
Abstract
Conservation genomics is an important tool to manage threatened species under current biodiversity loss. Recent advances in sequencing technology mean that we can now use whole genomes to investigate demographic history, local adaptation, inbreeding, and more in unprecedented detail. However, for many rare and elusive species only non-invasive samples such as faeces can be obtained, making it difficult to take advantage of whole genome data. We present a method to extract DNA from the mucosal layer of faecal samples to re-sequence high coverage whole genomes using standard laboratory techniques. We use wild collected faecal pellets collected from caribou (Rangifer tarandus), a species undergoing declines in many parts of its range in Canada and subject to comprehensive conservation and population monitoring measures. We compare four faecal genomes to two tissue genomes sequenced in the same run. Quality metrics were similar between faecal and tissue samples with the main difference being the alignment success of raw reads to the reference genome due to differences in low quality and endogenous DNA content, affecting overall coverage. One of our faecal genomes was only re-sequenced at low coverage (1.6 ×), however the other three obtained between 7 and 15 ×, compared to 19 and 25 × for the tissue samples. We successfully re-sequenced high-quality whole genomes from faecal DNA and are one of the first to obtain genome-wide data from wildlife faecal DNA in a non-primate species. Our work represents an important advancement for non-invasive conservation genomics.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Human induced global biodiversity loss, for example due to habitat destruction and/or climate change, is accelerating (Harrison et al. 2014; McMahon et al. 2014; Shafer et al. 2015; Brandies et al. 2019). Conservation genomics is one tool to help with the management of threatened taxa, particularly with recent advances in sequencing technologies and reducing costs (Perry et al. 2010; Shafer et al. 2015; Brandies et al. 2019). There are many articles outlining the advantages of genome-wide data for conservation including for estimating demographic histories, and for detecting genomic regions involved with local adaptation or inbreeding depression (e.g. Allendorf et al. 2010; Harrison et al. 2014; McMahon et al. 2014; Shafer et al. 2015). Different genomic methods have been developed, including reduced-representation sequencing (RRS), however there are clear advantages of having whole genome information (Fuentes-Pardo and Ruzzante 2017). Understanding adaptation is touted as one of the major advantages of genomics, however many adaptive traits are polygenic and may not be detected using RRS (Funk et al. 2012; McMahon et al. 2014; Shafer et al. 2015; Fuentes-Pardo and Ruzzante 2017; Brandies et al. 2019). Similarly, whole genomes can be used to determine the genetic basis of phenotypic traits or diseases of interest to conservation (Fuentes-Pardo and Ruzzante 2017; Brandies et al. 2019).
For many threatened taxa obtaining high-quality samples can be difficult, therefore advances in non-invasive genetics have been important for conservation initiatives as they allow the study of rare or elusive species without needing to handle, or sometimes even see, the target species (Smith and Wang 2014; Snyder-Mackler et al. 2016; Ozga et al. 2020). There are many types of non-invasive samples, including faeces, hair, urine, feathers, egg shells, and skin (Beja-Pereira et al. 2009; Smith and Wang 2014; Russello et al. 2015), however, faecal samples are commonly used since they are easy to obtain and can provide additional relevant information such as hormones, microbiome, and diet (Perry et al. 2010; Chiou and Bergey 2018). However, obtaining genome-wide data from non-invasive samples is challenging due to low host (endogenous) DNA in extractions, fragmented DNA, the presence of PCR inhibitors, and high levels of allelic dropout, all of which are particularly true for faecal DNA (Perry et al. 2010; Smith and Wang 2014; Snyder-Mackler et al. 2016; Chiou and Bergey 2018).
New, promising approaches have been developed to sequence genomic data from faecal samples. Most have used sequence capture methodologies which use DNA or RNA baits to hybridise to target DNA (Perry et al. 2010; Snyder-Mackler et al. 2016). For example, Perry et al. (2010) used a DNA capture protocol with custom baits to enrich megabases of nuclear genomic regions and the mitochondrial genome from chimpanzees. Snyder-Mackler et al. (2016) were the first to use genome-wide enrichment capture from RNA baits to enrich faecal DNA, which resulted in low-coverage (a mean of 0.493) data for baboons. However, capture methodologies can be expensive and time consuming, have high PCR duplication rates, and bias the resulting datasets towards particular regions of the genome (Chiou and Bergey 2018; Orkin 2020). A recent study by Ozga et al. (2020) tested different non-invasive samples from chimpanzees using both whole genome and exome capture methods, and found that urine had much higher success than faecal DNA, producing genome-wide data using the same extraction and sequencing methods as with high quality tissue samples (needing no extra methodological considerations). However, urine is not always easily collected for many taxa, and the capture method still does not give unbiased whole genome coverage.
Chiou and Bergey (2018) developed a cost-effective method they called ‘FaecalSeq’, which takes advantage of the difference in CpG-methylation densities between bacterial and vertebrate genomes to enrich host faecal DNA. They validated their FaecalSeq approach using double-digest restriction-site associated DNA sequencing (ddRADseq) to obtain genome-wide SNP data in baboons (Chiou and Bergey 2018). However, FaecalSeq still biases which genomic regions are captured based on methylation patterns, and can co-enrich non-target DNA such as from plant and animal food sources (Chiou and Bergey 2018; Ozga et al. 2020). Orkin (2020) obtained unbiased ‘uniform’ high and low coverage whole genome sequences from faecal DNA by collecting faecal samples from capuchin monkeys and using fluorescence activated cell sorting (FACS) to isolate mammalian cells from the faeces, as an alternative to enriching host DNA. They successfully reconstructed one high coverage (12 ×) and 15 low coverage (0.1–4 ×) re-sequenced genomes, however the use of FACS adds additional expense ($40 per sample to isolate the cells) and assumes the availability of FACS resources (Orkin 2020).
A study by Khan et al. (2020) re-sequenced whole genomes from hair and scat samples of tigers using standard laboratory techniques. They had good success with the hair samples, however the faecal genomes, which were extracted by swabbing the scat sample, had a low percentage of reads which mapped to the reference genome and a low percentage of coverage across the genome (Khan et al. 2020). Using a modified protocol whereby we carefully extract DNA from the mucosal layer of faecal pellets collected from wild caribou (Rangifer tarandus), we attempted to re-sequence four high coverage whole genomes also using only standard laboratory techniques. Caribou (known as reindeer in Europe and Asia) occur across Canada in different ecozones from the High Arctic to the boreal forests (Banfield 1961; COSEWIC 2011). In Canada there are four subspecies and 12 conservation units, known as Designatable Units (DUs; Banfield 1961; COSEWIC 2011). All 11 of the extant DUs are listed as at risk of extinction (COSEWIC 2014a, b, 2015a, b, 2017a, b), and many are threatened due to anthropogenic activities such as habitat destruction and climate change (Vors and Boyce 2009; Festa-Bianchet et al. 2011; Weckworth et al. 2018). Caribou is a keystone species for the ecosystem and is of cultural and economic significance to indigenous communities (Festa-Bianchet et al. 2011; Polfus et al. 2016), highlighting the need for population monitoring and conservation initiatives.
Genetic analyses using microsatellites and mitochondrial DNA sequences from winter collected faecal samples have been fundamental in understanding population structure and evolutionary history of Canadian caribou (e.g. Klütsch et al. 2012, 2016; Polfus et al. 2016, 2017; Horn et al. 2018), as well as for monitoring population trends (Hettinga et al. 2012; McFarlane et al. 2020), pedigree reconstruction and inbreeding estimations (e.g. McFarlane et al. 2018; Thompson et al. 2019). To advance this work, we aim to re-sequence whole genomes at high-coverage from non-invasively collected faecal samples. We extracted host DNA from the mucosal layer on the outside of the faecal pellets to increase the likelihood of extracting host DNA from the epithelial cells of the intestines (Ball et al. 2007) and compared those genomes to two tissue genomes sequenced in the same run to assess performance and bias in the resulting data. We also assessed the type and amount of non-host DNA sequences in each sample. We are one of the first to successfully re-sequence high-coverage whole genomes from faecal DNA of a non-primate species without host-DNA enrichment steps that could introduce bias.
Materials and methods
Sample collection and DNA extraction
Faecal pellets were collected during winter aerial surveys between 2004 and 2018 (Table 1). Pellets were collected from the snow, bagged, and kept frozen for shipping to Trent University. Tissue samples were collected during harvesting activities in 1994 and 2002 (Table 1) and stored in 1 × lysis buffer, which was a modified version of the Queen’s lysis buffer (Seutin et al. 1991) containing 4 M urea, 0.2 M NaCL, 0.5% sarkosyl, 10 mM EDTA, and 0.1 M Tris, pH 8.0.
For the faecal genomes, three individuals were boreal caribou; two from Cold Lake, Alberta, and one from Wood Buffalo National Park, Northwest Territories. The fourth was a central mountain caribou from A La Peche, Alberta. The samples were chosen to fill in sampling gaps for other conservation genomic analyses being undertaken for areas where we do not have tissue samples (Taylor et al. 2021). Multiple faecal samples from these locations had been genotyped at microsatellite loci and to select which faecal samples to re-extract for whole genome sequencing, we surveyed the raw genotype files and looked for those with the cleanest, highest peaks, to select those most likely to have the highest amounts of high-quality endogenous DNA. All chosen faecal samples successfully amplified for 10 microsatellite loci and showed high RFU peak scores (Supplementary Material S1 and S2).
DNA was extracted from the mucosal coat on the faecal pellets from four individuals. To do this, four faecal pellets were put into a tube with 1 ml of lysis buffer and gently rotated or washed for about 30 s. The faecal pellet and (after settling) any precipitate were discarded, following which 10 µl of proteinase K (provided in Qiagen DNAeasy Blood and Tissue Kits, Qiagen, Hilden, Germany) was added to the lysis buffer and the sample incubated at 65 °C for two hours. Another 10 µl of proteinase K was then added and the sample was left at 37 °C overnight. For each individual, we did this process twice. DNA extraction was then carried out using a DNAeasy Blood and Tissue Kit (Qiagen, Hilden, Germany). The DNA was eluted with 200 µl of TE, and the two extractions for each individual combined for a total of 400 µl. The samples were then run through a concentration column (Amicon Ultra-0.5 Centrifugal Filter Unit-30 K Device, Millipore Sigma, Burlington, MA, USA). To do this, the 400 µl of Qiagen extracted sample was loaded into the column and spun at 14,000×g for 5 min. Once the sample has been spun through the concentrator column, the column was flipped upside down into the final 1.5 ml collection tube and spun for 2 min at 1000×g giving the final volume at ~ 50 µl at a concentration factor of 12 ×.
Tissue samples from the Tay population in the Yukon (a northern mountain caribou) and the Fortymile caribou population (Grant’s caribou) straddling the Yukon and Alaska border were also extracted using a DNAeasy Blood and Tissue Kit (Qiagen, Hilden, Germany).
Quality control and sequencing
The DNA extractions were run on a 1.5% agarose gel, and quantified with a Qubit fluorometer (Thermo Fisher Scientific, MA, USA) using the High Sensitivity Assay Kit to ensure high DNA concentrations for sequencing. The samples were also analysed on a Nanodrop ND-8000 spectrophotometer (Nanodrop Technologies Inc., Wilmington, DE, USA) to assess purity. The DNA was normalized to 20 ng/µl at a final volume of 50 µl for the tissue samples and to 22 ng/µl at a final volume of 50 µl for the faecal samples and shipped to The Centre for Applied Genomics (TCAG) at the Hospital for Sick Children (Toronto, Ontario) for library preparation and sequencing. The library preparation was done using the Truseq PCR-free DNA Library Preparation Kit (Illumina, San Diego, CA, USA) following the manufacturers recommended protocol, using 700 ng of DNA as input material. Briefly, DNA was fragmented to 400 bp on average using sonication on a Covaris LE220 instrument (Covaris, Woburn, MA, USA). Fragmented DNA was end-repaired, A-tailed, and indexed Truseq Illumina adaptors with overhand-T added to the DNA. Libraries were validated on a Fragment Analyzer for four samples (24476, 40092, 27673, and 27773) and a Bioanalyzer for two samples (24461 and 34877), using a high sensitivity NGS kit to check for size and absence of primer dimers, and quantified by qPCR using Kapa Library Quantification Illumina/ABI Prism Kit protocol (KAPA Biosystems, Wilmington, MA, USA). Samples were also analysed using a TapeStation using a genomic DNA ScreenTape analysis (Agilent, Santa Clara, CA, USA). Validated libraries were pooled in equimolar quantities. The samples were run alongside 10 other samples being used for another study (for a total of 16 samples) on 8 lanes of an Illumina HiSeq X (Illumina, San Diego, CA, USA) 150 bp paired-end. All raw reads are available on the NCBI database (BioProject ID PRJNA694662).
Filtering reads and variant calling
Raw read files were checked using FastQC version 0.11.8 (Andrews 2010). We used Trimmomatic version 0.38 (Bolger et al. 2014) to trim adaptors and other Illumina sequences from the reads which can result from sequencing very short DNA fragments, as may be expected from lower quality DNA. We used the sliding window approach (4 base pairs at a time) to trim reads once the phred score dropped below 15. Any reads that were smaller than 36 bp were then discarded. Reads were aligned to the reference genome (Taylor et al. 2019) using Bowtie2 version 2.3.0 (Langmead and Salzberg 2012), and the SAM file converted to a BAM file using Samtools version 1.5 (Li et al. 2009). We then sorted the BAM files using Samtools, and then removed duplicate reads and added correct read group information to each BAM file using Picard version 2.17.3 (Available: http://broadinstitute.github.io/picard/). We then re-sorted the BAM file and built an index using Picard.
We called variants using Haplotype Caller in GATK version 3.8 (McKenna et al. 2010) and produced a variant call format (VCF) file for each caribou. Individual VCF files were combined using the Combine GVCFs function, and then we performed joint genotyping using Genotype GVCFs, both in GATK. Due to one faecal genome being of poor quality compared to the others (see Results), we also produced a joint VCF file with this individual removed. We used VCFtools version 0.1.14 (Danecek et al. 2011) to filter the VCF files to ensure quality. We did two rounds of filtering, firstly to remove indels and any site with a depth of less than 5 or more than 40 (double the depth we were aiming for across the genome), and removed any low-quality genotype calls (minGQ) and low quality sites (minQ), with scores below 20, which in VCFtools are changed to missing data. Secondly, we filtered to remove all missing data.
Quality assessments
Given the differences in alignment success to the reference genome (see “Results”), we examined the raw reads to see how much host vs non-host DNA was sequenced, and define the composition of the non-host DNA. To do this, we ran the raw reads through Kraken2 (Wood et al. 2019). We used reference libraries available through the kraken2-build command, using the RefSeq libraries archaea, bacteria, plasmid, viral, human, fungi, plant, and protozoa, the NCBI non-redundant nucleotide database, and UniVec_Core, the NCBI-supplied database of vector, adapter, linker, and primer sequences that may be contaminating sequencing projects and/or assemblies. We then also added our reference genome in the reference database.
We plotted the distribution of raw reads (after trimming with Trimmomatic), and the reads which were discarded during alignment to the reference genome, to see if the faecal samples showed a shift towards containing or discarding more shorter reads than the tissue samples. The average depth for each BAM file was calculated both before and after duplicate removal using Samtools and the BAM files (after duplicate removal) were also run through FastQC. We also ran each BAM file through BUSCO version 3.0.2 (Benchmarking Universal Single-Copy Orthologs; Waterhouse et al. 2018) to reconstruct 4,104 conserved mammalian genes to assess the completeness of each genome. As our reference genome reconstructed 3,820 (93.1%; Taylor et al. 2019) complete mammalian BUSCO genes, this represents an upper limit for our re-sequenced individuals. We used Picard to run some quality checks on the BAM files, using ‘CollectGcBiasMetrics’ to assess GC content and produce statistics regarding GC bias in the genomes, ‘CollectWgsMetrics’ to assess the fraction of reads that pass quality filters for each of the genomes, and ‘QualityScoreDistribution’ to output quality scores of all bases.
We did two population genomic analyses with the genomes to assess how they performed. Using both combined VCF files, we performed a principle component analyses in R version 3.4.4 (R Core Team 2015) using the packages vcfR (Knaus and Grünwald 2017) and Adegenet (Jombart 2008). We also used the populations module in Stacks version 2.4.1 (Catchen et al. 2013) to convert our VCF files into input files for Treemix version 1.13 (Pickrell and Pritchard 2012). We ran Treemix from 0 to 4 migration events, with three iterations of each. We grouped the SNPs in windows to account for possible linkage, using a block size of 100 for two of the iterations and 50 for one of the iterations (because to run the OptM package you must not have the same likelihood scores for each iteration). We plotted the resulting trees and residual plots in RStudio version 1.0.136 (RStudio Team 2015), and used the R package OptM (available here: https://cran.r-project.org/web/packages/OptM/index.html) to calculate the second order rate of change in the log-likelihood of the different migration events (the ad hoc statistic delta M). We also tried running without accounting for linkage due to the smaller number of SNPs in the VCF file with all 6 caribou, however the trees were the same.
Results
Genome quality assessments.
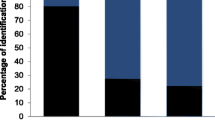
All extractions had relatively high concentrations before normalising, however with three of the faecal extractions having slightly lower Nanodrop scores than the 1.8 “ideal” ratio (Table 1). Gel images show fainter bands for a large fragment size for the faecal samples than for the tissue (Supplementary Material S3). Results from the TapeStation analysis vary between individuals, although the faecal samples show increased numbers of smaller fragment sizes (Supplementary Material S4 and S5 for full TapeStation and Fragment analyzer/Bioanalyzer results, respectively). All four faecal genomes and the two tissue genomes had comparable numbers of reads and percentage of reads retained after trimming (Table 1). However, the number of reads which successfully aligned to the reference genome was highly variable for the faecal genomes, ranging from 5 to 60%. In contrast, the two tissue genomes had very high alignment success at around 95–96%. Kraken2 indicated differences in the composition of the raw reads between samples, particularly between tissue and faecal. There was an increase in non-host DNA in the faecal samples, particularly bacteria which was not detected in the tissue samples but pertained to between 4.69 and 12.32% of the faecal samples (Fig. 1). Similarly, the amount of human, green plant, Sar (an eukaryotic clade which includes many parasites of animals), virus, and archaea DNA increased in the faecal samples, although still remained a small proportion of reads overall (Fig. 1). The number of unclassified reads was very different between tissue and faecal samples (Fig. 1).

Composition of the raw reads for each caribou as per Kraken2, showing the proportions of caribou, human, fungi, green plants, sar (an eukaryotic clade which includes many parasites of animals), bacteria, archaea, viruses, other eukaryotes, and the unclassified reads. Proportions of caribou reads are smaller and non-host and unclassified reads larger for faecal samples (a–d) compared to tissue (e–f) samples
The distributions of raw reads (after trimming) and the reads which were discarded during alignment were the same for faecal and tissue samples, and all heavily skewed towards reads of 149 and 150 bp (Supplementary Material S6-S11). To better see if the distribution of shorter reads was different between faecal and tissue, we re-did the plots removing reads of 149 and 150 bp, but still find no difference in fragment sizes (Supplementary Material S12-S17). Given the skew towards shorter fragment sizes of faecal samples showing in the gel images and TapeStation analysis (Supplementary Material S3-S4), this suggests that during the library preparation the shorter reads were lost (we discarded all reads below 36 bp during adapter trimming with Trimmomatic but don’t see a skew towards reads at 37–148 bp for the faecal samples). As the distribution of read sizes remains the same before alignment and for those reads which are discarded during alignment to the reference genome, even for the shorter reads, the alignment process does not appear to bias which reads are retained.
The different alignment success between samples resulted in varying depth of coverage overall for each individual. One faecal genome, from Wood Buffalo, had only an average depth of 1.63 after duplicate removal, the others achieving between 7 and 15X coverage. The tissue genomes were 19 and 25X coverage (Table 1). The depth did not drop significantly more for the faecal genomes after duplicate removal, however, indicating that they did not contain an inflated number of PCR or sequencing duplicates. FastQC results from the raw reads were good although with some lower quality bases at the ends of the reads both for the tissue and faecal samples. All FastQC results from the BAM files looked good, with the per base sequencing quality not dropping below 28 even at the ends of the reads, high per sequence quality scores and no detected duplication levels, overrepresented sequences, or adaptor content.
BUSCO successfully reconstructed 92–93% of the conserved mammalian genes for all genomes apart from Wood Buffalo which reconstructed 44.6% (Table 2 and Fig. 2). The GC distribution was the same between all 6 genomes (Fig. 3a–f). They all had similar mean base quality scores across regions of the genome with different percentage GC, although with windows of very high GC content dropping in quality score. Wood Buffalo, however, decreased dramatically in regions with high GC content (Fig. 3a–f). Normalised coverage also seemed to be affected by GC content in all genomes, with Wood Buffalo again dropping dramatically compared to the others (Fig. 3a–f). The BAM files all showed no adaptors or duplicates, as expected given prior filtering (Table 3). The percentage of bases with low quality scores and those in reads without a mapped read pair were all low and consistent between the genomes (Table 3). The percentage of bases with a low mapping score was higher, and slightly elevated in the Wood Buffalo genome (Table 3), showing the importance of quality filtering when producing the variant call format (VCF) file. Differences in coverage levels were also very apparent between the genomes. The percent of the genome sequenced (or the breadth of coverage across the genome) at a minimum of 1X was very high in all genomes at over 99% of coverage of the reference genome, apart from the Wood Buffalo genome which was at 72.3% (Table 3). The theoretical heterozygous single nucleotide polymorphism (SNP) sensitivity scores, which is an estimate of the sensitivity to detect heterozygous sites (between 0 and 1), also varied. The score was low for the Wood Buffalo genome, but was high for all other genomes apart from the lowest quality Cold Lake individual which was intermediate (Table 3). The quality score distribution of the base pairs in the BAM files was consistent between all individuals, with the vast majority showing high quality scores (Fig. 4a–f).

BUSCO results for each genome showing the number of conserved mammalian genes successfully reconstructed and in single copy (light blue), duplicate (dark blue), fragmented (yellow) or missing (red). All successfully reconstruct within 1% of all possible genes (given the reference genome), aside from the Wood Buffalo genome which only reconstructed 1829 (44.6%) complete and single copy genes

GC bias plots for each caribou showing the distribution of GC content in red bars along the bottom. For each window, the normalised coverage is shown in blue circles and the mean base quality (phred score) is shown by the green line

Quality score distributions for all base pairs in the BAM file for each genome
SNP calls and population genomic analyses
The VCF files with all six genomes before removing missing data contained 18,438,793 SNPs. However, the missing data was heavily skewed towards the faecal genome from Wood Buffalo which had 99% missing data (Table 2). This is potentially because we filtered for low quality genotype scores and sites, which are changed to missing data in VCFtools. After removing all missing data from the VCF file, only 25,390 SNPs remained. Additionally, the Wood Buffalo genome had an order of magnitude more private SNPs (Table 2). In the VCF file without the Wood Buffalo caribou, there were 18,261,032 SNPs before removing missing data. Missing data levels were quite high for one of the Cold Lake caribou (Table 2), however when removing all missing data 5,065,428 SNPs were still retained. The lower quality genome from Cold Lake had a slightly elevated number of private sites, indicating the potential for some errors due to quality affecting SNPs called for that individual (Table 2).
We used both VCF files with no missing data to do principal component analyses (PCAs) and Treemix analyses, to assess how well they would perform. The PCA using all six caribou showed a pattern that we expected (Taylor et al. 2020 and 2021), although with the boreal caribou (both Cold Lake and the Wood Buffalo caribou) quite separated from one another. The Central mountain caribou (A La Peche) separated from all others, and the Northern Mountain (Tay) and Grant’s caribou (Fortymile) clustering closer together which matches the geography of the sampling sites (Fig. 5a). The PCA without the Wood Buffalo caribou showed the two Cold Lake boreal caribou clustering closely, and with the Northern mountain and Grant’s caribou also clustering closely (Fig. 5b) which may be due to increased power from the greater number of SNPs used in the analysis. The Treemix analysis failed to build a tree when including the Wood Buffalo caribou, showing a large standard error bar (Fig. 6a). In contrast, when removing Wood Buffalo the analysis could reconstruct a phylogeny which grouped the Grant’s caribou (Fortymile) with Northern Mountain (Tay) as a separate clade to the other three as expected (Fig. 6b). When adding migration events, after 2 migration events no new migration events could be inferred. The OptM analysis gave 1 migration event as having the highest delta M, which showed a migration event from the ancestor of the Grant’s (Fortymile) and Northern Mountain (Tay) caribou into a Cold Lake caribou (Fig. 6c).

PCA with all six caribou included (a) and without the Wood Buffalo caribou (b)

Treemix analysis using all six genomes failed to show any topology and had a large standard error bar (a), however when removing the lowest quality genome a maximum Likelihood tree could be produced (b). Adding one migration event inferred migration from the ancestor of the clade containing the Grant’s and northern mountain caribou into a Cold Lake individual (c)
Discussion
We have successfully re-sequenced both high and low coverage whole genomes from faecal DNA using only standard laboratory protocols and sequencing. This represents an important contribution for non-invasive conservation studies to move from genetics to genomics and investigate questions such as the local adaptation of populations, particularly for caribou which are declining in many parts of their range (Vors and Boyce 2009; Festa-Bianchet et al. 2011; Weckworth et al. 2018).
As expected, the main difference between the tissue and faecal genomes was in the alignment success of reads to the reference. This appears to be due to a combination of differences in endogenous DNA content, along with reads which could not be classified in the faecal samples which could be due to several factors, including database incompleteness or biases (Pignatelli et al. 2008; Kibegwa et al. 2020), or potentially low quality reads (Fig. 1). Overall, the quality statistics of the BAM files were very similar between our tissue and faecal genomes, however, apart from the Wood Buffalo individual, with no sign of lowered per base sequencing quality, a skewed GC content, inflated numbers of PCR duplicates, percentage of bases in reads with lower mapping quality, skewed quality score distributions of base pairs, or the number of genes reconstructed in a BUSCO analysis, for example (Table 3, Figs. 2, 3a–f, and 4a–f).
We aimed for high coverage whole genomes, and the Wood Buffalo individual was only re-sequenced at low coverage. Another of our genomes, from the Cold Lake population, was also a bit low at around 7X coverage which may have affected the quality of genotyping as it has a slightly elevated number of private SNPs and a lower theoretical heterozygous SNP sensitivity. With a slight refinement in laboratory techniques, it may be possible to increase the likelihood of selecting samples with higher endogenous DNA content to ensure good sequencing. To select which samples to sequence, we looked at raw genotype peaks from microsatellite scores to assess quality and endogenous DNA content. Moving forward, we will likely only use samples collected in 2014 or later, as our extraction techniques were not as refined and sample quality not as good (our lowest quality genome was the only one collected before this time). We only selected samples with no missing microsatellite genotypes, which includes ~ 82% of the samples we have genotyped since 2014. However, a better sample selection method (or to use in combination) might be to use qPCR to screen for proportion of host DNA within extractions (Snyder-Mackler et al. 2016; Chiou and Bergey 2018; Hayward et al. 2020), or the PCR method developed by Ball et al. (2007). If used in combination with our DNA extraction technique, it is likely that genomes to the standard of our high-quality faecal genomes will be more consistently produced, further increasing the cost effectiveness of our method. We plan to use PCR quantification technique moving forward (Ball et al. 2007; Hayward et al. 2020), which should likely be standard practice for researchers choosing samples for sequencing.
We completed other important and standard checks including the measure of DNA concentration using a Qubit and purity using a Nanodrop. As we only did four samples we cannot do a quantitative analysis, but it is interesting that our best faecal genome, which attained almost the same coverage as our tissue samples, was also the only faecal sample to reach a Nanodrop reading of 1.8 (Table 1). Also, our faecal sample which could only be used as a low coverage genome, Wood Buffalo, had a very high Qubit reading compared with our ‘average’ faecal samples. At 80 ng/µl, it has reached the same concentrations as we see with our tissue extractions (Table 1). We wonder if a spuriously high DNA concentration may indicate high levels of bacterial DNA, and Wood Buffalo did have the highest levels of bacterial and other non-endogenous content in the raw reads (Fig. 1c).
With any genomic data produced from non-invasive samples, strict filtering and careful monitoring of data quality is essential. We performed extensive data quality assessments with our genomes to assess potential areas of bias (Tables 1, 2, 3 and Figs. 2, 3, 4). Filtering for low quality sites is standard practice with any whole genome data but is even more important with faecal samples as we expect higher genotyping errors with poorer quality DNA. We filtered low quality sites (both base and mapping quality) in VCFtools which is changed to missing data. Missing data filtering before further analyses is therefore crucial due to missing data levels being heavily skewed towards lower quality samples (Table 2). After removing the missing data, if we included the low quality faecal genome we ended up with an order of magnitude fewer SNPs in the VCF file than when we excluded it (25,390 vs 5,065,428). We also tested the performance of the genomes with two standard analyses, PCA and Treemix. Clearly the inclusion of the lower quality Wood Buffalo genome affected the results, especially the Treemix which completely failed to reconstruct a phylogeny (Fig. 6). As we only included one individual per population we did not use a minor allele frequency filter, although with multiple individuals per population in a larger dataset this could also be an important filtering step.
One potential drawback of our method, as well as many other methods being developed for producing genome-wide data from faecal samples (Perry et al. 2010; Snyder-Mackler et al. 2016; Orkin 2020), is the need for a reference genome. However, with reducing costs and increased availability of bioinformatics pipelines for non-model species (Fuentes-Pardo and Ruzzante 2017; Brandies et al. 2019), the availability of a reference genome is becoming less of an issue, especially with initiatives such as the CanSeq150 (www.cgen.ca/canseq150) and the Genome 10 K project (Koepfli et al. 2015).
Another advantage we had is the collection of faecal samples in winter from the snow. The fact that the samples are collected while frozen will mean lower degradation of DNA than if they had been collected, for example, in the tropics (Smith and Wang 2014). As such, for many taxa the collection of fresh faecal matter which is immediately frozen or appropriately stored would be highly beneficial. Additionally, the method we use will likely only work with faecal matter from certain taxonomic groups which produce faecal pellets.
One current challenge is that we sequence a proportion of non-endogenous DNA, lowering the coverage of the host genome (Fig. 1). Although one of our faecal genomes could not be used as a high coverage genome, all of them could be used as low-coverage genomes which are typically between 1 and 4 × per individual (Fuentes-Pardo and Ruzzante 2017). More individuals would be needed for genotype likelihood calls which may be cost prohibitive, although improved lab screening for samples with higher quality and higher amounts of endogenous DNA will improve sample selection, and therefore the number of samples which could be run on one sequencing lane, if low coverage genomes are the desired outcome. Sequencing whole genomes from few individuals per population for in-depth analyses (e.g. investigations of local adaptation or runs of homozygosity) to supplement traditional genetic methods may be most cost-effective for non-invasive monitoring of threatened taxa.
Overall, our method to extract DNA for whole genome re-sequencing from non-invasively collected faecal samples is an important step forward in our ability to study and monitor caribou using our already existing sample collection. We will now be able to re-sequence genomes from populations for which we had no existing tissue samples for comprehensive investigations of adaptation, inbreeding, and demographic histories of caribou across North America which will be invaluable knowledge to inform the conservation of this declining species. Further, our protocol for extracting DNA from the mucosal layer of faecal matter could be used in other taxa, especially if they have access to winter or freshly collected samples. Together with our thorough considerations of data quality and bias, we hope other research groups will be able to produce high-quality whole genome data for other rare or elusive species.
References
Allendorf FW, Hohenlohe PA, Luikart G (2010) Genomics and the future of conservation genetics. Nat Rev Genet 11:697–709
Andrews S (2010) FastQC: a quality control tool for high throughput sequence data. http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
Ball MC, Pither R, Manseau M, Clark J, Peterson SD, Kingston S, Morrill N, Wilson P (2007) Characterization of target nuclear DNA from faeces reduces technical issues associated with the assumptions of low-quality and quantity template. Conserv Genet 8:577–586
Banfield AWF (1961) A Revision of the Reindeer and Caribou, Genus Rangifer. National Museum of Canada, Bulletin No. 177, Queen’s Printer, Ottawa
Beja-Pereira A, Oliveira R, Alves PC, Schwartz MK, Luikart G (2009) Advancing ecological understandings through technological transformations in noninvasive genetics. Mol Ecol Resour 9:1279–1301
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Brandies P, Peel E, Hogg CJ, Belov K (2019) The value of reference genomes in the conservation of threatened species. Genes 10:846
Catchen J, Hohenlohe PA, Bassham S, Amores A, Cresko WA (2013) Stacks: an analysis tool set for population genomics. Mol Ecol 22:3124–3140
Chiou KL, Bergey CM (2018) Methylation-based enrichment facilitates low-cost, noninvasive genomic scale sequencing of populations from feces. Sci Rep 8:1975
COSEWIC (2011) Designatable units for caribou (Rangifer tarandus) in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2014a) COSEWIC assessment and status report on the Caribou Rangifer tarandus, Newfoundland population, Atlantic-Gaspésie population and Boreal population, in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2014b) COSEWIC assessment and status report on the Caribou Rangifer tarandus, Northern Mountain population, Central Mountain population and Southern Mountain population in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2015a) COSEWIC assessment and status report on the Peary Caribou Rangifer tarandus pearyi in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2015b) COSEWIC assessment process, categories and guidelines. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2016) COSEWIC assessment and status report on the Caribou Rangifer tarandus, Barren-ground population, in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2017a) COSEWIC assessment and status report on the Caribou Dolphin and Union population (Rangifer tarandus), in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
COSEWIC (2017b) COSEWIC assessment and status report on the Caribou Rangifer tarandus, Eastern Migratory population and Torngat Mountains population, in Canada. Committee on the Status of Endangered Wildlife in Canada, Ottawa
Danecek P, Auton A, Abecasis G et al (2011) The variant call format and VCFtools. Bioinformatics 27:2156–2158
Festa-Bianchet M, Ray JC, Boutin S, Côté SD, Gunn A (2011) Conservation of caribou (Rangifer tarandus) in Canada: An uncertain future. Can J Zool 89:419–434
Fuentes-pardo AP, Ruzzante DE (2017) Whole-genome sequencing approaches for conservation biology: advantages, limitations and practical recommendations. Mol Ecol 26:5369–5406
Funk WC, Mckay JK, Hohenlohe PA, Allendorf FW (2012) Harnessing genomics for delineating conservation units. TREE 27:489–496
Harrisson KA, Pavlova A, Telonis-Scott M, Sunnucks P (2014) Using genomics to characterize evolutionary potential for conservation of wild populations. Evol Appl 7:1008–1025
Hayward KM, Harwood MP, Lougheed SC, Sun Z, de Groot PVC, Jensen EL (2020) A real-time PCR assay to accurately quantify polar bear DNA in fecal extracts. PeerJ 8:e8884. https://doi.org/10.7717/peerj.8884
Hettinga PN, Arnason AN, Manseau M, Cross D, Whaley K, Wilson PJ (2012) Estimating size and trend of the North Interlake woodland caribou population using fecal-DNA and capture-recapture models. J Wildl Manag 76:1153–1164
Horn RL, Marques AJD, Manseau M, Golding B, Klütsch CFC, Abraham K, Wilson PJ (2018) Parallel evolution of site-specific changes in divergent caribou lineages. Ecol Evol 8:6053–6064
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Khan A, Patel K, Bhattacharjee S, Sharma S, Chugani AN, Sivaraman K, Hosawad V, Sahu YK, Reddy GV, Ramakrishnan U (2020) Are shed hair genomes the most effective noninvasive resource for estimating relationships in the wild? Ecol Evol 10:4583–4594
Kibegwa FM, Bett RC, Gachuiri CK, Stomeo F, Mujibi F (2020) A comparison of two DNA metagenomic bioinformatics pipelines while evaluating the microbial diversity in feces of Tanzanian small holder dairy cattle. BioMed Res Int. https://doi.org/10.1155/2020/2348560
Klütsch CFC, Manseau M, Wilson PJ (2012) Phylogeographical analysis of mtDNA data indicates postglacial expansion from multiple glacial refugia in woodland caribou (Rangifer tarandus caribou). PLoS ONE 7:e52661. https://doi.org/10.1371/journal.pone.0052661
Klütsch CFC, Manseau M, Trim V, Polfus JL, Wilson PJ (2016) The eastern migratory caribou: the role of genetic introgression in ecotype evolution. R Soc Open Sci 3:150469. https://doi.org/10.1098/rsos.150469
Knaus BJ, Grüwald NJ (2017) vcfr: a package to manipulate and visualize variant call format data in R. Mol Ecol Resour 17:44–53
Koepfli K, Paten B, O’Brien SJ (2015) The genome 10K project: a way forward. Annu Rev Anim Biosci 3:57–111. https://doi.org/10.1146/annurev-animal-090414-014900
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie2. Nat Methods 9:357–359
Li H, Handsaker B, Wysoker A et al (2009) The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079
McFarlane S, Manseau M, Flasko A, Horn RL, Arnason N, Neufeld L, Bradley M, Wilson PJ (2018) Genetic influences on male and female variance in reproductive success and implications for the recovery of severely endangered mountain caribou. Glob Ecol Conserv 16:e00451. https://doi.org/10.1016/j.gecco.2018.e00451
McFarlane S, Manseau M, Steenweg R, Hervieux D, Hegel T, Slater S, Wilson PJ (2020) An assessment of sampling designs using SCR analyses to estimate abundance of boreal caribou. Ecol Evol. https://doi.org/10.1002/ece3.6797
McKenna A, Hanna M, Banks E et al (2010) The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20:1297–1303
McMahon BJ, Teeling EC, Höglund J (2014) How and why should we implement genomics into conservation? Evol Appl 7:999–1007
Orkin JD (2020) Selection and local adaptation in capuchin monkeys revealed through fluorescence-activated cell sorting of feces (fecalFACS). Preprint. https://doi.org/10.1101/366112
Ozga AT, Webster TH, Gilby IC et al (2020) Urine as a high-quality source of host genomic DNA from wild populations. Preprint. https://doi.org/10.1101/2020.02.18.955377
Perry GH, Marioni JC, Melsted P, Gilad Y (2010) Genomic-scale capture and sequencing of endogenous DNA from feces. Mol Ecol 19:5332–5344
Pickrell JK, Pritchard JK (2012) Inference of population splits and mixtures from genome-wide allele frequency data. PLOS Genet 8:e1002967. https://doi.org/10.1371/journal.pgen.1002967
Pignatelli M, Aparicio G, Blanquer I, Hernández V, Moya A, Tamames J (2008) Metagenomics reveals our incomplete knowledge of global diversity. Bioinformatics 24:2124–2125
Polfus JL, Manseau M, Simmons D et al (2016) Łeghágots’enetę (learning together) the importance of indigenous perspectives in the identification of biological variation. Ecol Soc 21:18
Polfus JL, Manseau M, Klütsch CFC, Simmons D, Wilson PJ (2017) Ancient diversification in glacial refugia leads to intraspecific diversity in a Holarctic mammal. J Biogeogr 44:386–396
R Core Team (2015) R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna. https://www.R-project.org/
RStudio Team (2015) RStudio: integrated development for R. RStudio, Inc., Boston. http://www.rstudio.com/
Russello MA, Waterhouse MD, Etter PD, Johnson EA (2015) From promise to practice: pairing non-invasive sampling with genomics in conservation. PeerJ 3:e1106. https://doi.org/10.7717/peerj.1106
Seutin G, White BN, Boag PT (1991) Preservation of avian blood and tissue samples for DNA analyses. Can J Zool 69:82–90
Shafer ABA, Wolf JBW, Alves PC et al (2015) Genomics and the challenging translation into conservation practice. TREE 30:78–87
Smith O, Wang J (2014) When can noninvasive samples provide sufficient information in conservation genetics studies? Mol Ecol Resour 14:1011–1023
Snyder-mackler N, Majoros WH, Yuan ML et al (2016) Efficient genome-wide sequencing and noninvasively collected samples. Genetics 203:699–714. https://doi.org/10.1534/genetics.116.187492
Taylor RS, Horn RL, Zhang X, Golding GB, Manseau M, Wilson PJ (2019) The caribou (Rangifer tarandus) genome. Genes 10:540
Taylor RS, Manseau M, Horn RL, Keobouasone S, Golding GB, Wilson PJ (2020) The role of introgression and ecotypic parallelism in delineating intra-specific conservation units. Mol Ecol. https://doi.org/10.1111/mec.15522
Taylor RS, Manseau M, Klutsch CFC, Polfus JL, Steedman A, Hervieux D, Kelly A, Larter NC, Gamberg M, Schwantje H, Wilson PJ (2021) Population dynamics of caribou shaped by glacial cycles before the Last Glacial Maximum. Mol Ecol. https://doi.org/10.1111/mec.16166
Thompson LM, Klütsch CFC, Manseau M, Wilson PJ (2019) Spatial differences in genetic diversity and northward migration suggest genetic erosion along the boreal caribou southern range limit and continued range retraction. Ecol Evol 9:7030–7046
Vors LS, Boyce MS (2009) Global declines of caribou and reindeer. Glob Chang Biol 15:2626–2633
Waterhouse RM, Seppey M, Simão FA, Manni M, Ioannidis P, Kliotchnikov G, Kriventseva EV, Zdobnov E (2018) BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol Biol Evol 35:543–548
Weckworth BV, Hebblewhite M, Mariani S, Musiani M (2018) Lines on a map: conservation units, meta-population dynamics, and recovery of woodland caribou in Canada. Ecosphere 9:e02323. https://doi.org/10.1002/ecs2.2323
Wood DE, Lu J, Langmead B (2019) Improved metagenomic analysis with Kraken 2. Genome Biol 20:257. https://doi.org/10.1186/s13059-019-1891-0
Acknowledgements
We thank Austin Thompson for help with lab work. We thank Melinda-Lee Baker for help with literature searches, and The Centre for Applied Genomics (TCAG) at the Hospital for Sick Children (Toronto, Ontario) for library preparation and sequencing, to the facilities of the Shared Hierarchical Academic Research Computing Network (SHARCNET: www.sharcnet.ca) and Compute Canada/ Calcul Canada gme-665-ab.Compute Canada (RRG gme-665-ab), and Amazon Cloud Computing for high-performance computing services. We thank all those involved in sample collections. The tissue samples were obtained from the National Wildlife Research Centre specimen bank (Ottawa, ON). Faecal samples were collected by biologists with the Alberta government, Jasper and Wood Buffalo National Parks.
Funding
Funding was provided through an NSERC Collaborative Research & Development (CRD) Grant, NSERC Grant RGPIN-2015-04477, Manitoba Hydro, Saskatchewan Power, and Weyerhaeuser Inc.
Author information
Authors and Affiliations
Contributions
RST helped with study design, did the bioinformatics, and wrote the manuscript, MM conceived the study, acquired funding, and edited the manuscript, BR helped with study design, did the laboratory work, and edited the manuscript, SK did some of the bioinformatics and edited the manuscript, PG and CM did the Kraken analysis and edited the manuscript, and PJW conceived the study, acquired funding, and edited the manuscript.
Corresponding author
Ethics declarations
Conflict of interests
The authors declare that they have no conflict of interests.
Data availability
All raw reads will have been uploaded onto the NCBI database, BioProject ID PRJNA694662 (will be released upon acceptance).
Code availability
Used publically available software.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Taylor, R.S., Manseau, M., Redquest, B. et al. Whole genome sequences from non-invasively collected caribou faecal samples. Conservation Genet Resour 14, 53–68 (2022). https://doi.org/10.1007/s12686-021-01235-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12686-021-01235-2