
Abstract
Competitive endogenous RNAs (ceRNAs) and tumor-infiltrating immune cells play essential roles in colorectal cancer (CRC) tumorigenesis. However, their prognostic role in elderly patients with CRC is unclear. Gene expression profiles and clinical information for elderly patients with CRC were downloaded from The Cancer Genome Atlas. Univariate, LASSO, and multivariate Cox regression analyses were utilized for screening key ceRNAs and prevent overfitting. A total of 265 elderly patients with CRC were included. We constructed a novel ceRNA network consisting of 17 lncRNAs, 35 miRNAs, and 5 mRNAs. We established three prognosis predictive nomograms based on four key ceRNAs (ceRNA nomogram), five key immune cells (immune cell nomogram), and their combination (ceRNA-immune cell nomogram). Among them, the ceRNA-immune cell nomogram had the best accuracy. Furthermore, the areas under the curve of the ceRNA-immune cell nomogram were also significantly greater than the TNM stage at 1 (0.818 vs. 0.693), 3 (0.865 vs. 0.674), and 5 (0.832 vs. 0.627) years. Co-expression analysis revealed that CBX6 was positively correlated with activated dendritic cells (R = 0.45, p < 0.01), whereas negatively correlated with activated mast cells (R =− 0.43, p < 0.01). In conclusion, our study constructed three nomograms to predict prognosis in elderly patients with CRC, among which the ceRNA-immune cell nomogram had the best prediction accuracy. We inferred that the mechanism underlying the regulation of activated dendritic cells and mast cells by CBX6 might play a crucial role in tumor development and prognosis of elderly patients with CRC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Colorectal cancer (CRC) is a prevalent gastrointestinal malignancy worldwide [1]. The highest percentages of morbidity and mortality of CRC mainly pertain to elderly patients ( ≥ 65 years old), with nearly 70% of cases diagnosed in those older than age 65 [2, 3]. Compared to young patients with CRC, elderly patients exhibit several key differences. Firstly, the incidence of colorectal cancer increases with age, making it more prevalent among the elderly population [4]. Additionally, elderly patients often present with different clinical features, including a greater proportion of advanced-stage disease at diagnosis, and a higher prevalence of comorbidities [5]. Last but not least, elderly patients with CRC undergoing surgery are more likely to have a poor prognosis and survival rates than younger patients [6, 7]. Thus, it is necessary to identify new prognostic markers and explore the potential mechanism of poor prognosis in elderly patients with CRC, thereby improving their survival and leading to better management and treatment of the disease.
In 2011, the hypothesis of competitive endogenous RNA (ceRNA), proposed by Salmena et al., held that all types of RNA transcripts, such as lncRNAs, mRNAs, and other RNAs, can act as ceRNAs via microRNA response elements that bind to miRNAs [8]. Based on this hypothesis, an increasing number of studies further confirmed the significance of the lncRNA-miRNA-mRNA regulatory network in the initiation and progression of various tumors. For example, Wang et al. [9] elucidated that cAMP-responsive element-binding protein regulates the expression of HULC by competitively binding endogenous miR-372 in liver cancer. In addition, Wang et al. [10] found a novel ceRNA regulatory network in gallbladder cancer. The ceRNA network has also been suggested as a promising prognosis biomarker in cancer, including pancreatic cancer, melanoma, and other tumors [11, 12].
The tumor microenvironment is a complex network containing many cells, such as tumor, mesenchymal, and infiltrating immune cells [13]. To date, numerous studies have revealed the crucial role of tumor and tumor-infiltrating immune cells in the progression of cancer [14] and their tight association with prognosis [15, 16]. The communication between tumor and tumor-infiltrating immune cells is often regulated by the ceRNA network [8]. Some studies have recently demonstrated that the ceRNA network and tumor-infiltrating immune cells have an excellent predictive value for the prognosis of soft tissue sarcoma and mesothelioma [17, 18]. Therefore, it is essential to understand the role of the ceRNA network and tumor-infiltrating immune cells in elderly patients with CRC.
This study aimed to construct and validate nomograms based on the ceRNA network and tumor-infiltrating immune cells for prognosis prediction in elderly patients with CRC. We constructed a ceRNA network based on the different gene expression profiling and calculated the proportions of immune cells between normal and tumor samples in elderly patients with CRC. Accordingly, three nomograms based on key ceRNAs, key immune cells, or their combination were established to predict the 1-, 3-, and 5-year overall survival (OS) in elderly patients with CRC. We used the area under the curve (AUC) of the receiver operating characteristic (ROC) analysis, concordance index (C-index), and calibration curves to assess the accuracy, discrimination, and calibration of these three nomograms. In addition, a correlation analysis of key ceRNAs and immune cells was performed to explore the potential mechanism underlying poor prognosis in elderly patients with CRC.
2 Methods
2.1 Data acquisition
We downloaded the gene expression profiling data of 312 samples from elderly patients with CRC (aged ≥ 65) with complete clinical data (including 252 tumor and 60 adjacent normal samples) from The Cancer Genome Atlas (TCGA) (https://tcga-data.nci.nih.gov/tcga/). After filtering out genes that were not differentially expressed between tumor and normal samples, differential expression analysis of mRNAs, lncRNAs, and miRNAs was performed using the “DESeq2” package in R software. The threshold was set at false discovery rate (FDR) < 0.05 and |log2fold change (FC)| > 1.
2.2 Construction of the ceRNA network
To improve prediction accuracy, we downloaded experimentally verified information on miRNA-mRNA and lncRNA-miRNA interactions from the starBase database (http://starbase.sysu.edu.cn/) [19]. Subsequently, the eligible lncRNAs, miRNAs, and mRNAs were chosen based on hypergeometric detection and correlation analysis results with a significance threshold of p < 0.05. These selected molecules were used to construct the ceRNA network, which was visualized using Cytoscape v.3.5.1 software [20].
2.3 Survival analysis and nomogram of key ceRNAs
The prognostic value of all members in the ceRNA network was assessed using Kaplan-Meier survival analysis and the Cox proportional hazard model. LASSO Cox regression was employed to identify and eliminate potential overfitting factors within the ceRNA network. Subsequently, based on the expression levels and coefficients of each feature in the Cox model, a risk score was calculated for elderly patients with CRC as follows:
where k is the number of key genes included in the ceRNA network,\(\beta i\) is the coefficient per gene, and \(Si\) is the level of gene expression [21, 22]. Patients were categorized into high- and low-risk groups based on the median risk score as the cut-off value. Subsequently, survival analysis was conducted between the high-risk group and low-risk group. We then established a nomogram based on the Cox proportional hazard model to predict the survival rate of elderly patients with CRC. The accuracy, discrimination, and calibration of the nomogram in predicting the 1-, 3-, and 5-year OS were evaluated using the C-index, AUC of ROC analysis, and calibration curves.
2.4 Immune landscape assessment
To further explore the relationship between the onset of colorectal cancer and the tumor microenvironment in elderly patients with CRC, the CIBERSORT algorithm was employed to estimate the relative abundance of 22 immune cells in CRC samples obtained from elderly patients. Samples with a p-value less than 0.05 were defined statistically significant and could be included in Wilcoxon rank-sum tests between normal and tumor samples.
2.5 Survival analysis and nomogram of key immune cells
We identified immune cell types associated with prognosis using Kaplan-Meier survival and Cox regression analyses. LASSO Cox regression was applied to prevent the overfitting of immune cells. Similar to the method previously described, the risk score of each patient was calculated according to the coefficient and expression levels of each immune cell in the Cox model, followed by the classification of patients into high- and low-risk groups based on their median risk score. We then selected the immune cells in the model to establish a nomogram. The prediction efficiency of the nomogram was reflected through the C-index, ROC analysis, and calibration curves. Finally, to explore whether combining key ceRNAs and immune cells results in improved prediction accuracy, we constructed a combined nomogram and verified its accuracy, discrimination, and calibration using the same methodology.
2.6 External validation of the protein expression level corresponding to mRNAs
To validate the accuracy of the results from the bioinformatics analysis and reduce cohort bias, we detected the levels of protein expression corresponding to key mRNAs in the ceRNA signature at the tissue level using the Human Protein Atlas database [23].
2.7 Statistical analysis
The R version 4.0.3 was used for all statistical analyses (packages: GDCRNATools, DESeq2, ggplot2, RMS, glmnet, survminer, and timeROC). The Wilcoxon test was employed to compare two independent nonparametric samples. The chi-square test was utilized for comparing categorical variables. Correlations between variables were analyzed using Spearman’s correlation coefficient. A p < 0.05 was considered statistically significant.
3 Results
3.1 Identification of significant differentially expressed genes
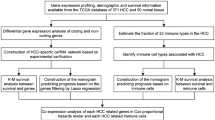
This study’s experimental design and analytical process are shown in Fig. 1. We included 265 elderly patients with primary CRC from the TCGA database (Table 1). By employing differential expression analysis between tumor and normal samples, we identified a total of 2991 mRNAs (Supplementary Fig. 1A, B), 223 lncRNAs (Supplementary Fig. 1C, D), and 367 miRNAs (Supplementary Fig. 1E, F) that met the criteria of p < 0.05 and |log2FC| > 1.

Flow chart of the experimental design and analysis process. ceRNA, competitive endogenous RNA; CRC, colorectal cancer
3.2 Construction of a ceRNA network and survival analysis
Based on the differentially expressed lncRNAs, miRNAs, and mRNAs, we constructed a ceRNA network consisting of 17 lncRNAs, 35 miRNAs, and 5 mRNAs, all of which met the criteria of the hypergeometric test and correlation analysis (p < 0.05) (Fig. 2A and Supplementary Table 1). Subsequently, we explored the prognostic value of the genes in the ceRNA network in elderly patients with CRC. We performed Kaplan-Meier survival analysis and found that the expression levels of LDLRAD3 (p = 0.032), LMNB2 (p = 0.042), NMB (p = 0.005), SNTB1 (p = 0.038), SOX4 (p = 0.022), TRAF5 (p = 0.047), SNHG16 (p = 0.049) and has-mir-1271-5p (p = 0.032) could influence the prognosis in elderly patients with CRC (Fig. 2B–H).

Construction of a ceRNA network and survival analysis in elderly patients with CRC. A ceRNA network consisting of 17 lncRNAs, 35 miRNAs, and 5 mRNAs (A). Kaplan-Meier survival curves of LDLRAD3 (B), LMNB2 (C), NMB (D), SNTB1 (E), SOX4 (F), TRAF5 (G), SNHG16 (H), and has-mir-1271-5p (I). p < 0.05. ceRNA, competitive endogenous RNA; CRC, colorectal cancer; lncRNA: long non-coding RNA; miRNA: microRNA; mRNA, messenger RNA
3.3 Development of a ceRNA signature and construction of a nomogram based on key genes in the ceRNA signature
Using univariate Cox regression analysis, we detected four key ceRNA genes (Table 2). In addition, LASSO Cox regression indicated the lack of overfitting among these four key genes, which could be incorporated into a multivariate stepwise regression (Fig. 3A–B). We thus constructed the ceRNA prognostic signature involving four key genes (CD276, CBX6, NMB, and has-miR-1271-5p) using a multivariate stepwise regression method (Fig. 3C). Consecutively, we extracted the coefficient and expression values of the four ceRNAs to calculate the risk score for each patient using the following formula:
risk score = (0.245 × level of CD276) + (0.194 × level of CBX6) + (0.321 × level of NMB) + (0.262 × level of miR-1271-5p).
We calculated the risk scores for each patient according to the above formula, and divided them into a high- (n = 132) and low-risk (n = 133) group based on the median risk score (0.984). Using Kaplan-Meier survival curve analysis, we observed that the high-risk group had worse OS than the low-risk group (hazard ratio (HR) = 3.272, 95% confidence interval (95% CI) = 1.894–5.654, p < 0.001) (Fig. 3D).
To establish a quantitative method for predicting prognosis in elderly patients with CRC, we established a nomogram (ceRNA nomogram) based on the four key ceRNAs (Supplementary Fig. 2A). We noticed that the C-index of the nomogram was 0.665. We then applied both ROC and calibration curve analyses, which indicated this nomogram did not possess good accuracy (1-year AUC: 0.743, 3-year AUC: 0.764, 5-year AUC: 0.761) (Supplementary Fig. 2B) and calibration (Supplementary Fig. 2C–E).

Development of a ceRNA signature for prognosis prediction. Tenfold cross-validation was performed to select the tuning parameter (lambda) in the LASSO Cox model based on the minimum criteria for OS (A). The LASSO coefficient profiles of survival-related ceRNAs (B). Forest plot of four key ceRNAs based on multivariate Cox regression analysis (C). Kaplan-Meier curves of OS for patients in the high- and low-risk groups (D). p < 0.05. ceRNA, competitive endogenous RNA; HR, hazard ratio; LASSO, the least absolute shrinkage and selection operator method; OS, overall survival
3.4 Composition of infiltrating immune cells between tumor and normal samples in elderly patients with CRC
We performed the CIBERPORT algorithm to calculate the proportions of the 22 immune cell types in each sample of elderly patients with CRC (Fig. 4A), and visualized the level of expression of immune cells in cancer and paired normal samples using a heatmap (Fig. 4B). Wilcoxon rank-sum test revealed a significant increase in the proportion of Palma cells (p < 0.001), regulatory T cells (p < 0.001), gamma delta T cells (p < 0.001), resting NK cells (p = 0.002), monocytes (p = 0.002), resting dendritic cells (p = 0.040), activated dendritic cells (p < 0.001), and neutrophils (p = 0.006) in normal samples compared with those in tumor samples. However, we found that memory B cells (p = 0.006), CD8 T cells (p = 0.001), memory resetting CD4 T cells (p = 0.001), activated NK cells (p = 0.002), M2 macrophages (p = 0.028), and activated mast cells (p < 0.001) were significantly more abundant in tumor than in normal samples (Fig. 4C). These results collectively suggested that the altered abundance of immune cells in elderly patients with CRC might disturb the tumor microenvironment.

Composition of infiltrating immune cells between tumor and normal samples. Percentage stacked bar chart (A) and heatmap (B) showing the distribution of the 22 immune cells assessed by the CIBERSORT algorithm in CRC samples from elderly patients. Violin plots were used to visualize the differences in immune cell abundance between tumor and normal samples. Tumor samples are represented in red, while normal samples are represented in blue (C). p < 0.05. CIBERSORT, cell type identification by estimating relative subsets of RNA transcripts; CRC colorectal cancer
3.5 Development of an immune cell signature and construction of a nomogram based on key cells in the immune cell signature
Using univariate Cox regression analysis of the 22 immune cells, we identified five immune cells that influenced prognosis. We did not detect any overfitting among the five immune cells, as indicated by the LASSO Cox regression analysis (Fig. 5A–B). We constructed the immune cell signature involving a total of five key immune cells (memory activated CD4 T cells, activated dendritic cells, M0 macrophages, M1 macrophages, and activated mast cells) using a multivariate stepwise regression method (Fig. 5C). Subsequently, patients were divided into high- and low-risk groups based on the median risk score obtained from the immune cell signature. Using Kaplan-Meier survival analysis, we noticed that elderly patients with CRC in the high-risk group had worse OS compared with those in the low-risk group (HR = 7.226, 95% CI = 2.145–24.345, p < 0.001) (Fig. 5D). The heatmap showed that the levels of activated mast cells and M0 macrophages were higher, whereas those of activated dendritic cells, activated memory CD4 T cells, and M1 macrophages were lower in the high-risk group than in low-risk group (Fig. 5E).
We accordingly constructed a nomogram (immune cell nomogram) based on these five key immune cells in the immune cell signature to predict prognosis in elderly patients with CRC (Supplementary Fig. 3A). We detected that the C-index of the nomogram was 0.736. We then applied both ROC and calibration curve analyses, which manifested an acceptable accuracy (1-year AUC: 0.671, 3-year AUC: 0.688, 5-year AUC: 0.668) (Supplementary Fig. 3B) and calibration of the nomogram (Supplementary Fig. 3C–E).

Development of an immune cell signature for prognosis prediction. Tenfold cross-validation was performed to select the tuning parameter (lambda) in the LASSO Cox model based on the minimum criteria for OS (A). The LASSO coefficient profiles of survival-related immune cells (B). Forest plot of five key immune cells based on multivariate Cox regression analysis (C). Kaplan-Meier curves of OS for patients in high- and low-risk groups (D). Heatmap (E) showing the distribution of the five key immune cells in the high- and low-risk groups. p < 0.05. ceRNA, competitive endogenous RNA; HR, hazard ratio; LASSO, the least absolute shrinkage and selection operator method; OS, overall survival
3.6 Correlation analysis of key ceRNAs with immune cells
To further explore whether ceRNAs and immune cells were related, we visualized the co-expression patterns of four key ceRNAs and 22 immune cells using a correlation heatmap (Fig. 6A). Pearson correlation analysis revealed that CBX6 was positively correlated with activated dendritic cells (R = 0.45, p < 0.01), whereas it was negatively correlated with activated mast cells (R =− 0.43, p < 0.01) (Fig. 6B–C). Overall, we found that these markers were strongly correlated with the overall level of the tumor immune microenvironment in elderly patients with CRC.

Correlation analysis of four key ceRNAs with immune cells. Correlation matrix of the four key ceRNAs with 22 tumor-infiltrating immune cells. Red represents positive correlation, whereas blue represents negative correlation (A). CBX6 was positively correlated with activated dendritic cells (B), whereas it was negatively correlated with activated mast cells (C). p < 0.05. ceRNA, competitive endogenous RNA.
3.7 Survival analysis and construction of a nomogram based on ceRNA-immune cell signature
We found that the ceRNA and immune cell signatures could independently predict prognosis in elderly patients with CRC. Therefore, assuming that combining these two signatures could result in better prediction accuracy, we integrated the four key ceRNAs and five key immune cells to develop a ceRNA-immune cell signature. According to the median risk score of the signature, patients were divided into high- and low-risk groups. Through Kaplan-Meier survival analysis, it was observed that elderly patients with CRC in the high-risk group had a poorer OS compared to those in the low-risk group (HR = 7.650, 95% CI = 2.271–25.770, p < 0.001) (Supplementary Fig. 4). Then, we constructed a combined nomogram (ceRNA-immune cell nomogram), including the four key ceRNAs and five key immune cells (Fig. 7A). We accordingly observed that the calibration curves for 1-, 3-, and 5-years OS were close to the standard curve, indicating good model performance (Fig. 7B–D). The results of C-index (C-index = 0.807 vs. 0.665 vs. 0.736) and AUCs (1-year AUC: 0.818 vs. 0.720 vs. 0.666, 3-year AUC: 0.865 vs. 0.731 vs. 0.689, 5-year AUC: 0.832 vs. 0.736 vs. 0.627) showed that the ceRNA-immune cell nomogram had better accuracy and consistency than either the immune cell nomogram or the ceRNA nomogram alone (Fig. 7E–G). Although the TNM stage is the most widely used clinical prognosis-related predictor, we noticed that the AUCs of the ceRNA-immune cell nomogram were significantly greater than those of the TNM stage at 1 (0.818 vs. 0.693), 3 (0.865 vs. 0.674), and 5 (0.832 vs. 0.627) years, indicating that our ceRNA-immune cell nomogram had better prognostic accuracy (Fig. 7E–G).

Construction of a nomogram based on the ceRNA-immune cell signature. Nomogram constructed by combining the four key ceRNAs and five key immune cells (A). The red dot represents an example of a single elderly patient. Calibration plots for predicting the 1- (B), 3- (C), and 5-years (D) OS. Comparison of the AUC values of the three nomograms and TNM stages based on time-dependent ROC curve analysis at 1- (E), 3- (F), and 5-years (G) OS. AUC, area under the curve; ceRNA, competitive endogenous RNA; OS, overall survival; ROC, receiver operating characteristic
3.8 External validation of the protein expression level corresponding to mRNAs
The expression level of mRNAs (CD276, NMB, and CBX6) in the ceRNA signature was confirmed by the sequencing data in TCGA. As already known, mRNA performs its function through protein; we thus used the Human Protein Atlas database for external validation of the expression of proteins corresponding to these three mRNAs in normal and tumor tissues of elderly patients with CRC. Our results revealed higher expression of the CD276 and NMB proteins in tumor than in normal tissues (Fig. 8A, B), whereas CBX6 was only highly expressed in normal intestinal mucosal tissues (Fig. 8C). The results are consistent with our analysis at the mRNA level.

External validation of protein expression levels corresponding to mRNAs in the Human Protein Atlas database. Comparison of the level of protein expression of CD276 (A), NMB (B) and CBX6 (C) in normal and tumor tissues.
4 Discussion
New cases of CRC mainly occur in elderly patients [24, 25]. Various molecular and genetic biomarkers, including protein-coding genes, non-coding genes, and immune cells, are utilized for predicting the prognosis and identifying potential treatment targets in CRC [21, 26, 27]. Among these biomarkers, ceRNAs and tumor-infiltrating immune cells have recently emerged as potential candidates. However, little research has focused on their action in elderly patients with CRC.
Therefore, we aimed to identify and analyze a novel ceRNA network and related immune cells for prognosis prediction and clinical treatment guidance in elderly patients with CRC. We found differently expressed ceRNAs and immune cells between normal and tumor samples in elderly patients with CRC. Subsequently, we established three prognosis predictive nomograms based on four key ceRNAs (ceRNA nomogram), five key immune cells (immune cell nomogram) or their combination (ceRNA-immune cell nomogram). Among them, the ceRNA-immune cell nomogram had the best accuracy and consistency compared with others, which might be used to predict the 1-, 3-, and 5-year OS of patients and guide their clinical treatment.
In recent years, increasing studies have revealed the involvement of ceRNAs in the tumorigenesis and their potential as promising predictive prognosis biomarkers [28]. The present study used bioinformatic analysis to identify the ceRNA networks. Using univariate, LASSO, and multivariate Cox regression analyses, we developed a ceRNA prognostic signature involving four key genes (CD276, CBX6, NMB, and has-miR-1271-5p). As a key member of the B7 superfamily, CD276 is highly expressed in hepatocellular carcinoma, lung cancer, and adrenocortical carcinoma [29,30,31]. Takashima et al. [32] also found that CD276 was effective for the prognosis of glioblastoma multiforme. The expression of the CBX family proteins has been reported in a variety of malignancies [33]. Furthermore, CBX6 is known to regulate gene expression, cell replication, and differentiation, and has been demonstrated to play a significant role in hepatocellular carcinoma [34, 35]. NMB is a single transmembrane protein expressed in differentiated immune cells. Metz et al. [36] demonstrated that NMB has properties of enhancing tumorigenesis and might thus be the core component in the development of malignant tumors. In addition, has-miR-1271-5p was found to play a critical role in cancer development [37, 38]. These findings were consistent with those of our study.
Previous studies have revealed that tumor-infiltrating immune cells play a vital role in determining the prognosis of solid tumors [39, 40]. In our study, using the method as that used for constructing the ceRNA signature, we constructed an immune cell prognostic signature involving five immune cells (activated memory CD4 T cells, activated dendritic cells, M0 macrophages, M1 macrophages, and activated mast cells). Notably, the activated memory CD4 T cells were identified as independent prognostic factors. Novy et al. [41] found that activated memory CD4 T cells influence tumor growth by affecting the function of CD8 T cells. Several studies have also found that activated memory CD4 T cells are associated with prognoses in many cancers, such as breast cancer, cervical cancer, non-small cell lung cancer, and pancreatic adenocarcinomas [42,43,44,45].
Our co-expression analysis revealed that the expression of CBX6 was positively associated with activated dendritic cells (R = 0.45, p < 0.01), but negatively associated with activated mast cells (R =-0.43, p < 0.01). Previous studies also found that the expression of CBX6 was significantly correlated with the infiltration of dendritic cells in carcinoma [46, 47]. Concomitantly, a series of studies have reported that the number of mast cells in the tumor is related to prognosis [48,49,50]. In our study, we showed that the expression of CBX6 was closely related to the infiltration of mast cells, suggesting that CBX6 might reflect not only disease prognosis but also immune status. Thus, we deduced that the two pairs and their relevant mechanisms would play essential roles in the prognosis of elderly patients with CRC. We presumed that CBX6 might regulate the populations of activated dendritic cells and activated mast cells in the tumor immune microenvironment, in turn affecting the development of tumors in elderly patients with CRC.
As mentioned above, both ceRNAs and immune cells play an important role in tumor development and prognosis. We here found that the ceRNA and immune cell nomograms could independently predict prognosis in elderly patients with CRC. Therefore, assuming that their combination could result in better prediction accuracy, we developed a ceRNA-immune cell nomogram. We found this had better accuracy and discrimination than the others, as evaluated based on the C-index, ROC analysis, and calibration curves. The ceRNA-immune cell nomogram would be valuable to elderly patients with CRC and colorectal surgeons because it could facilitate the accurate evaluation of the 1-, 3-, and 5-year OS after surgical treatment and provide more comprehensive information for the improved guidance of personalized clinical treatment.
Inevitably, our study has some limitations. As this analysis was conducted retrospectively using public databases and it is challenging to avoid selection bias in such settings, further external cohorts are required to validate these results. Moreover, in vivo and in vitro experiments are needed to further study the mechanism of prognostic ceRNAs and immune cells in elderly patients with CRC.
5 Conclusions
In conclusion, our study constructed three nomograms based on the ceRNA network and tumor-infiltrating immune cells to predict prognosis in elderly patients with CRC. Among them, the ceRNA-immune cell nomogram had the best prediction accuracy. Moreover, our study inferred that the mechanism underlying the regulation of activated dendritic cells and activated mast cells by CBX6 might playa crucial role in tumor development and prognosis in elderly patients with CRC.
Data availability
The gene expression profiling data were downloaded from The Cancer Genome Atlas (TCGA) (https://tcga-data.nci.nih.gov/tcga/). The datasets used and/or analyzed in the current study are available from the corresponding author upon reasonable request.
Code availability
The code used in the current study are available from the corresponding author upon reasonable request.
Abbreviations
- AUC:
-
Area under the curve
- ceRNA:
-
Competitive endogenous RNA
- C-index:
-
Concordance index
- CRC:
-
Colorectal cancer
- FC:
-
Fold change
- FDR:
-
False discovery rate
- HR:
-
Hazard ratio
- IQR:
-
Interquartile range
- LASSO:
-
Least absolute shrinkage and selection operator method
- lncRNA:
-
Long non-coding RNA
- miRNA:
-
MicroRNA
- mRNA:
-
Messenger RNA
- OS:
-
Overall survival
- ROC:
-
Receiver operating characteristic
- SD:
-
Standard deviation
- TCGA:
-
The Cancer Genome Atlas
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394–424.
Edwards BK, Howe HL, Ries LAG, Thun MJ, Rosenberg HM, Yancik R, Wingo PA, Jemal A, Feigal EG. Annual report to the nation on the status of cancer,1973–1999, featuring implications of age and aging on U.S. cancer burden. Cancer. 2002;94:2766–92.
Sanoff HK, Goldberg RM. Colorectal cancer treatment in older patients. Gastrointest Cancer Res: GCR. 2007;1:248–53.
Atkin W, Wooldrage K, Parkin DM, Kralj-Hans I, MacRae E, Shah U, Duffy S, Cross AJ. Long term effects of once-only flexible sigmoidoscopy screening after 17 years of follow-up: the UK Flexible Sigmoidoscopy Screening randomised controlled trial. Lancet 2017;389:1299–311.
Frizis H, Papadopoulos A, Akritidis G, Frizis HR, Hatzitheoharis G. Are there any differences in colorectal cancer between young and elderly patients? Tech Coloproctol. 2004;8(Suppl 1):147–s148.
Numata K, Ono Y, Toda S, Kamioka Y, Suematsu H, Sawazaki S, Tsuchida K, Higuchi A, Saeki H, Rino Y, Masuda M, Matsukawa H. Modified Glasgow Prognostic score and Carcinoembryonic Antigen Predict Poor Prognosis in Elderly Patients with Colorectal Cancer. Oncol Res Treat. 2020;43:125–33.
Itatani Y, Kawada K, Sakai Y. Treatment of Elderly Patients with Colorectal Cancer. Biomed Res Int. 2018;2018:2176056.
Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–8.
Wang J, Liu X, Wu H, Ni P, Gu Z, Qiao Y, Chen N, Sun F, Fan Q. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res. 2010;38:5366–83.
Wang S-H, Ma F, Tang Z-H, Wu X-C, Cai Q, Zhang M-D, Weng M-Z, Zhou D, Wang J-D, Quan Z-W. Long non-coding RNA H19 regulates FOXM1 expression by competitively binding endogenous mir-342-3p in gallbladder cancer. J Exp Clin Cancer Res: CR. 2016;35:160.
Wang W, Lou W, Ding B, Yang B, Lu H, Kong Q, Fan W. A novel mRNA-miRNA-lncRNA competing endogenous RNA triple sub-network associated with prognosis of pancreatic cancer. Aging. 2019;11:2610–27.
Cheng Y, Liu C, Liu Y, Su Y, Wang S, Jin L, Wan Q, Liu Y, Li C, Sang X, Yang L, Liu C, Wang X, Wang Z. Immune Microenvironment related competitive endogenous RNA network as powerful predictors for Melanoma Prognosis based on WGCNA Analysis. Front Oncol. 2020;10:577072.
Yuan Y, Jiang Y-C, Sun C-K, Chen Q-M. Role of the tumor microenvironment in tumor progression and the clinical applications (review). Oncol Rep. 2016;35:2499–515.
Giraldo NA, Sanchez-Salas R, Peske JD, Vano Y, Becht E, Petitprez F, Validire P, Ingels A, Cathelineau X, Fridman WH, Sautès-Fridman C. The clinical role of the TME in solid cancer. Br J Cancer. 2019;120:45–53.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pagès C, Tosolini M, Camus M, Berger A, Wind P, Zinzindohoué F, Bruneval P, Cugnenc P-H, Trajanoski Z, Fridman W-H. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–4.
Geiger J, Hutchinson R, Hohenkirk L, McKenna E, Chang A, Mulé J. Treatment of solid tumours in children with tumour-lysate-pulsed dendritic cells. Lancet 2000;356:1163–5.
Huang R, Meng T, Chen R, Yan P, Zhang J, Hu P, Zhu X, Yin H, Song D, Huang Z. The construction and analysis of tumor-infiltrating immune cell and ceRNA networks in recurrent soft tissue sarcoma. Aging. 2019;11:10116–43.
Huang R, Wu J, Zheng Z, Wang G, Song D, Yan P, Yin H, Hu P, Zhu X, Wang H, Lv Q, Meng T, Huang Z, Zhang J. The construction and analysis of ceRNA Network and patterns of Immune Infiltration in Mesothelioma with Bone Metastasis. Front Bioeng Biotechnol. 2019;7:257.
Li J-H, Liu S, Zhou H, Qu L-H, Yang J-H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–7.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Li X, Xie M, Yin S, Xiong Z, Mao C, Zhang F, Chen H, Jin L, Lan P, Lian L. Identification and validation of a six Immune-Related genes signature for Predicting prognosis in patients with stage II colorectal Cancer. Front Genet. 2021;12:666003.
Zhang M, Wang X, Chen X, Zhang Q, Hong J. Novel Immune-Related gene signature for risk stratification and prognosis of Survival in Lower-Grade Glioma. Front Genet. 2020;11:363.
Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Ã, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA-K, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist P-H, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J. Tissue-based map of the human proteome. Science. 2015;347:1260419.
Liposits G, Eshøj HR, Möller S, Winther SB, Skuladottir H, Ryg J, Hofsli E, Shah C-H, Poulsen L, Berglund Ã, Qvortrup C, Österlund P, Glimelius B, Sorbye H, Pfeiffer P. Quality of life in vulnerable older patients with metastatic colorectal cancer receiving palliative chemotherapy-the randomized NORDIC9-study. Cancers. 2021;13:2604.
Douaiher J, Ravipati A, Grams B, Chowdhury S, Alatise O, Are C. Colorectal cancer-global burden, trends, and geographical variations. J Surg Oncol. 2017;115:619–30.
Olsson L, Hammarström M-L, Israelsson A, Lindmark G, Hammarström S. Allocating colorectal cancer patients to different risk categories by using a five-biomarker mRNA combination in lymph node analysis. PLoS ONE. 2020;15:e0229007.
Liu S, Cao Q, An G, Yan B, Lei L. Identification of the 3-lncRNA signature as a prognostic biomarker for colorectal cancer. Int J Mol Sci. 2020;21:9359.
Jiang J, Bi Y, Liu X-P, Yu D, Yan X, Yao J, Liu T, Li S. To construct a ceRNA regulatory network as prognostic biomarkers for bladder cancer. J Cell Mol Med. 2020;24:5375–86.
Cheng R, Wang B, Cai X-R, Chen Z-S, Du Q, Zhou L-Y, Ye J-M, Chen Y-L. CD276 promotes vasculogenic mimicry formation in Hepatocellular Carcinoma via the PI3K/AKT/MMPs pathway. Onco Targets Ther. 2020;13:11485–98.
Zhang C, Hao X. Prognostic significance of CD276 in non-small cell lung cancer. Open Med (Wars). 2019;14:805–12.
Liang J, Liu Z, Pei T, Xiao Y, Zhou L, Tang Y, Zhou C, Wu K, Zhang F, Zhang F, Yin X, Chen N, Wei X, Lu Y, Zhu Y. Clinicopathological and prognostic characteristics of CD276 (B7-H3) expression in adrenocortical carcinoma. Dis Markers. 2020;2020:5354825.
Takashima Y, Kawaguchi A, Hayano A, Yamanaka R. CD276 and the gene signature composed of GATA3 and LGALS3 enable prognosis prediction of glioblastoma multiforme. PLoS ONE. 2019;14:e0216825.
Zheng H, Jiang W-H, Tian T, Tan H-S, Chen Y, Qiao G-L, Han J, Huang S-Y, Yang Y, Li S, Wang Z-G, Gao R, Ren H, Xing H, Ni J-S, Wang L-H, Ma L-J. Zhou W-P: CBX6 overexpression contributes to tumor progression and is predictive of a poor prognosis in hepatocellular carcinoma. Oncotarget. 2017;8:18872–84.
Bernstein E, Duncan EM, Masui O, Gil J, Heard E, Allis CD. Mouse polycomb proteins bind differentially to methylated histone H3 and RNA and are enriched in facultative heterochromatin. Mol Cell Biol. 2006;26:2560–9.
Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney GA, Li Z, Vedadi M, Schapira M, Min J, Arrowsmith CH. Recognition and specificity determinants of the human cbx chromodomains. J Biol Chem. 2011;286:521–9.
Metz RL, Patel PS, Hameed M, Bryan M, Rameshwar P. Role of human HGFIN/nmb in breast cancer. Breast Cancer Res: BCR. 2007;9:R58.
Liu X, Ma L, Rao Q, Mao Y, Xin Y, Xu H, Li C, Wang X. MiR-1271 inhibits ovarian cancer growth by targeting cyclin G1. Med Sci Monitor: Int Med J Expl Clin Res. 2015;21:3152–8.
Zhang B, Chen J, Cui M, Jiang Y. LncRNA ZFAS1/miR-1271-5p/HK2 promotes Glioma Development through regulating proliferation, Migration, Invasion and apoptosis. Neurochem Res. 2020;45:2828–39.
Ge P, Wang W, Li L, Zhang G, Gao Z, Tang Z, Dang X, Wu Y. Profiles of immune cell infiltration and immune-related genes in the tumor microenvironment of colorectal cancer. Biomed Pharmacother = Biomed Pharmacother. 2019;118:109228.
Meng J, Liu Y, Guan S, Fan S, Zhou J, Zhang M, Liang C. The establishment of immune infiltration based novel recurrence predicting nomogram in prostate cancer. Cancer Med. 2019;8:5202–13.
Novy P, Quigley M, Huang X, Yang Y. CD4 T cells are required for CD8 T cell survival during both primary and memory recall responses. J Immunol. 2007;179:8243–51.
Zhang S-C, Hu Z-Q, Long J-H, Zhu G-M, Wang Y, Jia Y, Zhou J, Ouyang Y, Zeng Z. Clinical implications of Tumor-Infiltrating Immune cells in breast Cancer. J Cancer. 2019;10:6175–84.
Wang J, Li Z, Gao A, Wen Q, Sun Y. The prognostic landscape of tumor-infiltrating immune cells in cervical cancer. Biomed Pharmacother = Biomed Pharmacother. 2019;120:109444.
Li J, Li X, Zhang C, Zhang C, Wang H. A signature of tumor immune microenvironment genes associated with the prognosis of non–small cell lung cancer. Oncol Rep. 2020;43:795–806.
Katsuta E, Qi Q, Peng X, Hochwald SN, Yan L, Takabe K. Pancreatic adenocarcinomas with mature blood vessels have better overall survival. Sci Rep. 2019;9:1310.
Li Q, Pan Y, Cao Z, Zhao S. Comprehensive Analysis of Prognostic Value and Immune Infiltration of Chromobox Family Members in Colorectal Cancer. Front Oncol. 2020;10:582667.
Zhu Y, Pu Z, Li Z, Lin Y, Li N, Peng F. Comprehensive Analysis of the expression and prognosis value of Chromobox Family Members in Clear Cell Renal Cell Carcinoma. Front Oncol. 2021;11:700528.
Hempel Sullivan H, Maynard JP, Heaphy CM, Lu J, De Marzo AM, Lotan TL, Joshu CE, Sfanos KS. Differential mast cell phenotypes in benign versus cancer tissues and prostate cancer oncologic outcomes. J Pathol. 2021;253:415–26.
Rajput AB, Turbin DA, Cheang MC, Voduc DK, Leung S, Gelmon KA, Gilks CB, Huntsman DG. Stromal mast cells in invasive breast cancer are a marker of favourable prognosis: a study of 4,444 cases. Breast Cancer Res Treat. 2008;107:249–57.
Hempel HA, Cuka NS, Kulac I, Barber JR, Cornish TC, Platz EA, De Marzo AM, Sfanos KS. Low Intratumoral mast cells are Associated with a higher risk of prostate Cancer recurrence. Prostate. 2017;77:412–24.
Acknowledgements
The authors thank all the staff members of the School of Nursing and Health Sciences, Henan University. We acknowledge the contributions of the TCGA project team.
Funding
This work was supported by the Henan Provincial Science and Technology Research Project [grant number 192102310517]. XL received funding from the China Scholarship Council [grant number 202208440010].
Author information
Authors and Affiliations
Contributions
ZT analyzed the data, designed the study, and wrote the manuscript. BL, CK, and WW analyzed the data and critically revised the manuscript. XL and YD participated in the conception of the study, designed the study, and revised the manuscript critically. All authors reviewed and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tao, Z., Li, B., Kang, C. et al. Construction of a novel nomogram based on competing endogenous RNAs and tumor-infiltrating immune cells for prognosis prediction in elderly patients with colorectal cancer. Discov Onc 14, 125 (2023). https://doi.org/10.1007/s12672-023-00742-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12672-023-00742-y