
Abstract
Many viruses appear each year. Some of these viruses result in severe disease and even death. The frequency of epidemics and pandemics is growing at an alarming rate. The lack of virus-specific etiopathogenic drugs necessitates the search for new tools for the complex treatment of severe viral diseases and their late complications. Small noncoding RNAs and their antagonists may be effective therapeutic tools for preventing virus-induced damage to targeted epithelial cells and surrounding tissues in the manifestation stage. Moreover, sncRNAs could interfere with the virus-interacting host genes that trigger the malignant transformation of target cells as a late complication of severe viral diseases.
Similar content being viewed by others


1 Introduction
Over 5630 confirmed virus species exist on Earth, and at least 320,000 undiscovered viruses infect mammalian hosts. Every year, many human virus-induced diseases emerge. Viruses can cause numerous disorders affecting all human organs and systems, injuring target cells. Viruses cause irreversible changes and death, minimal host morphology or function changes, and mediate adaptive reactions, depending on their virulence and pathogenicity. In the near future, the number of virus-induced infections will increase as environmental conditions change (for example, technologic progress, technogenic catastrophes and changes in climate and sun activity) due to the ability of viruses to develop new genetic modifications allowing them to adapt to new living conditions. Human population increases and overcrowding facilitate viral transmission from person to person and cause epidemics and pandemics.
2 Viruses and Immunity
Viruses have tropism for different cells depending on their capsid structure. Viruses activate the innate and adaptive immune systems in the host. The final outcome of viral infection may be full elimination, induction of an abortive response or latent, persistent, restricted or productive infection. All these outcomes depend on host immune system functionality and virus pathogenicity. In the worst situation, immune cells become targets for viruses, or a primarily defective immune system becomes incapable of recognizing viral antigens. Viral targeting of immune cells may provoke the development of haematologic neoplasms (acute lymphocytic leukaemia or chronic lymphocytic leukaemia) and/or secondary (acquired) immunodeficiency. The majority of infections can be effectively cured, but not when the therapeutic target is virus-injured immune system cells that have altered functionality due to the infection. Viruses that have such effects on immune cells include the measles virus, which injures T-lymphocytes (decreases proliferation) and dendritic cells; the influenza A virus, which induces neutrophil degranulation with further destruction of surrounding tissues (especially in the lungs); human immune deficiency virus (HIV), which injures CD4+ cells due to their depletion; human T cell lymphotropic virus-1 (HTLV-1), which alters T cells and NK cells (induces inhibition of cytokine-producing functions in T cells and decreases natural killer (NK) cell activity); cytomegalovirus, which causes immune senescence by decreasing the number of naïve T cells and increasing the accumulation of memory T cells; and Epstein–Barr virus, which depletes B cells (X-linked lymphoproliferative syndrome) and induces the development of gammopathy and autoimmune reactions [1].
Cytokine storm syndrome (CSS) is the main virus-induced pathogenic mechanism in the host that results in tissue destruction (e.g. in the lungs, liver and kidneys). The pathogenesis of CSS is associated with two mechanisms: one is the depletion in number or decrease in functionality of some types of T lymphocytes, and the other is connected with the activation of macrophages, NK cells and dendritic cells (DCs) by pathogens and the release of deadly levels of proinflammatory cytokines, such as C-X-C motif chemokine ligand 8 (CXCL8), C-X-C motif chemokine ligand 9 (CXCL9), C-X-C motif chemokine ligand 10 (CXCL10), C-C motif chemokine ligand 2 (CCL2), C-C motif chemokine ligand 3 (CCL3) and C-C motif chemokine ligand 5 (CCL5), along with cytokines, such as interleukin-1β (IL-1β), interleukin 6 (IL-6), interleukin 12 (IL-12), interleukin 18 (IL-18), interleukin 23 (IL-23), IFN-α, IFN-γ, TNF-α and transforming growth factor beta (TGF-β). In the case of Middle East respiratory syndrome coronavirus (MERS-CoV), increased expression levels of proinflammatory cytokines, such as IFN-α and IL-6, and chemokines, including CCL-5, CXCL-8 and CXCL-10, have been reported [5]. An extremely high rate of CCR6 T-helper 17 (Th17) lymphocytes has been detected in the peripheral blood of COVID-19 (coronavirus disease 2019) patients, sustaining a Th17-type cytokine storm [2,3,4,5]. Some known viruses can induce CSS, such as Epstein–Barr virus [6, 7], influenza virus (H5N1, H1N1, H7N9) [8], measles [9], Ebola virus [10], Dengue virus [11], Lassa virus [12,13,14], Marburg virus [15, 16], yellow fever virus [17], Crimean-Congo haemorrhagic fever virus [18], human immunodeficiency virus [19], MERS-CoV [20] and severe acute respiratory syndrome coronavirus (SARS-CoV) [21,22,23].
CSS induces secondary immunodeficiency. Virus-induced secondary immunodeficiency has temporary or long-term characteristics. Measles infection causes several weeks of immunosuppression after disease onset [9]. Influenza infection also temporarily suppresses immune responses. T-cell lymphotrophic-1 and human immune deficiency viruses induce long-term immunodeficiency [8].
3 RNA Viruses and CSS
RNA viruses are the most contagious among humans. SARS coronaviruses, the Ebola virus and MERS-CoV have caused pandemics. SARS-CoV and MERS-CoV viral infections caused global epidemics in 2003 and 2012, respectively, characterized by high fatality, pathogenicity and contagiousness rates.
SARS-CoV-2 (severe acute respiratory syndrome coronavirus-2), responsible for the recent pandemic, causes the multisymptomatic illness COVID-19. This virus is a member of the Group IV Baltimore classification of RNA viruses. This group also includes hepatitis C virus (HCV), West Nile virus, dengue virus and rhinovirus. SARS-CoV-2 is a single-stranded RNA virus belonging to the family Coronaviridae and is most closely related to SARS-CoV and MERS-CoV. The SARS-CoV-2 RNA sequence has 89.1% identity to SARS-CoV and 50% identity to MERS-CoV.
CSS in COVID-19 is associated with elevated production of interleukin-1β, interleukin-6, interleukin-10 and related factors. Its pathways are connected with the induction of STAT2, SUZ12, JUN, STAT1, MEF2A, RAD21, STAT3, BCL11A, NFE2 and BATF expression in peripheral blood mononuclear cells. Activation of these factors results in tissue damage and multiple organ failure [24].
4 Molecular Targets for SARS-CoV and SARS-CoV-2
While no effective antiviral drugs are currently available to treat COVID-19, detecting the molecular targets of SARS-CoV-2 could help find an effective treatment [25]. The pathophysiology of this deadly pathogen is complex, and a Calu-3-specific human-SARS-CoV-2 interactome (CSI) has been identified. Recently, in CSI, nodes corresponding to hubs and bottlenecks, including respiratory chain complex I proteins and immunophilins (CoV nonstructural protein 1 — Nsp1), were identified as targets of SARS-CoV-2 [26], and 332 SARS-CoV-2 interacting proteins (SIPs) have been detected in human tissues [27]. The pathophysiology of COVID-19 is also associated with several signalling pathways, including (1) eIF2 signalling/translation (induction of MXI1, BRCA1, ELF1, SIN3A, E2F4, IRF1, GABPB1, HMGN3, ETS1, SP2, POLR2A, ELK4, CHD2 and CCNT2); (2) inhibition of the ARE-mediated mRNA degradation pathway; (3) the protein ubiquitination pathway; (4) T cell receptor regulation of apoptosis; (5) the NER pathway; (6) RNA degradation; and (7) the retinoic acid-mediated apoptosis signalling pathway (expression of IRF1, CCNT2, BRCA1, MXI1, CHD2, POLR2A, SIN3A, E2F4 and HEY1). ETS1, ELK4, HMGN3, SP2, IRF1 and GABPB1) [28].
5 Carcinogenesis and Coronaviruses
Almost 150 types of viruses have carcinogenic potential in humans. Presumably, coronaviruses are not an exception. Severe viral infection likely provokes the development of cancers in target organs as a late complication. There are several reasons to make this conclusion. Massive damage to lung tissue due to an oxidative burst induced by virus-activated cells of the native immune system (ARDS — acute respiratory distress syndrome) and inhibition of adaptive immune responses may provoke local malignant transformation of normal cells [29, 30]. There is evidence that in a certain percentage of patients with severe manifestations of coronavirus infection, the virus may persist in target cells, with a higher probability of symptom recurrence [31,32,33]. Oxidative burst is accompanied by injury of the lung, kidney, liver, brain and other organs (CSS) by pathogen-activated released components, and insufficient immune defence plus the possibility of virus persistence in target cells may be a reason for the development of malignant transformation in target organs.
Moreover, the direct action of SARS-CoV-2 on cancer-inducing proteins, such as pRb and p53, was recently demonstrated [34]. Khan and others [35] have suggested that COVID-19 has oncogenic potential. In an in vitro experimental model of glioblastoma, SARS-CoV-2 had very high affinity to EGFR, VEGFR and c-MET receptors expressed on glial cells, which are strongly related to gliomagenesis. Moreover, SARS-CoV-2 may cross the blood–brain barrier, increasing the risk of injury to cells of the nervous system and causing many neurological symptoms [36].
Interestingly, SARS-CoV-2 tyrosine kinase activity was recently detected [37]. Tyrosine kinases can induce the malignant transformation of multiple cell types [38].
6 Tyrosine Kinase Inhibitors as Potentially Active Antiviral Therapeutic Tools
Passamonti and others [39] demonstrated that tyrosine kinase inhibitors were effective in SARS-CoV-2 in patients with CML. Patients with CML who were infected with SARS-CoV-2 and treated with tyrosine kinase inhibitors overcame COVID-19 and survived. In these patients, decreases in TNF-alpha and interleukin-6 levels were detected. These effects may be connected with the regulation of NF-kB pathways by tyrosine kinase inhibitors [39,40,41].
In in vitro investigations, imatinib and dasatinib (ABL kinase inhibitors) inhibited both SARS-CoV and MERS-CoV replication, while nilotinib inhibited only SARS-CoV replication [42]. The mechanism of the antiviral effects of imatinib against SARS-CoV and MERS-CoV is based on inhibition of the early stages of the virus life cycle and viral replication by targeted knockdown of ABL2 in vitro [43]. Recently, imatinib, similar to 17 other FDA-approved drugs, was reported to inhibit SARS-CoV-2, SARS-CoV and MERS-CoV with the same IC50 values in vitro [44, 45]. Moreover, there was a 4-log increase in the antiviral activity of imatinib against SARS-CoV-2 in vitro [46,47,48].
The effect of ibrutinib (Bruton’s tyrosine kinase inhibitor) was also clinically evaluated in six SARS-CoV-2 patients with Waldenström’s macroglobulinemia. These patients received different doses of ibrutinib: five received high doses (420 mg/day), and one received a low dose (140 mg/day). The patients receiving the higher dose had a more favourable COVID-19 course with milder symptoms, and hospitalization was not necessary. The patient who received the lower dose had symptoms with poorer outcomes and required hospitalization [37, 49].
Src tyrosine kinases regulate complex signalling pathways, including the EGF receptor (EGFR) pathway, Ras/Raf/MEK, PI3K/AKT and JAK/STAT. The antiviral mechanism of targeted tyrosine kinase inhibitors is unclear [50]. Recent studies revealed that dasatinib (Bcl-Abl and Src-tyrosine kinase inhibitor) is highly effective against SARS-CoV and MERS-CoV [51]. Saracatinib is an inhibitor of multiple Src tyrosine kinase (SFK) family members, including Fyn and Lyn. Saracatinib inhibited the early stages of the MERS-CoV life cycle in in vitro experiments in Huh-7 cells [52].
7 Small Noncoding RNAs and Viruses
Small noncoding RNAs are key regulators of multiple functions in organisms, and they play key roles in the pathophysiology of different diseases. Recently, the expression patterns of different microRNAs in SARS-CoV, SARS-CoV-2, influenza and measles have been determined. SARS-CoV has 53 unique miRNAs, influenza has 227, SARS-CoV-2 has 98 and measles has 42. Viral miRNAs may interact with different host genes to regulate host immune system responses, facilitating viral invasion, replication and dissemination [53].
SARS-CoV-2 invasion changes the expression levels of different host miRNAs. Downregulation of hsa-miR-1-3p, hsa-miR-17-5p, hsa-miR-199a-3p, hsa-miR-429, hsa-miR-15a-5p and hsa-miR-20a-5p expression has been observed. The interaction network enabled the identification of 51 miRNAs that interact with 77 transcription factor genes, including HMOX1, DNMT1, PLAT, GDF1 and ITGB1. These miRNAs are involved in modulating antiviral activity by the induction and epigenetic control of IFN-α2b (interferon-α2b) [54]. Both SARS-CoV-1 and SARS-CoV-2 miRNAs interact with host miRNA genes that participate in TGF-β signalling, adherens junction function and AMPK signalling. SARS-CoV miRNAs mostly regulate host miRNAs that control the VEGFα-VEGFR2 and EGF/EGFR signalling pathways. Host miRNAs targeted by SARS-CoV-2 miRNAs regulate the mTOR, Wnt and p38MAPK pathways [55].
8 RNA Interference as a Therapeutic Antiviral Mechanism
The use of viral-targeted RNA interference as a treatment for SARS-CoV infection began in 2003. In that year, the viral sequence of SARS-CoV was determined [56]. At that time, siRNAs and shRNAs were used to inhibit SARS-CoV replication in vitro. The main targets for RNA interference were RNA polymerase and spike protein genes [57,58,59,60]. Viruses have a high mutation rate, and the search for virus-targeting specific shRNAs or siRNAs is a “one-way ticket” for antiviral therapy. Despite the high therapeutic efficiency of one virus-specific siRNA or shRNA against a select virus strain, in the case of large mutational rearrangements of viral genetic material with changes in viral pathogenicity and the appearance of other virus strains, this siRNA or shRNA may become ineffective. Investigating new virus-interfering sequences will require additional funding, time and intellectual resources. Identifying the main therapeutic targets for severe viral diseases necessitates the identification of key virus-induced pathways in the host organism.
9 miR-155 and Viral Infections
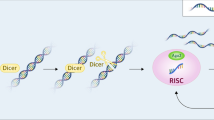
SncRNAs, as coordinators of host–virus interactions, may be preferable tools for the treatment of severe viral infections and their late complications. Recently, the role of miR-155 in the regulation of anti-SARS-CoV-2 and other viral infection immune responses was investigated. Increased levels of miR-155 were detected in patients with numerous viral diseases and SARS-CoV-2 patients [61, 62]. MiR-155 is a regulator of various immune functions. A decrease in miR-155 expression increases Peli1 expression in CD4+ T cells and inhibits c-Rel due to compromised CD40L expression in CD4+ T cells and the impaired proliferation of antigen-specific CD4+ T cells at the late cell differentiation stage. MiR-155 is a key factor for DC maturation and activation. Mature DCs obtained from miR-155-deficient mice exhibited defects in DC morphology and decreased upregulation of CD40 and CD86 [63]. MiR-155 plays a significant role in the regulation of different types of active immune cells, including T and B cells, DCs and NK cells. Moreover, increased miR-155 expression was observed during the activation of T cells, B cells, DCs and macrophages. Additionally, miR-155 regulates the expression of immune modulators and regulators such as Ship1, Socs1, Jarid2, Ets1, PU.1 and Fosl2 [63] (Fig. 1).

The use of the sncRNAs/DDMC complex as an antiviral therapeutic tool. The DDMC polymer vector is a carrier for sncRNAs or antago-miRs. It protects active RNAs from biodegradation in blood and cells. The complex of sncRNAs and DDMC slowly decays in the cytoplasm, releasing sncRNAs or their antago-miRs. SncRNAs induce RNA interference mechanisms. The effects and mechanisms regulated by miR-155 and antago-miR-155 in virus-targeted cells are associated with severe viral manifestations and late complications
The role of miR-155 in the activation and regulation of immune responses to viruses has recently been revealed [64, 65]. This miRNA may act as a “double-edged sword” in different viral infections. In experiments with human NK cells infected with a lentivirus, it was observed that in cells treated with IL-12 and IL-18, miR-155 levels increased as interleukin concentrations increased. MiR-155 expression levels also correlated with IFN-γ release from NK cells [66]. Patients infected with RSV had highly elevated levels of miR-155 in macrophages [67]. miR-155 was found to control leukotriene B4 pathways via leukotriene receptor 1 signalling. The antago-miR against miR-155 prevented both the LTB4-mediated decrease in SOCS-1 and the increase in MyD88 levels in macrophages by controlling cell activation [68]. Lind et al. [69] showed that immunity to lymphocytic choriomeningitis virus was lacking in miR-155-deficient mice. Significantly, miR-155 was overexpressed in EBV-infected resting primary human B cells, increasing the survival of lymphoblastic cells. It was hypothesized that miR-155 is important for EBV persistence [70].
Epithelial cells of the respiratory tract are the main targets in RV infection. miR-155 can regulate innate immune responses against RV-1B, resulting in targeting of the genetic components of RV [71]. miR-155 knockdown can stimulate RV replication [72, 73].
Recently, it was observed that the treatment of SARS-CoV-2-infected tg-mice hACE2 (a mouse model of SARS-CoV-2 infection; mice transgenic for human angiotensin I-converting enzyme 2 receptor) with antago-miR-155 improved the clinical manifestations and increased animal survival rates. In antago-miR-155-treated tg-mice, hACE2 cells infected with SARS-CoV-2 exhibited reduced levels of proinflammatory cytokines and induced antiviral and anti-inflammatory cytokine responses in the lungs [74, 75]. Woods and colleagues [76] revealed that pulmonary oedema (ARDS) in A/WSN/33 (H1N1) influenza-infected 155-KO mice was significantly reduced compared with that in virus-infected control mice [76, 77].
In in vivo experiments, we recently obtained full recovery of mice with virus-induced src-tyrosine kinase sarcoma as a model of virus-induced tumours in humans. We used a slow-biodegradable complex of antago-miR-155 with a polymer carrier for treatment. We did not detect src gene expression in the sectioned tissues of mice with virus-induced sarcoma treated with antago-miR-155, and we did not find any tumours or tumour cells [78].
Our in vitro experiments also achieved irreversible transformation of AML M0-M1 cancer cells into noncancerous stem cells and their differentiation into other types of cells after adding different cell differentiating factors to the medium [79].
10 Conclusion
sncRNAs and their antago-miRs may be effective tools for the complex treatment of severe viral diseases. Primary manifestations of severe viral infections may be prevented by using sncRNAs, which play key roles in the epigenetic regulation of immune cells (reduction of the affected area by decreasing the release of injury factors by virus-induced target immune cells after treatment with sncRNAs). MiR-155 regulates the expression of proinflammatory cytokines. Using antago-miR-155 inhibits the hyperreactivity of the immune system and prevents the development of CSS. The persistence of viruses in cells, oxidative bursts, immunologic disturbances and activation of tumorigenic pathways by viruses may provoke late severe postviral complications and even the development of cancers in target organs. SncRNAs may prevent some virus-induced host carcinogenic pathways and may be an effective tool for treating late severe complications of viral diseases. Moreover, sncRNAs are not associated with severe side effects, unlike tyrosine kinase inhibitors.
Abbreviations
- ABL2:
-
tyrosine-protein kinase
- ABL2:
-
Abelson-related gene
- AML:
-
acute myeloid leukaemia
- ARE:
-
adenylate-uridylate-rich elements (AU-rich elements)
- BATF:
-
basic leucine zipper transcription factor, ATF-like
- BCL11A:
-
B-cell lymphoma/leukaemia 11A
- BRCA1:
-
breast cancer type 1 susceptibility protein
- CCNT2:
-
cyclin-T2
- CD:
-
cluster of differentiation
- CHD2:
-
chromodomain-helicase-DNA-binding protein 2
- CML:
-
chronic myelogenous leukaemia
- DNMT1:
-
DNA (cytosine-5)-methyltransferase 1
- EGF:
-
epidermal growth factor
- ELK4:
-
ETS domain-containing protein Elk-4
- ETS1:
-
protein C-ets-1
- Fosl2:
-
Fos-related antigen 2
- GABPB1:
-
gamma-aminobutyric acid receptor subunit beta-1
- GDF1:
-
growth differentiation factor 1
- HEY1:
-
hairy/enhancer-of-split related with YRPW motif protein 1
- HMGN3:
-
high mobility group nucleosome-binding domain-containing protein 3
- HMOX1:
-
heme oxygenase 1 gene
- IC50:
-
half maximal inhibitory concentration
- IRF1:
-
interferon regulatory factor
- ITGB1:
-
integrin beta-1
- JAK/STAT:
-
Janus kinases/signal transducer and activator of transcription proteins
- Jarid2:
-
protein Jumonji, alpha-ketoglutarate-dependent hydroxylase superfamily
- MEF2A:
-
myocyte-specific enhancer factor 2A
- MXI1:
-
MAX-interacting protein 1
- MyD88:
-
myeloid differentiation primary response 88
- NER:
-
nucleotide excision repair
- NF-kB:
-
nuclear factor kappa-light-chain-enhancer of activated B cells
- p38MAPK:
-
p38 mitogen-activated protein kinases
- PI3K/AKT:
-
PI3K/AKT/mTOR pathway is an intracellular signalling pathway important in regulating the cell cycle
- PLAT:
-
tissue plasminogen activator
- POLR2A:
-
DNA-directed RNA polymerase II subunit RPB1
- RAD21:
-
double-strand-break repair protein rad21
- Ras/Raf/MEK (MAPK/ERK pathway):
-
mitogen-activated protein kinases/extracellular signal-regulated kinases pathway
- Ship1:
-
Src homology 2 (SH2) domain containing inositol polyphosphate 5-phosphatase 1
- shRNAs:
-
small hairpin RNA
- SIN3A:
-
paired amphipathic helix protein Sin3a
- siRNAs:
-
small interfering RNA
- Socs1:
-
suppressor of cytokine signalling 1
- STAT:
-
signal transducer and activator of transcription
- SUZ12:
-
polycomb protein SUZ12
- TGF-β:
-
transforming growth factor beta
- TNF-alpha:
-
tumour necrosis factor alpha
References
Bonagura, V. R., & Rosenthal, D. W. (2020). Infections that cause secondary immune deficiency. In Stiehm’s immune deficiencies (pp. 1035–1051). Elsevier. https://doi.org/10.1016/B978-0-12-816768-7.00049-1
Z, Xu, L. Shi, Y. Wang, J. Zhang, L. Huang, C. Zhang, S. Liu, P. Zhao, H. Liu, L. Zhu, Pathological findings of COVID-19 associated with acute respiratory distress syndrome. The Lancet Respiratory Medicine 8(4) (2020) 420-422;
Li, G., Fan, Y., Lai, Y., Han, T., Li, Z., Zhou, P., Pan, P., Wang, W., Hu, D., Liu, X., Zhang, Q., & Wu, J. (2020). Coronavirus infections and immune responses. Journal of Medical Virology, 92, 424–432.
Immunity to microbes. (2007). Injurious effects of immune responses. In A. K. Abbas, A. H. Lichtman, & S. Pillai (Eds.), Cellular and molecular immunology (pp. 354–355). Elsevier.
Tisoncik, J. R., Korth, M. J., Simmons, C. P., Farrar, J., Martin, T. R., & Katze, M. G. (2012 Mar). Into the eye of the cytokine storm. Microbiology and Molecular Biology Reviews, 76(1), 16–32.
Verbist, K. C., & Nichols, K. E. (2019). Cytokine storm syndromes associated with Epstein–Barr virus. In R. Cron & E. Behrens (Eds.), Cytokine storm syndrome. Springer. https://doi.org/10.1007/978-3-030-22094-5_15
Sutkowski, N., Palkama, T., Ciurli, C., Sekaly, R. P., Thorley-Lawson, D. A., & Huber, B. T. (1996). An Epstein-Barr virus-associated antigen. The Journal of Experimental Medicine, 184(3), 971–980.
Simon, F. F., McCorrister, S., Hu, P., Chong, P., Silaghi, A., Westmaccot, G., Coombs, K. M., & Kobasa, D. (2015). Highly pathogenic H5N1 and novel H7N9 influenza A viruses induce more profound proteomic host responses than seasonal and pandemic H1N1 strains. Journal of Proteome Research, 14(11), 4511–4523.
Griffin, D. E. (2016). The immune response in measles: Virus control, clearance and protective immunity. Viruses, 8(10), 282.
Leroy, E. M., Becquart, P., Wauquier, N., & Baize, S. (2011). Evidence for Ebola virus superantigen activity. J.Virol., 4041–4042.
Gagnon, S. J., Leporati, A., Green, S., Kalayanarooj, S., Vaughn, D. W., Stephens, H. A., Suntayakorn, S., Kurane, I., Ennis, F. A., & Rothman, A. L. (2001). T cell receptor Vbeta gene usage in Thai children with Dengue virus infection. Am.J.Trop.Med.Hyg., 64(1-2), 41–48.
Baillet, N., Reynard, S., Perthame, E., Hortion, J., Journeaux, A., Mateo, M., Carnec, X., Schaeffer, J., Picard, C., Barrot, L., Barron, S., Vallve, A., Duthey, A., Jacquot, F., Boehringer, C., Jouvion, G., Pietrosemoli, N., Legendre, R., Dillies, M. A., et al. (2021). Systemic viral spreading and defective host responses are associated with fatal Lassa fever in macaques. Communications Biology, 4, 27.
Prescott, J., Marzi, A., Safronetz, D., Robertson, S. J., Feldmann, H., & Best, S. M. (2017). Immunobiology of Ebola and Lassa virus infections. Nature Reviews. Immunology, 17, 195–207.
Willard, K. A., Alston, J. T., Acciani, M., & Brindley, M. A. (2019). Identification of residues in lassa virus glycoprotein subunit 2 that are critical for protein function. Pathogens, 8(1), 1.
Bixler, S. L., & Goff, A. J. (2015). The role of cytokines and chemokines in filovirus infection. Viruses, 7(10), 5489–5507.
Leroy, E. M., Gonzalez, J.-P., & Baize, S. (2011). Ebola and Marburg haemorrhagic fever viruses: Major scientific advances, but a relatively minor public health threat for Africa. Clinical Microbiology and Infection, 17(7), 964–976.
Cong, Y., McArthur, M. A., Cohen, M., Jahrling, P. B., Janosko, K. B., Josleyn, N., Kang, K., Zhang, T., & Holbrook, M. (2016). Characterisation of yellow fever virus infection of human and non-human primate antigen presenting cells and their interaction with CD4+ T cells. PLoS Neglected Tropical Diseases, 10(5), e0004709.
Bertolotti-Ciarlet, A., Smith, J., Strecker, K., Paragas, J., Altamura, L. A., McFalls, J. M., Frias-Staheli, N., Garcia-Sastre, A., Schmaljohn, C. S., & Doms, R. W. (2005). Cellular localization and antigenic characterization of Crimean-congo hemorrhagic fever virus glycoproteins. Journal of Virology, 79(10), 6152–6161.
Muema, D. M., Akilimali, N. A., Ndumnego, O. C., Rasehlo, S. S., Durgiah, R., Ojwach, D. B. A., Ismail, N., Dong, M., Moodley, A., Dong, K. L., Ndhlovu, Z. M., Mabuka, J. M., Walker, B. D., Mann, J. K., & Ndung'u, T. (2020). Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Medicine, 18, 81.
Kim, E. S., Choe, P. G., Park, W. B., Oh, H. S., Kim, E. J., Nam, E. Y., Na, S. H., Kim, M., Song, K.-H., Bang, J. H., Park, S. W., Kim, H. B., Kim, N. J., & Oh, M.-D. (2016). Clinical progression and cytokine profiles of Middle East respiratory syndrome coronavirus infection. Journal of Korean Medical Science, 31, 1717–1725.
Brown, M., & Bhardwaj, N. (2021). Super(antigen) target for SARS-CoV-2. Nature Reviews. Immunology, 21, 72.
Maggi, E., Canonica, G. W., & Moretta, L. (2020). COVID-19: Unanswered questions on immune response and pathogenesis. The Journal of Allergy and Clinical Immunology, 146(1), 18–22.
Zhong, J., Tang, J., Ye, C., & Dong, L. (2020). The immunology of COVID-19: Is immune modulation an option for treatment? The Lancet Rheumatolology, 2, e428–e436.
Cascella, M., Rajnik, M., Cuomo, A., Dulebohn, S. C., & Di Napoli, R. (2021). Features, evaluation and treatment coronavirus (COVID-19). [Updated 2021 Sep 2]. in: StatPearls [Internet]. StatPearls Publishing https://www.ncbi.nlm.nih.gov/books/NBK554776/
Vitali, F., Cohen, L. D., Demartini, A., Amato, A., Eterno, V., Zambelli, A., & Bellazzi, R. (2016). A network-based data integration approach to support drug repurposing and multi-target therapies in triple negative breast cancer. PLoS ONE, 11, e0162407.
Pfefferle, S., Schöpf, J., Kögl, M., Friedel, C. C., Müller, M. A., Carbajo-Lozoya, J., Stellberger, T., von Dall'Armi, E., Herzog, P., Kallies, S., Niemeyer, D., Ditt, V., Kuri, T., Züst, R., Pumpor, K., Hilgenfeld, R., Schwarz, F., Zimmer, R., Steffen, I., et al. (2011). The SARS-coronavirus-host interactome: Identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathogens, 7, e1002331.
Gordon, D. E., Jang, G. M., Bouhaddou, M., Xu, J., Obernier, K., White, K. M., O’Meara, M. J., Rezelj, V. V., Guo, J. Z., Swaney, D. L., Tummino, T. A., Hüttenhain, R., Kaake, R. M., Richards, A. L., Tutuncuoglu, B., Foussard, H., Batra, J., Haas, K., Modak, M., et al. (2020). A SARS-CoV-2-human protein-protein interaction map reveals drug targets and potential drug-repurposing. Nature, 583, 459–468. https://doi.org/10.1038/s41586-020-2286-9
Kumar, N., Mishra, B., Mehmood, A., Athar, M., & Mukhtar, M. S. (2020). Integrative network biology framework elucidates molecular mechanisms of SARS-CoV-2 pathogenesis. iScience, 23(9), 101526. https://doi.org/10.1016/j.isci.2020.101526 https://doi.org/10.1101/2020.04.09.033910, 2020.
Mehta, P., McAuley, D. F., Brown, M., Sanchez, E., Tattersall, R. S., & Manson, J. J. (2020). COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet, 395(10229), 1033–1034.
Ritchie, A. I., & Singayagam, A. (2020). Immunosuppression for hyperinflammation in COVID-19: A double-edged sword? Lancet, 395(10230), 1111.
Kochi, A. N., Tagliari, A. P., Forleo, G. B., Fassini, G. M., & Tondo, C. (2020 May). Cardiac and arrhythmic complications in patients with COVID-19. Journal of Cardiovascular Electrophysiology, 31(5), 1003–1008. https://doi.org/10.1111/jce.14479
Heneka, M. T., Golenbock, D., Latz, E., Morgan, D., & Brown, R. (2020). Immediate and long-term consequences of COVID-19 infections for the development of neurological disease. Alzheimer's Research & Therapy, 12, 69.
Klok, F. A., Kruip, M. J. H. A., van der Meer, N. J. M., Arbous, M. S., Gommers, D. A. M. P. J., Kant, K. M., Kaptein, F. H. J., van Paassen, J., Stals, M. A. M., Huisman, M. V., & Endeman, H. (2020). Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thrombosis Research, 191, 145–147. https://doi.org/10.1016/j.thromres.2020.04.013
Alpalhao, M., Ferreira, J. A., & Filipe, P. (2020). Persistent SARS-CoV-2 infection and the risk for cancer. Medical Hypotheses, 143, 109882.
Khan, I., & Hatiboglu, M. A. (2020). Can COVID-19 induce glioma tumorogenesis through binding cell receptors? Medical Hypotheses, 144, 110009.
Rhea, E. M., Logsdon, A. F., Hansen, K. M., Williams, L. M., Reed, M. J., Baumann, K. K., Holden, S. J., Raber, J., Banks, W. A., & Erickson, M. A. (2021). The S1 protein of SARS-CoV-2 crosses the blood-brain barrier in mice. Nature Neuroscience, 24, 368–378.
Purcaru, O.-S., Artene, S.-A., Barcan, E., Silosi, C. A., Stanciu, I., Danoiu, S., Tudorache, S., Tataranu, L. G., & Dricu, A. (2021). The interference between SARS-CoV-2 and tyrosine kinase receptor signaling in cancer. International Journal of Molecular Sciences, 22, 4830.
Pottier, C., Fresnais, M., Gilon, M., Jerusalem, G., Longuespee, R., & Sounni, N. E. (2020). Tyrosine kinase inhibitors in cancer: Breakthrough and challenges of targeted therapy. Cancers, 12, 731–748.
Passamonti, F., Cattaneo, C., Accani, L., Bruna, R., Cavo, M., Merli, F., Angelucci, E., Krampera, M., Cairoli, R., Porta, M. G. D., et al. (2020). Clinical characteristics and risk factors associated with COVID-19 severity in patients with haematologial malignancies in Italy: A retrospective, multicenter, cohort study. The Lancet Haematology, 7(10), E737–E745.
Morales-Ortega, A., de Tena, J. G., Frutos-Perez, B., Jaenes-Barrios, B., Farfan-Sedano, A. I., Canales-Albendea, M. A., & Bernal-Bello, D. (2021). COVID-19 in patients with hematological malignancies: Considering the role of tyrosine kinase inhibitors. Cancer, 127(11), 1937–1938.
Semih, B., Nur, Y. T., Sinuan, D. M., Serdal, K., Burhan, T., & Fevzi, A. (2020). Tyrosine kinase inhibitors and COVID-19. Journal of Oncology Pharmacy Practice, 26(8), 2072–2073.
Dyall, J., Coleman, C. M., Hart, B. J., Venkataraman, T., Holbrook, M. R., Kindrachuk, J., Johnson, R. F., Olinger, G. G., Jahrling, P. B., Laidlaw, M., Johansen, L. M., Lear-Rooney, C. M., Glass, P. J., Hensley, L. E., & Frieman, M. B. (2014). Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrobial Agents and Chemotherapy, 58(8), 4885–4893.
Coleman, C. M., Sisk, J. M., Mingo, R. M., Nelson, E. A., White, J. M., & Frieman, M. B. (2016). Abelson kinase inhibitors are potent inhibitors of severe acute respiratory syndrome coronavirus and Middle East respiratory syndrome coronavirus fusion. J.Virol., 90(19), 8924–8933.
Jeon, S., Ko, M., Lee, J., Choi, I., Byun, S.Y., Park, S., Shum, D., Kim, S. (n.d) Identification of antiviral drug candidates against SARS-CoV-2 from FDA-approved drugs, bioRxiv: https://doi.org/10.1101/2020.03.20.999730
Weston, S., Coleman, C.M., Haupt, R., Logue, J., Matthews, K., M.B.Frieman, Broad anti-coronaviral activity of FDA approved drugs against SARS-CoV-2 in vivo, bioRxiv: https://doi.org/10.1101/2020.03.25.008482
Weisberg, E., Parent, A., Yang, P. L., Sattler, M., Liu, Q., Wang, J., Meng, J., Buhrlage, S. J., Gray, N., & Griffin, J. D. (2020). Repurposing of kinase inhibitors for treatment of COVID-19. Pharmaceutical Research, 37(9), 167.
Cascella, M., Rajnik, M., Cuomo, A., Dulebohn, S. C., & Di Napoli, R. (2021). Features, evaluation and treatment coronavirus (COVID-19), 2021 Sep 2. In StatPearls [Internet]. StatPearls Publishing.
Wu, F., Zhao, S., Yu, B., Chen, Y.-M., Wang, W., Song, Z.-G., Hu, Y., Tao, Z.-W., Tian, J.-H., Pei, Y.-Y., Yuan, M.-L., Zhang, Y.-L., Dai, F.-H., Liu, Y., Wang, Q.-M., Zheng, J.-J., Xu, L., Holmes, E. C., & Zhang, Y.-Z. (2020). A new coronavirus associated with human respiratory disease in China. Nature, 579, 265–269.
Treon, S. P., Castillo, J. J., Skarbnik, A. P., et al. (2020). The BTK inhibitor ibrutinib may protect against pulmonary injury in COVID-19–infected patients. Blood, 135(21), 1912–1915.
Nabavi, S. F., Habtemariam, S., Clementi, E., Berindan-Neagoe, I., Cismaru, C. A., Rasekhian, M., Banach, M., Izadi, M., Bagheri, M., Bagheri, M. S., & Nabavi, S. M. (2020). Lessons learned from SARS-CoV and MERS-CoV: FDA-approved Abelson tyrosine-protein kinase 2 inhibitors may help us combat SARS-CoV-2. Archives of Medical Science, 16(3), 519–521.
Rivera-Torres, J., & San Jose, E. (2019). Src tyrosine kinase inhibitors: New perspectives on their immune, antiviral, and senotherapeutic potential. Frontiers in Pharmacology, 10, 1011.
Shin, J. S., Jung, E., Kim, M., Baric, R. S., & Go, Y. Y. (2018). Saracatinib inhibits Middle East respiratory syndrome-coronavirus replication in vitro. Viruses, 10(6), 283.
Abu-Izneid, T., AlHajri, N., Ibrahim, A. M., Javed, M. N., Salem, K. M., Pottoo, F. H., & Kamal, M. A. (2021). Micro-RNAs in the regulation of immune response against SARS-CoV-2 and other viral infections. Journal of Advanced Research, 30, 133–145.
Sardar, R., Satish, D., & Gupta, D. (2020). Identification of novel SARS-CoV-2 drug targets by host micro-RNAs and transcription factors co-regulatory interaction network analysis Front. Genet., 11(571274), 1–9.
Khan, M. A. A. K., Sany, M. R. U., Islam, M. S., & Islam, A. B. M. M. K. (2020). Epigenetic regulator miRNA pattern differences among SARS-CoV, SARS-CoV-2, and SARS-CoV-2 world-wide isolates delineated the mystery behind the epic pathogenicity and distinct clinical characteristics of pandemic COVID-19. Frontiers in Genetics, 11(765), 1–17.
Haasnoot, J., Berkhout B. (2006). RNA interference: Its use as antiviral therapy, Handbook Exp.Pharmacol. 173 117–50 Springer-Verlag, Berlin, Heidelberg
He, M. L., Zheng, B., Peng, Y., Peiris, J. S., Poon, L. L., Yuen, K. Y., Lin, M. C., Kung, H. F., & Guan, Y. (2003). Inhibition of SARS-associated coronavirus infection and replication by RNA interference. JAMA, 290, 2665–2666.
Lu, A., Zhang, H., Zhang, X., Wang, H., Hu, Q., Shen, L., Schaffhausen, B. S., Hou, W., & Li, L. (2004). Attenuation of SARS coronavirus by a short hairpin RNA expression plasmid targeting RNA-dependent RNA polymerase. Virol., 324, 84–89.
Wang, Z., Ren, L., Zhao, X., Hung, T., Meng, A., Wang, J., & Chen, Y. G. (2004). Inhibition of severe acute respiratory syndrome virus replication by small interfering RNAs in mammalian cells. Journal of Virology, 78, 7523–7527.
Zhang, Y., Li, T., Fu, L., Yu, C., Li, Y., Xu, X., Wang, Y., Ning, H., Zhang, S., Chen, W., Babiuk, L. A., & Chang, Z. (2004). Silencing SARS-CoV Spike protein expression in cultured cells by RNA interference. FEBS Letters, 560, 141–146.
Badry, A., Jaspers, V. L., & Waugh, C. A. (2020). Environmental pollutants modulate RNA and DNA virus-activated miRNA-155 expression and innate immune system responses: Insights into new immune-modulative mechanisms. Journal of Immunotoxicology, 17(1), 86–93.
Dickey, L. L., Hanley, T. M., Huffaker, T. B., Ramstead, A. G., O'Connell, R. M., & Lane, T. E. (2017). MicroRNA 155 and viral-induced neuro-inflammation. Journal of Neuroimmunology, 308, 17–24.
Pashangzadeh, S., Motallebnezhad, M., Vafashoar, F., Khalvandi, A., & Mojtabavi, N. (2021). Implications the role of miR-155 in the pathogenesis of autoimmune diseases. Frontiers in Immunology, 12(669382), 1–14.
Kemp, V., Laconi, A., Cocciolo, G., Berends, A. J., Breit, T. M., & Verheije, A. (2020). miRNA repertoire and host immune factor regulation upon avian coronavirus infection in eggs. Archives of Virology, 165, 835–843.
Tsai, C.-Y., Allie, S. R., Zhang, W., & Usherwood, E. J. (2013). MicroRNA miR-155 affects antiviral effector and effector memory CD8 T cell differentiation. Journal of Virology, 87(4), 2348–2351.
Trotta, R., Chen, L., Ciarlariello, D., Josyula, S., Mao, C., Costinean, S., Yu, L., Butchar, J. P., Tridandapani, S., Croce, C. M., & Caligiuri, M. A. (2012). miR-155 regulates IFN-γ production in natural killer cells. Blood, 119(15), 3478–3485.
Leon-Icaza, S. A., Zheng, M., & Rosas-Taraco, A. G. (2019). MicroRNAs in viral acute respiratory infections: Immune regulation, biomarkers, therapy, and vaccines. ExRNA, 1(1), 1.
Wang, Z., Filgueiras, L. R., Wang, S., Serezani, A. P. M., Peters-Golden, M., Jancar, S., & Serezani, C. H. (2014). Leukotriene B4 enhances the generation of pro-inflammatory microRNAs to promote MyD88-dependent macrophage activation. Journal of Immunology, 192(5), 2349–2356.
Lind, E. F., Elford, A. R., & Ohashi, P. S. (2013). MicroRNA 155 is required for optimal CD8+ T cell response to acute viral and intracellular bacterial challenges. Journal of Immunology, 190(3), 1210–1216.
Linnstaedt, S. D., Gottwein, E., Skalsky, R. L., Luftig, M. A., & Cullen, B. R. (2010 Nov). Virally induced cellular microRNA miR-155 plays a key role in B-cell immortalization by Epstein-Barr virus. Journal of Virology, 84(22), 11670–11678.
Megremis, S., Taka, S., Oulas, A., Kotoulas, G., Iliopoulos, I., & Papadopoulos, N. G. (2014). O20-human rhinovirus replication-dependent induction of micro-RNAs in human bronchial epithelial cells. Clinical and Translational Allergy, 4(1), O20.
Bondanese, V. P., Francisco-Garsia, A., Bedke, N., Davies, D. E., & Sanchez-Elsner, T. (2014). Identification of host miRNAs that may limit human rhinovirus replication. World Journal of Biological Chemistry, 5(4), 437.
Mirzaei, R., Mahdavi, F., Badrzadeh, F., Hosseini-Fard, S. R., Heidary, M., Jeda, A. S., Mohammadi, T., Roshani, M., Yousefimashouf, R., Keyvani, H., Darvishmotevalli, M., Sani, M. Z., & Karampoor, S. (2021). The emerging role of microRNAs in the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. International Immunopharmacology, 90(107204), 1.
Soni, D. K., Cabrera-Luque, J., Kar, S., Sen, C., Devaney, J., Biswas, R. (n.d). Suppression of miR-155 attenuates lung cytokine storm induced by SARS-CoV-2 infection in human ACE2-transgenic mice, bioRxiv 2020 https://doi.org/10.1101/2020.12.17.423130
Gangemi, S., & Tonacci, A. (2021). AntagomiRs: A novel therapeutic strategy for challenging COVID-19 cytokine storm. Cytokine & Growth Factor Reviews, 58, 111–113.
Woods, P. S., Doolittle, L. M., Rosas, L. E., Nana-Sinkam, S. P., Tili, E., & Davis, I. C. (2020). Increased expression of microRNA-155-5p by alveolar type II cells contributes to development of lethal ARDS in H1N1 influenza A virus-infected mice. Virol., 545, 40–52.
Pociask, D. A., Robinson, K. M., Chen, K., McHugh, K. J., Clay, M. E., Huang, G. T., Benos, P. V., Janssen-Heininger, Y. M. W., Kolls, J. K., Anathy, V., & Alcorn, J. F. (2017). Epigenetic and transcriptomic regulation of lung repair during recovery from influenza infection. The American Journal of Pathology, 187, 851–863.
Klimenko, O. V., & Sidorov, A. (2019). The full recovery of mice (Mus Musculus C57/BL/6 strain) from virus-induced sarcoma after treatment with a complex of DDMC delivery system and SncRNAs. Non-coding RNA Research, 4(2), 69–78.
Klimenko, O. V., & Shtilman, M. I. (2013). Transfection of Kasumi-1 cells with a new type of polymer carriers loaded with miR-155 and antago-miR-155. Cancer Gene Therapy, 20, 237–241.
Author information
Authors and Affiliations
Contributions
OVK planned, designed, performed and wrote the manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The author declares no competing interests.
Additional information
Key Points
• The number of severe viral infections and associated complications is increasing.
• Symptomatic therapy is sometimes not enough and/or is not effective for treating severe viral diseases.
• Etiopathogenic therapy has not been well studied in the context of viral diseases.
• Using tyrosine kinase inhibitors to treat coronavirus infections is effective but is associated with severe side effects.
• Some sncRNAs may effectively treat severe viral disease manifestations and late complications; antago-miR-155 may be effective for the complex therapy of severe coronavirus manifestations (ARDS) and late complications (viral tyrosine kinase-induced cancers).
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Klimenko, O.V. Perspectives on the Use of Small Noncoding RNAs as a Therapy for Severe Virus-Induced Disease Manifestations and Late Complications. BioNanoSci. 12, 994–1001 (2022). https://doi.org/10.1007/s12668-022-00977-z
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12668-022-00977-z