanocladus longifolius populations supports its postglacial recruitment from extra-regional locations, rather than long-term persistence in situ in the Antarctic. However, due to a general lack of variation across the range, it is difficult to propose a location of its refugia and reconstruct its dispersal route. In contrast to homogeneous non-coding DNA regions, in representatives of D. longifolius from the maritime Antarctic, a missense mutation in the gene encoding plastid ribosomal protein S4 was detected. This variation could reflect an adaptive response to different light conditions correlated with observed variation in ecological forms: deeply submerged aquatic in the Antarctic vs. shallow aquatic in other regions.
References
Allegrucci, G., Carchini, G., Todisco, V., Convey, P., & Sbordoni, V. (2006). A molecular phylogeny of Antarctic Chironomidae and its implications for biogeographical history. Polar Biology, 29(4), 320–326.
Allen, B. (2014). Maine mosses Drummondiaceae–Polytrichaceae. Memoirs of the New York Botanical Garden, 111(1−15), 1–607.
Bartish, I. V., Aïnouche, A., Jia, D., Bergstrom, D., Chown, S. L., Winkworth, R. C., & Hennion, F. (2012). Phylogeny and colonization history of Pringlea antiscorbutica (Brassicaceae), an emblematic endemic from the South Indian Ocean Province. Molecular Phylogenetics and Evolution, 65(2), 748–756.
Bednarek-Ochyra, H., & Ochyra, R. (1998). Racomitrium lamprocarpum (Müll. Hal.) Jaeg.—an addition to the moss flora of Îles Kerguelen and the Subantarctic. Journal of Bryology, 20(2), 525–528.
Biersma, E. M., Jackson, J. A., Hyvönen, J., Koskinen, S., Linse, K., Griffiths, H., & Convey, P. (2017). Global biogeographic patterns in bipolar moss species. Royal Society Open Science, 4(7), 170147.
Biersma, E. M., Jackson, J. A., Bracegirdle, T. J., Griffiths, H., Linse, K., & Convey, P. (2018). Low genetic variation between South American and Antarctic populations of the bank-forming moss Chorisodontium aciphyllum (Dicranaceae). Polar Biology, 41(4), 599–610.
Birkenmajer, K., Ochyra, R., Olsson, I. U., & Stuchlik, L. (1985). Mid-holocene radiocarbon-dated peat at Admiratly, Bay, King George Island (South Shetland islands, West Antarctica). Bulletin of the Polish Academy of Sciences, Earth Sciences, 33(1−2), 7–13.
Burgess, J. S., Spate, A. P., & Shevlin, J. (1994). The onset of deglaciation in the Larsemann Hills, Eastern Antarctica. Antarctic Science, 6(4), 491–495.
Convey, P. (2013). Antarctic ecosystems. In S. A. Levin (Ed.), Encyclopedia of biodiversity. Ed. 2 (Vol. 1, pp, 179−188). San Diego: Elsevier.
Convey, P., Gibson, J. A., Hillenbrand, C. D., Hodgson, D. A., Pugh, P. J., Smellie, J. L., & Stevens, M. I. (2008). Antarctic terrestrial life – challenging the history of the frozen continent? Biological Reviews, 83(2), 103–117.
Convey, P., Stevens, M. I., Hodgson, D. A., Smellie, J. L., Hillenbrand, C. D., Barnes, D. K., et al. (2009). Exploring biological constraints on the glacial history of Antarctica. Quaternary Science Reviews, 28(27), 3035–3048.
Darriba, D., Taboada, G. L., Doallo, R., & Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nature Methods, 9(8), 772–772.
De Wever, A., Leliaert, F., Verleyen, E., Vanormelingen, P., Van der Gucht, K., Hodgson, D. A., et al. (2009). Hidden levels of phylodiversity in Antarctic green algae: further evidence for the existence of glacial refugia. Proceedings of the Royal Society of London B: Biological Sciences, 276(1673), 3591–3599.
Deng, S., Wang, C., Philippis, R., Zhou, X., Ye, C., & Chen, L. (2016). Use of quantitative PCR with the chloroplast gene rps4. Biology and Fertility of Soils, 52(5), 595–599.
Dierßen, K. (2001). Distribution, ecological amplitude and phytosociological characterization of European bryophytes. Bryophytorum Bibliotheca, 56, 1–289.
Du Rietz, G. E. (1940). Problems of bipolar plant distribution. Acta Phytogeographica Suecica, 13, 215–282.
Fraser, C. I., Nikula, R., Ruzzante, D. E., & Waters, J. M. (2012). Poleward bound: biological impacts of southern hemisphere glaciation. Trends in Ecology & Evolution, 27(8), 462–471.
Gouy, M., Guindon, S., & Gascuel, O. (2009). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Molecular Biology and Evolution, 27(2), 221–224.
Green, T. G. A., Sancho, L. G., Türk, R., Seppelt, R. D., & Hogg, I. D. (2011). High diversity of lichens at 84°S, Queen Maud Mountains, suggested preglacial survival of species in the Ross Sea region, Antarctica. Polar Biology, 34(8), 1211–1220.
Griffiths, J. R. (1971). Reconstruction of the South-west Pacific margin of Gondwanaland. Nature, 234, 203–207.
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. In Nucleic Acids Symposium Series (Vol. 41, no. 41, pp. 95−98). [London]: Information Retrieval Ltd., c1979–c2000.
Hall, K. (2004). Quaternary glaciation of the sub-Antarctic Islands. Developments in Quaternary Sciences, 2, part C, 339−345.
Harris, E. H., Boynton, J. E., & Gillham, N. W. (1994). Chloroplast ribosomes and protein synthesis. Microbiological Reviews, 58(4), 700–754.
Hedenäs, L. (1997). The Drepanocladus s. str. species with excurrent costae (Amblystegiaceae). Nova Hedwigia, 64(3), 535–548.
Hedenäs, L. (1998). An overview of the Drepanocladus sendtneri complex. Journal of Bryology, 20(1), 83–102.
Hedenäs, L. (2003). The European species of the Calliergon-Scorpidium-Drepanocladus complex: including some related or similar species. Meylania, 28, 1–116.
Hedenäs, L. (2008). Molecular variation in Drepanocladus aduncus s.l. does not support recognition of more than one species in Europe. Journal of Bryology, 30(2), 108–120.
Hedenäs, L. (2011). Incongruence among morphological species circumscriptions and two molecular datasets in Sarmentypnum (Bryophyta: Calliergonaceae). Taxon, 60(6), 1596–1606.
Hedenäs, L., & Rosborg, C. (2008). Pseudocalliergon is nested within Drepanocladus (Bryophyta: Amblystegiaceae). Lindbergia, 33, 67–74.
Hill, M. O., Bell, N., Bruggeman-Nannenga, M. A., Brugués, M., Cano, M. J., Enroth, J., Flatberg, K. I., Frahm, J. P., Gallego, M. T., Garilleti, R., Guerra, J., Hedenäs, L., Holyoak, D. T., Hyvönen, Ignatov, M. S., Lara, F., Mazimpaka, V., Muñoz, J., & Söderström, L. (2006). An annotated checklist of the mosses of Europe and Macaronesia. Journal of Bryology, 28(3), 198–267.
Hills, S. F., Stevens, M. I., & Gemmill, C. E. (2010). Molecular support for Pleistocene persistence of the continental Antarctic moss Bryum argenteum. Antarctic Science, 22(6), 721–726.
Ignatov, M. S., Afonina, M. O., Ignatova, E. A., Abolina, A., Akatova, T. V., Baisheva, E. Z., et al. (2006). Check-list of mosses of East Europe and North Asia. Arctoa, 15, 1–130.
Ingólfsson, Ó., Hjort, C., Björck, S., & Lewis Smith, R. I. (1992). Late Pleistocene and Holocene glacial history of James Ross Island, Antarctic Peninsula. Boreas, 21(3), 209–222.
Kato, K., Arikawa, T., Imura, S., & Kanda, H. (2013). Molecular identification and phylogeny of an aquatic moss species in Antarctic lakes. Polar Biology, 36(11), 1557–1568.
Kumar, S., Stecher, G., & Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution, 33(7), 1870–1874.
La Farge, C., Williams, K. H., & England, J. H. (2013). Regeneration of Little Ice Age bryophytes emerging from a polar glacier with implications of totipotency in extreme environments. Proceedings of the National Academy of Sciences, 110(24), 9839–9844.
Lewis, A. R., Marchant, D. R., Ashworth, A. C., Hedenäs, L., Hemming, S. R., Johnson, J. V., et al. (2008). Mid-Miocene cooling and the extinction of tundra in continental Antarctica. Proceedings of the National Academy of Sciences, 105(31), 10676–10680.
Lewis, L. R., Behling, E., Gousse, H., Qian, E., Elphick, C. S., Lamarre, J. F., Bêty, J., Liebezeit, J., Rozzi, R., & Goffinet, B. (2014). First evidence of bryophyte diaspores in the plumage of transequatorial migrant birds. Peer Journal, 2, e424.
Lewis, L. R., Biersma, E. M., Carey, S. B., Holsinger, K., McDaniel, S. F., Rozzi, R., & Goffinet, B. (2017). Resolving the northern hemisphere source region for the long-distance dispersal event that gave rise to the South American endemic dung moss Tetraplodon fuegianus. American Journal of Botany, 104(11), 1651–1659.
Li, S. P., Ochyra, R., Wu, P. C., Seppelt, R. D., Cai, M. H., Wang, H. Y., & Li, C. S. (2009). Drepanocladus longifolius (Amblystegiaceae), an addition to the moss flora of King George Island, South Shetland Islands, with a review of Antarctic benthic mosses. Polar Biology, 32(10), 1415–1425.
Light, J. J., & Heywood, R. B. (1973). Deep-water mosses in Antarctic lakes. Nature, 242(5399), 535–536.
Manuell, A. L., Quispe, J., & Mayfield, S. P. (2007). Structure of the chloroplast ribosome: novel domains for translation regulation. PLoS Biology, 5(8), e209.
McGaughran, A., Stevens, M. I., & Holland, B. R. (2010). Biogeography of circum-Antarctic springtails. Molecular Phylogenetics and Evolution, 57(1), 48–58.
McGaughran, A., Stevens, M. I., Hogg, I. D., & Carapelli, A. (2011). Extreme glacial legacies: a synthesis of the Antarctic springtail phylogeographic record. Insects, 2(2), 62–82.
Mortimer, E., van Vuuren, B. J., Lee, J. E., Marshall, D. J., Convey, P., & Chown, S. L. (2011). Mite dispersal among the Southern Ocean Islands and Antarctica before the last glacial maximum. Proceedings of the Royal Society of London B: Biological Sciences, 278(1709), 1247–1255.
Muñoz, J., Felicísimo, Á. M., Cabezas, F., Burgaz, A. R., & Martínez, I. (2004). Wind as a long-distance dispersal vehicle in the Southern Hemisphere. Science, 304(5674), 1144–1147.
Murray, B. M. (1988). The genus Andreaea in Britain and Ireland. Journal of Bryology, 15(1), 17–82.
Ochyra, R. (1986). On the taxonomic position of Sciaromium lacustre Herz & Rich in Rich. Journal of Bryology, 14(1), 109–115.
Ochyra, R., & Buck, W. R. (2003). Arctoa fulvella, new to Tierra del Fuego, with notes on trans-American bipolar bryogeography. The Bryologist, 106(4), 532–538.
Ochyra, R. & Matteri, C. M. (2001). Amblystegiaceae. In S. A. Guerrera, I. J. Gamundi de Amos & C. M. Matteri (Eds.), Flora criptogámica de Tierra del Fuego (Tomo 14, Fasciculo 10, pp. 1−96). Buenos Aires: Consejo Nacional de Investigaciones Científicas y Técnicas de la Republica Argentina.
Ochyra, R., & Zander, R. H. (2002). The genera Didymodon and Bryoerythrophyllum (Pottiaceae) in Antarctica. Journal of Bryology, 24(1), 33–44.
Ochyra, R., Sérgio, C., & Schumacker, R. (1988). Racomitrium lamprocarpum (C. Muell.) Jaeg., an austral moss disjunct in Portugal, with taxonomic and phytogeographic notes. Bulletin du Jardin Botanique National de Belgique, 58(1–2), 225–258.
Ochyra, R., Lewis Smith, R. I., & Bednarek-Ochyra, H. (2008a). The illustrated moss flora of Antarctica. Cambridge: Cambridge University Press.
Ochyra, R., Bednarek-Ochyra, H., & Lewis Smith, R. I. (2008b). New and rare moss species from the Antarctic. Nova Hedwigia, 87(3–4), 457–477.
Peat, H. J., Clarke, A., & Convey, P. (2007). Diversity and biogeography of the Antarctic flora. Journal of Biogeography, 34(1), 132–146.
Pisa, S., Biersma, E. M., Convey, P., Patiño, J., Vanderpoorten, A., Werner, O., & Ros, R. M. (2014). The cosmopolitan moss Bryum argenteum in Antarctica: recent colonisation or in situ survival? Polar Biology, 37(10), 1469–1477.
Priddle, J. (1979). Morphology and adaptation of aquatic mosses in an Antarctic lake. Journal of Bryology, 10(4), 517–529.
Rambaut, A., Suchard, M. A., Xie, D., & Drummond, A. J. (2014). Tracer v1.6. https://beast.bio.ed.ac.uk/Tracer.
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., ... Huelsenbeck, J. P. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology, 61(3), 539−542.
Sabovljevic, M., Bijelovic, A., & Dragicevic, I. (2003). In vitro culture of mosses: Aloina aloides (K.F.Schultz) Kindb., Brachythecium velutinum (Hedw.) B.S. & G., Ceratodon purpureus (Hedw.) Brid., Eurhynchium praelongum (Hedw.) B.S. & G. and Grimmia pulvinata (Hedw.) Sm. Turkish Journal of Botany, 27(6), 441–446.
Schofield, W. B. (1974). Bipolar disjunctive mosses in the Southern Hemisphere, with particular reference to New Zealand. The Journal of the Hattori Botanical Laboratory, 38, 13–32.
Schofield, W. B., & Crum, H. A. (1972). Disjunctions in bryophytes. Annals of the Missouri Botanical Garden, 59, 174–202.
Schuster, R. M. (1983). Phytogeography of the bryophyta. In R. M. Schuster (Ed.), New manual of bryology (Vol. 1, pp. 463–626). Nichinan: The Hattori Botanical Laboratory.
Sharma, M. R., Wilson, D. N., Datta, P. P., Barat, C., Schluenzen, F., Fucini, P., & Agrawal, R. K. (2007). Cryo-EM study of the spinach chloroplast ribosome reveals the structural and functional roles of plastid-specific ribosomal proteins. Proceedings of the National Academy of Sciences, 104(49), 19315–19320.
Shaw, A. J., Werner, O., & Ros, R. M. (2003). Intercontinental Mediterranean disjunct mosses: morphological and molecular patterns. American Journal of Botany, 90(4), 540–550.
Shaw, A. J., Holz, I., Cox, C. J., & Goffinet, B. (2008). Phylogeny, character evolution, and biogeography of the Gondwanic moss family Hypopterygiaceae (Bryophyta). Systematic Botany, 33(1), 21–30.
Stamatakis, A. (2006). RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22(21), 2688–2690.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics, 30, 1312–1313.
Stamatakis, A., Hoover, P., & Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Systematic Biology, 57(5), 758–771.
Stech, M., Sim-Sim, M., Esquível, M. G., Fontinha, S., Tangney, R., Lobo, C., et al. (2008). Explaining the ‘anomalous’ distribution of Echinodium (Bryopsida: Echinodiaceae): independent evolution in Macaronesia and Australasia. Organisms Diversity and Evolution, 8(4), 282–292.
Stevens, M. I., & Hogg, I. D. (2003). Long-term isolation and recent range expansion from glacial refugia revealed for the endemic springtail Gomphiocephalus hodgsoni from Victoria Land, Antarctica. Molecular Ecology, 12(9), 2357–2369.
Streimann, H., & Klanzenga, N. (2002). Catalogue of Australian mosses. Flora of Australia Supplementary Series, 17(1−5), 1–259.
Tahar, S. B., Bottomley, W., & Whitfeld, P. R. (1986). Characterization of the spinach chloroplast genes for the S4 ribosomal protein, tRNAThr (UGU) and tRNASer (GGA). Plant Molecular Biology, 7(1), 63–70.
Van der Putten, N., Stieperaere, H., Verbruggen, C., & Ochyra, R. (2004). Holocene palaeoecology and climate history of South Georgia (sub-Antarctica) based on a macrofossil record of bryophytes and seeds. The Holocene, 14(3), 382–392.
Van der Putten, N., Verbruggen, C., Ochyra, R., Spassov, S., de Beaulieu, J.-L., De Dapper, M., et al. (2009). Peat bank growth, Holocene palaeoecology and climate history of South Georgia (sub-Antarctica), based on a botanical macrofossil record. Quaternary Science Reviews, 28(1–2), 65–79.
Van der Putten, N., Verbruggen, C., Ochyra, R., Verleyen, E., & Frenot, Y. (2010). Subantarctic flowering plants: pre-glacial survivors or post-glacial immigrants? Journal of Biogeography, 37(3), 582–592.
Van Zanten, B. O. (1976). Preliminary report on germination experiments designed to estimate the survival chances of moss spores during aerial trans oceanic long range dispersal in the Southern Hemisphere, with particular reference to New Zealand. The Journal of the Hattori Botanical Laboratory, 41, 133–140.
Van Zanten, B. O., & Pócs, T. (1981). Distribution and dispersal of bryophytes. Advances in Bryology, 1, 479–562.
Vanderpoorten, A., Hedenäs, L., Cox, C. J., & Shaw, A. J. (2002a). Phylogeny and morphological evolution of the Amblystegiaceae (Bryopsida). Molecular Phylogenetics and Evolution, 23(1), 1–21.
Vanderpoorten, A., Hedenäs, L., Cox, C. J., & Shaw, A. J. (2002b). Circumscription, classification, and taxonomy of Amblystegiaceae (Bryopsida) inferred from nuclear and chloroplast DNA sequence data and morphology. Taxon, 51(1), 115–122.
Vanderpoorten, A., Goffinet, B., Hedenäs, L., Cox, C. J., & Shaw, A. J. (2003). A taxonomic reassessment of the Vittiaceae (Hypnales, Bryopsida): evidence from phylogenetic analyses of combined chloroplast and nuclear sequence data. Plant Systematics and Evolution, 241(1), 1–12.
Vanderpoorten, A., Devos, N., Goffinet, B., Hardy, O. J., & Shaw, A. J. (2008). The barriers to oceanic island radiation in bryophytes: insights from the phylogeography of the moss Grimmia montana. Journal of Biogeography, 35(4), 654–663.
Vujičić, M., Sabovljević, A., Šinžar-Sekulić, J., Skorić, M., & Sabovljević, M. (2012). In vitro development of the rare and endangered moss Molendoa hornschuchiana (Hook.) Lindb. ex Limpr.(Pottiaceae, Bryophyta). Hort Science, 47(1), 84–87.
Wagstaff, S. J., Breitwieser, I., & Ito, M. (2011). Evolution and biogeography of Pleurophyllum (Astereae, Asteraceae), a small genus of megaherbs endemic to the subantarctic islands. American Journal of Botany, 98(1), 62–75.
Ward, M. (1960). Some techniques in the culture of mosses. The Bryologist, 63(4), 213–217.
Yamaguchi, K., Prieto, S., Beligni, M. V., Haynes, P. A., McDonald, W. H., Yates, J. R., & Mayfield, S. P. (2002). Proteomic characterization of the small subunit of Chlamydomonas reinhardtii chloroplast ribosome identification of a novel S1 domain–containing protein and unusually large orthologs of bacterial S2, S3, and S5. The Plant Cell, 14(11), 2957–2974.
Żarnowiec, J. (2001). A taxonomic monograph of the Drepanocladus aduncus group (Bryopsida: Amblystegiaceae). Bielsko-Biała: Bielsko-Biała (Poland), Łódź Technical University.
Acknowledgements
The authors thank Rodney D. Seppelt, Arundel, Australia, for his kind revision of the English language and useful comments; Tomasz Suchan, Kraków, and an anonymous reviewer, for constructive comments which helped to improve this manuscript; Lars Hedenäs, Stockholm, and Michail S. Ignatov, Moscow, for kindly providing us with the specimens used for our study; Marian Wysocki and Katarzyna Biłyk, Kraków, for help with illustrative material; Monika Stryjak-Bogacka and Magdalena Tomaszewicz, Kraków, for their assistance in molecular laboratory work.
Funding
This study was funded by the National Science Centre, Poland, grant number: NCN 2015/17/B/N28/02475/ and, partly, through the statutory fund of the W. Szafer Institute of Botany of the Polish Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Supplementary Figure 1
Micromorphological differences between Drepanocladus longifolius and D. capillifolius. 1a – Alar cells and mid-leaf cells of D. longifolius – microscopic slides. 1b – Leaves, mid-leaf cells and alar cells of D. longifolius – line drawings. 1c – Alar cells of D. capillifolius – microscopis slides. 1d – Leaves, mid-leaf cells and alar cells of D. capillifolius – line drawings. (PDF 3947 kb)
Supplementary Table 1
(XLSX 16 kb)
Supplementary Table 2
(XLSX 16 kb)
Supplementary Table 3
(XLSX 9 kb)
Supplementary Table 4
(XLSX 16 kb)
Supplementary File 1
Concatenated alignment of cpDNA sequences: atpB-rbcL + rpL16 + trnG + trnL-trnF. (FAS 86 kb)
Supplementary File 2
Alignment of nDNA sequences: ITS. (FAS 27 kb)
Supplementary File 3
Alignment of cpDNA sequences: atpH-atpI. (FAS 9 kb)
Supplementary File 4
Alignment of cpDNA sequences: rps4. (FAS 13 kb)
Supplementary File 5
Protein aligment of cpDNA sequences: rps4. (FAS 4 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Saługa, M., Ochyra, R., Żarnowiec, J. et al. Do Antarctic populations represent local or widespread phylogenetic and ecological lineages? Complicated fate of bipolar moss concepts with Drepanocladus longifolius as a case study. Org Divers Evol 18, 263–278 (2018). https://doi.org/10.1007/s13127-018-0372-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13127-018-0372-8