
Abstract
The integrity of wells, which are key components for CO2 sequestration, depends mainly on the seal between the wellbore cement and the geologic formation. To identify the reaction products that may alter the cement/caprock interface, batch experiments and computer modelling were conducted and analysed. Over time, the dissolution and precipitation of minerals alters the physical properties of the interface, including its tightness. One main objective of the simulation was thus to analyse the evolution of the porosity of cement and caprock over time. The alteration of the cement/caprock interface was identified as a complex problem and differentiated depending on rock type. The characteristic feature of a cement/shale contact zone is the occurrence of a highly carbonated, compacted layer within the shale, which in turn causes cement/shale detachment. In the case of a cement/anhydrite interface, the most important reaction is severe anhydrite dissolution. Secondary calcite precipitation takes place in deeper parts of the rock. The cement/rock contact zone is prone to rapid mineral dissolution, which contributes to increased porosity and may alter the well integrity. Comparison of computer simulations with autoclave experiments enabled the adjustment of unknown parameters. This enhances the knowledge of these particular assemblages. Overall, a good match was obtained between experiments and simulations, which enhances confidence in using models to predict longer-term evolution.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The principal function of well cementing is to support the casing as well as to restrict fluid movement between the well and formation rocks. The need to understand the reactivity of well cement in CO2-saturated brines is emphasised by many researchers who study the safety of carbon dioxide storage (Huet et al. 2010; Kutchko et al. 2007, 2009; Um et al. 2011). The experiments performed by Jung and Um (2013) demonstrate that preferential cement alteration can occur along the cement/steel and cement/rock interfaces and highlight the importance of further investigation of cement degradation along the interfaces to ensure permanent geologic carbon storage.
The injected CO2 reacts with the formation rocks and wellbore cement, changing their mineral composition and porosity and the integrity of the materials. Carbon dioxide dissolves into the saline pore water, which causes its acidification and leads to geochemical changes. The dissolution of some minerals and the precipitation of new ones can potentially change the porosity and permeability of the reservoir rocks. Similar reactions are also possible within caprocks, which are an important element of the wellbore system sealing capacity, preventing fluids from escaping the sequestration reservoir (Carey et al. 2007). Geochemical changes upon CO2 injection are susceptible to reservoir conditions (Czernichowski-Lauriol et al. 2004); hence, the important issue is to study the potential geochemical reactions that are possible during the process of sequestration. The geological conditions are essential in the selection of the proper storage reservoir for CO2 sequestration. The assessment of the sealing efficiency of a storage site is a key factor in evaluating the storage site performance (Gherardi and Audigane 2013).
Wellbore cement alteration under sequestration conditions is an important problem explored in a number of studies over the last decades. Cement phases are known to be thermodynamically stable only under alkaline pH conditions but may become unstable under neutral to acid pH conditions. The acidification of water is not only caused by CO2 injection but is also influenced by neutral to acidic pH fluids from caprocks and reservoir rocks. The extensively performed experimental and numerical studies have focused on the cement–brine or cement–rock–brine interactions involving reservoir rocks and caprocks. The rocks examined in contact with wellbore cement were: basalt (Jung and Um 2013; Jung et al. 2014), granite (Soler and Mäder 2010), limestone (Duguid and Scherer 2010; Gherardi et al. 2012), shale and sandstone (Carroll et al. 2011), clay-rich argillite (Gherardi and Audigane 2013) and siltstone (Fischer et al. 2013). The most variable group of reservoir rocks is sandstones, which is the result of various mineral compositions and diagenesis levels. Therefore, the results for any sandstone rock cannot be directly related to the sandstone at all.
Quartz sandstones are usually expected to be geochemically stable (Wertz et al. 2014). The experiments and simulations performed on a sandstone from the Altmark Gas Reservoir (Huq et al. 2015) indicate that the dissolution of rock cements, including anhydrite and clay minerals, governs the overall geochemical processes taking place at the very early injection period. Carey et al. (2007) analysed shale caprock obtained from a 30-year-old CO2-exposed well and found limited alteration of the shale.
Wertz et al. (2013) stated that the major changes occur at the well/caprock interface, while porosity reallocation at the well/reservoir interface is less marked. On the caprock side, they noticed rapid, complete porosity clogging, which was enhanced by higher diffusion rates. The authors indicate that this may have effect in reducing the mechanical strength of rocks near the well/caprock interface.
The research performed to date indicates that the experimental investigations are not sufficient to predict the reaction processes between rocks, brine and wellbore materials because of the restricted reaction time under laboratory conditions. The reaction time is much too short in comparison with the real reaction time in any carbon sequestration project (Erickson et al. 2015). To establish real-time results, the modelling of geochemical processes is needed.
The aim of the simulations carried out in this study was to examine the geochemical evolution of sealing rocks at the contact with wellbore cement under sequestration conditions. The determination of porosity evolution in cement and caprock, as well as at the interface of these two materials, was also the goal of the investigation. The important question is the comparison of cement/rock interface performance in the case of different lithologies under sequestration conditions. The autoclave reactor experiments (Labus and Such 2016; Lorek et al. 2016), as well as the ToughReact code, were used to estimate the geochemical effects and porosity evolution of cement and caprock in contact with acidified brine. To allow for the proper collation of the modelling results to the reactor experiment findings, the simulation time was restricted to 2 years.
Materials and methods
The rock samples studied here were anhydrite and shale, representing rocks of comparatively low porosities. The lower Silurian shale was derived from a drilled well (from depth of about 2000 m) in the Baltic Sea region (Łeba Elevation). It is of aleuritic-pelitic fraction, finely laminated, and exhibiting a well-developed cleavage. The shale sample is composed of clay minerals (mostly chlorite), quartz, calcite, dolomite, feldspars, muscovite, pyrite and organic matter (Table 1). The Permian (Zechstein) anhydrite was taken from the Fore-Sudetic Monocline. It is layered, grey rock of the fine crystalline structure. The main component mineral is anhydrite; isolated gypsum and magnesite crystals are also present. Gypsum, as a component of anhydrite sample, is not included in Table 1, as it was not detected by means of XRD, but this mineral was stated with the use of SEM/EDS measurement.
The rocks were combined with wellbore cement to investigate the processes taking place at the cement/rock interface. The wellbore cement for the composite samples was prepared with the use of Portland cement (CEM I 32.5) at a water-to-cement ratio of 0.52. Rock cores, of a diameter of 2.5 cm, were cut lengthwise in half and supplemented with cement grout (Lorek et al. 2016). Grout was prepared in accordance with the procedure given in the relevant standards (DIN EN 10426). The samples were cured in an autoclave at 50 °C, at a pressure of 17 MPa. The cores were sliced and placed in the reactor. After each stage of the experiment (described below), the composed cement-rock samples’ slices were washed with distilled water and dried. For SEM analysis, the samples were embedded in epoxy resin to prevent sample from its disintegration.
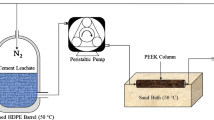
The composite samples (Fig. 1) were exposed, in an autoclave reactor, to a CO2-saturated brine under the static conditions simulating the underground environment: 50 °C and 10 MPa. The autoclave reactor is divided into 2 cells (4 dm3 each of them). The composite cement–anhydrite samples were put into one cell, and the composite cement–shale samples into the second one. In the reactor, the samples were fully covered with fluid—synthetic brine of 105 g/l concentration of NaCl, saturated with carbon dioxide (0.8 mol CO2/kg of solution). The initial pH of the solution was 3.4 (3.15 for the simulated acid brine). The experiment was performed in three stages of different duration: I—30 days, II—60 days, and III—200 days. Fluid pH was measured in the beginning of the experiment, and after each, mentioned above, stage.

Dimensions of slice of cement–rock cylindrical sample
The mineral compositions of the rocks were determined with the use of a polarising microscope, as well as with the X-ray diffraction method (Bruke-AXS Advance D8), and by SEM analysis with the use of an FEI Quanta-650 FEG Scanning Electron Microscope equipped with these analysers: EDX, WDA and EBSD. The mineral compositions, obtained from XRD measurement, with Rietveld refinement, of examined samples are given in Table 1. The formulae of the identified minerals are ideal, theoretical compositions of the stated minerals’ pure phases. For modelling purposes, the initial mass fraction (wt%) was transformed into a volume fraction, taking into account the pore volume in the rock (bulk sample). This value [vol%] is also provided in Table 1. The initial porosities of the rock samples were determined by mercury intrusion porosimetry (MIP) and are 14.4% for shale and 10% for anhydrite. It should be noted, however, that MIP method provides data for interconnected pores (effective porosity), and hence the actual porosity of the samples might be higher.
Initial cement composition for computer simulation was reconstituted from oxides (according to THERMODDEM database terminology). The assumed mineral composition of hydrated cement, which was used for the simulation, is given in Table 2. As with the rock’s composition, the composition of the cement was converted into a volume fraction, including pores (these values were used in the simulated model). The assumed initial porosity of the wellbore cement was nearly 28%.
Experimental results
Cement alteration
The cement alteration after subsequent stages of the experiment was examined with use of XRD analysis and SEM/EDS observations. After stage I, the content of ettringite and hydrotalcite decreases. All the amount of portlandite has reacted to form calcium carbonates (calcite and vaterite). At the last stage of the experiment (after 200 days), the XRD pattern shows the occurrence of abundant calcite (Lorek and Labus 2015). Observed under SEM, the carbonated zone in cement part of the sample has a thickness of about 1 mm.
Cement/anhydrite interface
Anhydrite is a rock formed mostly of mineral anhydrite, and hydrated form—gypsum. Both these minerals are easily dissolved in CO2-saturated brine under experiment conditions. In the outer zone of the sample and on the cement/rock interface, the significant increase in porosity is visible. The intense dissolution of anhydrite, and to a lesser extent—gypsum, is visible in Fig. 2. The remaining (not dissolved) mineral is magnesite (dark grey in Fig. 2). Moreover, the secondary precipitation of calcite on the interface of cement/rock is noted.

SEM image of the anhydrite part of the sample after 200 days of the reaction (Labus and Lorek 2015). The dissolution of anhydrite (A—light grey) is progressing from the bottom of the image. The remaining mineral is magnesite (M—dark grey). On the edge of the sample calcite (C) is precipitating. White crystals are pyrite. The objects of lenticular shape are the remnants of fossilised molluscs
Cement/shale interface
In the autoclave experiment, it was observed that the slate structure of the rock facilitates its fracturing, which enables fluid migration within the shale and in the space between rock and cement. Figure 3 presents the cement/shale rock interface after stage III of the reaction (200 days). At the contact with the wellbore cement, the shale is compacted and enriched in CaCO3 derived from the cement part of the sample. This zone is of 50–100 µm thick, and finely laminated, maintaining slate structure of shale. Beneath this dense zone, compressed as a result of compaction, the split between the cement and rock is present.

SEM image of cement/shale interface after 200 days of the autoclave reaction (stage III)
The minerals originally present in the shale rock generally do not show any changes. Most of these minerals are considered stable from geochemical point of view (quartz, muscovite). SEM observations after all the stages of the autoclave experiment also show the presence of iron sulphide, pyrite (FeS2), which could be expected to alter in the acid environment of CO2-saturated brine.
Modelling approach
To simulate the geochemical interactions at the cement/rock interface, the ToughReact numerical simulator was used (Xu et al. 2004). The alteration mechanisms were observed on a simplified 2D model, involving cement and rock in contact with brine water. The initial water salinity assumed during the simulation was 105 g/l NaCl (as in the autoclave experiment), with an additional comparison point at 10.5 g/l NaCl. Simulations were performed under isothermal (50 °C) and isobaric (10 MPa) conditions to mimic the autoclave experiment conditions. CO2 concentration of initial brine was 29 g/l. Pore water pH is derived from the primary mineral composition of the material. Cement pore waters are alkaline (pH 11), whereas shale and anhydrite pore waters have close to neutral pH of 6.4.
The simplified model is comprised of 160 cells of 1 mm × 1 mm size (Fig. 4). The rock/water ratio (1/35) is calculated based on the sample volume compared to the fluid volume in the reactor experiment. As the main objective of the investigation is the comparison of the obtained results from real experiment and computer simulation, the implemented rock/water ratio was calculated in such a way as to reflect accurately as possible the experimental conditions, although it is rather high for real formation rock system. The effective diffusion coefficient was assumed to be 10−10 m2/s for cement, 5 × 10−10 m2/s for shale (Wertz et al. 2013), and 7 × 10−12 m2/s for anhydrite, which is much tighter.

2D grid used for the simulation with initial pH. On the X-axis are 10 cement cells and 10 rock cells along the profile of the sample to mimic the average distance from the interface to the tightened edge. The Z-axis represents half of the height of the sample (7 mm), due to axial symmetry. R/W—rock/water ratio applied in the simulation
The thermodynamic database THERMODDEM (Blanc et al. 2012) was used to determine the thermodynamic equilibrium, element speciation, and mineral saturation indices. The ToughReact code (Xu et al. 2004) computes the rate law for mineral dissolution and precipitation according to Lasaga equation (Lasaga 1981):
where r n is a kinetic rate of mineral n (mol s−1), k is the rate constant (mol m−2 s−1) depending on the temperature, A n is the specific reactive surface area (m2 kg −1w ), and Ω n is the saturation index of the mineral n. The empirical parameters θ and η are adjustable positive numbers, determined from experiments or usually taken as 1. The dependence of the rate constant k on the temperature is calculated by means of the Arrhenius equation (Lasaga 1981):
where E a (J mol−1) is the activation energy, k 25 (mol m−2 s−1) is the rate constant at 25 °C, R (J K−1 mol−1) is the universal gas constant, and T (K) is the absolute temperature. Mineral precipitation and dissolution rates can be influenced by different mechanisms, for example acid or carbonate mechanisms (Palandri and Kharaka 2004).
where the superscripts or subscripts N, A and C indicate neutral, acid and carbonated mechanisms, respectively, and a is the activity of the corresponding species. The kinetic parameters of the minerals which were used in the simulations are given in Table 3. Parameters of C3FH6 are not provided, as this phase is assumed to be in equilibrium. The ettringite exchange surface depends on cement formation and is usually not a fixed value. Hence, in this simulation it has been increased, so that the simulation was closer to the experimental result. The value of exchange surface for ettringite was changed from 980 cm2/g (Baur et al. 2004) to 20,000 cm2/g (like CSH) and is marked in Table 3 with an asterisk (*).
Computer simulation results
Cement alteration
The cement alteration was mostly caused by the acid water environment. The initial pH of the cement pore waters is assumed to be near 11, as is that of the cement material itself. Under the influence of CO2-rich brine, the pH of the fluid in cement dramatically decreases (black curve in Fig. 5). The most soluble component of wellbore cement is Portlandite, but as a consequence of brine penetration into the cement, the other phases are also dissolved. The sudden decrease in pH is followed by a rapid dissolution of CSH and C3FH6. Portlandite dissolution starts in the outermost cells of the simulated sample, but rapidly propagates to the deeper cells. The degradation is similar in successive layers of cement but it is delayed in time (Fig. 5). Each Portlandite/C3FH6 layer buffers pH while dissolving and protects layers below. When it is dissolved, the Portlandite/C3FH6 layer below starts dissolving as well. The other phases which are also dissolving are CSH 1.6 and katoite. CSH 1.6, initially of over 30 vol%, nearly completely disappears after 100 days of simulation. Katoite content, from approximately 5%, drops to approximately 2% after 200 days.

Wellbore cement evolution as a function of time in two adjacent layers (the position is shown in Fig. 6). The vertical lines show the delay of Portlandite dissolution (approximately 3 days) between the layers

Spatial evolution of calcite distribution within cement and rock (anhydrite) parts of the sample
The dissolution of the above-mentioned cement components results in the porosity increase at the initial stage of the simulation. On the graphs shown in Fig. 3, the pore area is represented by the white background. After the increase in porosity (from the initial 28% up to approximately 30%) at the beginning of simulation, it subsequently decreases as a result of calcite and silica gel precipitation. After the assumed 200 days of the simulation, the initial cement porosity (28%) decreased to approximately 15%.
When comparing the results of cement alteration in the completed computer simulation to the results of the reactor experiment, the process runs analogously in both cases. It proceeds in a generally known way; portlandite and amorphous phases (CSH, C3FH6) dissolve, while calcite and silica gel precipitate. The difference between experiment and simulation is that ettringite is less soluble in all simulations. The results of XRD mineral phase determination within the cement part of the samples indicate complete dissolution of ettringite over the reaction time. The initial ettringite value of about 11% drops after 30 days of the experiment to approximately 1.5% and in the next stages completely disappears. This phenomenon is explained by the instability of this mineral under pH of 9.8 (Stegemann et al. 2012). In course of computer simulation, however, ettringite was partially dissolved, from the value of 11% to about 6%.
Cement/anhydrite interface
On the first days of simulation started the dissolution of calcite in the cells which are in contact with brine, whereas in the inner cells of the cement part of the sample, the amount of calcite is increasing. In course of the experiment, the precipitated CaCO3 is not only concentrated in the deeper part of cement but also consequently intrudes the rock part of the sample. Figure 4 shows the spatial evolution of calcite concentration in the simulated cement–rock sample. The rock part of the sample is anhydrite, containing no calcite at the beginning (0 days). At stages I (30 days) and II (60 days), calcite penetrates the deeper parts of the anhydrite rock, but after 200 simulated days, it moves into the cells where the initial components of the rock have partially disappeared—the mineral anhydrite has been dissolved. The concentration of calcite on the “rock side” of the sample goes up to a maximal value of 3%. In addition to calcite, trace amounts of silica (which is diluted from the cement) also intrude the rock.
Despite the fact that intruding calcite and silica are filling the voids in anhydrite, a significant increase in the porosity is noted at the cement/caprock interface. On the cement side of the modelled sample, the porosity is instead decreasing as a result of calcite precipitation. After a simulated time of 1 year, the initial porosity of the cement (28%) is reduced to approximately 15%. On the rock side of the modelled sample, intense dissolution of anhydrite takes place. The maximum porosity increase is possible even up to nearly 30% (Fig. 7). This phenomenon is caused by severe anhydrite dissolution, which is confirmed by SEM observations of the cement–anhydrite sample after 200 days of the reaction in the autoclave (Fig. 2).

Dissolution of anhydrite (mineral) within anhydrite (rock); 200-day computer simulation of the cell on the cement/rock interface
Cement/shale interface
The main findings of the computer simulation confirm the SEM observations of the composite cement–shale samples which were subjected to the autoclave experiment. In course of the simulation, the initial composition of the shale is rather stable. The initial pore water in the shale quickly mixes with the cement pore waters and acidic brine, and after 30 days, the primary pH of 6.4 stabilises at the level of 5. The first, and very fast, occurring reaction is calcite dissolution; in 30 days of simulation, calcite nearly completely disappears. The dissolution front propagates from the outer cells, which are in contact with acid brine. After 30 days, the reverse process of calcite precipitation starts, which propagates from the cement/shale contact zone, progressing deeper into the rock and upwards towards the outer part of the sample. In Fig. 8, we can notice the appearance of calcite at the end of the simulation (200 days), as this figure presents an X-axis-slice at the level of Z = −4. The other mineral that is subject to dissolution is dolomite, however to a lesser extent. The initial value of over 6% decreases to 3% in 60 days, 0.8% in 200 days, and nearly completely disappears after 2 years of modelling. The contents of the other minerals in the shale mostly remain at the same level (including pyrite, as it was observed in autoclave experiment).

Cement/shale interface evolution (X-slice on the Z = −4 level)
The alteration of the cement part of the sample is consistent with that taking place in the cement/anhydrite sample characterised in paragraph 5.2. Any migration of the shale components to the cement part of the sample was observed, contrary to the results presented by Carroll et al. (2011), who report the precipitation of smectite when cement reacts with shale and CO2.
Discussion
The modelled materials, wellbore cement and rock (either anhydrite or shale), are very different. Cement evolves very quickly; the effects are visible in the simulation processes after an interval of a few days, while in the case of rocks the evolution is rather slow, extending over a year’s time. This material diversity generates a problem with simulation and interpretation.
The most important and well-known phenomenon of cement degradation is its leaching, characterised by the rapid dissolution of the portlandite, CSH and C3FH6 phases. It is caused by the decrease in pH in pore fluid. The wellbore cement begins to evolve as soon as it is saturated with pore water of pH close to neutral (Gaucher and Blanc 2006). This saturation occurs if the cement is in contact with formation rocks of significantly lower pH. In the model considered, simulations show a stabilisation of the cement pore water pH after approximately 60–80 days (depending on the distance of the cell from acid water), once buffering materials have all dissolved. The dissolution of portlandite is obviously followed by calcite precipitation in the cement pore area. We did not observe the secondary precipitation of any primary cement components, as was reported by Gherardi and Audigane (2013). Their simulation revealed the secondary precipitation of some phases, predominantly represented by CSH 1.6 and ettringite, which is explained by the high pH of the cement pore waters.
The calcite precipitation in the outer part of the cement part of the sample leads to clogging of the pore space and eventually decreases primary porosity. Too much precipitation may actually fracture the cement. Thus, poro-mechanical models are required to complete wellbore cement assessment, in addition to this geochemical analysis. The alteration of the cement/caprock interface is a complex problem, caused by the heterogeneity of the materials (rocks and cement) involved. The performance of this zone is dependent on a range of parameters connected to the lithologic type of the rock.
The calcite precipitation present in the inner cells of the cement may be dependent on the salinity of brine. Being aware of the high salinity used in the model, simulations under lower salinity conditions were performed as well. The lower concentration of salt (10.5 g/l) results in insignificantly lower calcite precipitation. However, due to the sensitivity of the ToughReact saturation model to salinity, a delayed difference was observed for ongoing geochemical processes. Though it does not affect the general results of this paper, this salinity impact could be a topic for a further benchmark study.
The main product of wellbore cement alteration, calcite, not only precipitates inside cement but also partially moves into the rock part of the sample. The higher pH of the cement waters is a reason for the one-way diffusion of Ca2+ ion into the rock pore space. The OH− ions diffuse from the cement, causing still higher pH on the cement side of the sample. It is worth mentioning, however, that the pH tends to persist at an almost even level in the whole simulated system and significant differences between brine, rock and cement were compensated after approximately 80 days of simulated reaction. The pH compensation is achieved by the high value of the porosity of the cement and also by the relatively high porosity of the rocks. Each simulation requires some simplification of the model. In present simulations, it is assumed full saturation of pore space with water, which increases the chemical resistance of the system. High porosity contributes to the faster fluid flow, which causes a rapid pH adjustment and buffers the reactions.
It is worth to underline here that the experiments (and consequently the modelling) were done with a high fluid/rock ratio, which may not be the case in real downhole conditions. One should be aware of the fact that in real storage sites a free CO2 phase will concentrate below the reservoir/caprock interface and reactions at the cement–caprock interface will be triggered predominantly by CO2–rock–cement interactions, not by interactions with CO2-saturated brine. The availability of brine may be restricted to the irreducible pore filling.
The calcite-enriched zone, which was also reported by Wertz et al. (2013), is very characteristic of the cement/shale interface. In our study, this phenomenon was confirmed by the real-time experiment in the autoclave reactor. Compaction of highly carbonated shale at the cement/rock interface is clearly visible; this enlarges both the split between cement and rock and the porous zone within the wellbore cement. As a result of the performed modelling, the reduced porosity of cement is stated after simulated 200 days on the cement/shale interface, whereas Wertz et al. (2013) found an increase, up to 40%, of cement porosity near the interface with the shale rock. The other minerals (including pyrite) of the shale are geochemically stable over the timescale of the simulation.
In the case of anhydrite, the evolution of the cement/rock interface is different. However, degradation of the cement part of the sample proceeds in the same way, the diffusion of Ca2+ ions from cement to rock is deeper. This causes the porosity decrease in the inner part of the anhydrite rock, while at the cement/rock interface the porosity dramatically increases. The reason for this situation is very intense dissolution of the mineral anhydrite, which is also documented by the results of autoclave experiment.
The process of anhydrite dissolution was also reported by Fischer et al. (2010) for the Stuttgart Formation, the sandstone reservoir for the pilot storage site at Ketzin, Germany. In that case, anhydrite was a part to sandstone cement, and its dissolution causes the weakening of the rock structure. Minor dissolution of anhydrite was also reported by Liu et al. (2012) for the Mount Simon sandstone in the Midwestern United States. It is conceivable that gypsum may replace anhydrite at the temperatures of these experiments (40 and 56 °C, respectively), but no gypsum was reported in either study (Kaszuba et al. 2013). In the case of our experiment (both real-time and computer simulation), there was also no precipitation of gypsum observed, but rather its dissolution.
The significant discrepancy between the results of the experiment in the reactor and a computer simulation was different solubility of ettringite within cement body. This indicates that the used in simulation exchange surface for this mineral was still too low, even though it has been significantly increased (up to 20,000 cm2/g) for the needs of applied simulations. It should be noted here that the thermodynamic data for cement phases in general, and S-bearing materials in particular, at higher temperatures, are not well identified so far (Blanc et al. 2010, 2012; Gherardi and Audigane 2013).
Conclusions
Autoclave experiments and simulations were performed for wellbore cement/shale and cement/anhydrite interfaces. The cement/shale interface experiment and simulations both confirmed established results. The cement/anhydrite study provided novel insights into CO2 sequestration research.
Geochemical evolution of Portland cement in this simulation is very fast and dominated by portlandite and CSH phase dissolution. The precipitating calcite fills the pore space and eventually decreases primary porosity. If not completely clogged, reduced porosity may enhance cement tightness. The mineralogical transformations within the examined caprocks cause the porosity variations, which is confirmed by experimental results. The situation may be different, however, in a real well–rock system, where some confining pressure counteracts to the increased porosity. Dissolution of minerals due to fluid–rock interactions may thus not result in additional porosity, but may lead to compaction of the cement/caprock interface.
The enhanced reactivity of the cement and caprock was noted at the cement/rock interface. This zone is prone to fast mineral dissolution, which contributes to the porosity increase. This phenomenon is very rapid at simulation scale. The consequence of increased porosity in the contact zone is the potential for increased fluid flow in the case of positive porosity–permeability–diffusion feedback. This could in turn enhance pH compensation and subsequent acceleration of the lowering of the reaction rate of geochemical changes.
Calcite and silica, precipitating in cement, intrude the porous zone of the caprocks which are in contact with cement. The diffusion takes place only in this direction, which means that there are no phases intruding from rock into wellbore cement.
It has been demonstrated that there are important differences in the performance of cement/shale and cement/anhydrite interfaces. The characteristic feature of the cement/shale contact zone is the occurrence of a highly carbonated, compacted layer within the shale, which in turn causes cement/shale detachment. In the case of the cement/anhydrite interface, the most important reaction influencing porosity increase is severe anhydrite dissolution. Secondary precipitation of calcite takes place in the deeper parts of the rock, whereas at the cement/anhydrite contact zone pore space emerges, which is not beneficial in terms of well integrity.
The numerical model was benchmarked against the laboratory experiments. Calcite precipitation and the experimentally observed dissolution of cement components were confirmed by the model. In the real-time experiment, however, ettringite dissolved much faster than expected, which led us to increase its exchange surface in the model. The reactions occurring in the examined caprocks, as well as the porosity evolution, were properly predicted by the simulations. In addition, the model enables tracing cement/rock interface geochemical alteration for an extended period (2 years) compared to the real-time experiment. After 1 year, however, the intensity of mineral and textural changes decreases significantly.
References
Baur I, Keller P, Mavrocordatos D, Wehrli B, Johnson CA (2004) Dissolution–precipitation behaviour of ettringite, monosulfate, and calcium silicate hydrate. Cem Concr Res 34(2):341–348
Blanc P, Bourbon X, Lassin A, Gaucher EC (2010) Chemical model for cement-based materials: thermodynamic data assessment for phases other than C–S–H. Cem Concr Res 40(9):1360–1374
Blanc P, Lassin A, Piantone P, Azaroual M, Jacquemet N, Fabbri A, Gaucher EC (2012) Thermoddem: a geochemical database focused on low temperature water/rock interactions and waste materials. Appl Geochem 27(10):2107–2116
Carey JW, Wigand M, Chipera SJ, Woldegabriel G, Pawar R, Lichtner PC, Wehner SC, Raines MA, Guthrie GD Jr (2007) Analysis and performance of oil well cement with 30 years of CO2 exposure from the SACROC Unit West Texas USA. Int J Greenh Gas Control 1:75–85
Carroll SA, McNab WW, Torres SC, Singleton M, Zhao P (2011) Wellbore integrity in carbon sequestration environments 1: experimental study on cement—sandstone/shale-brine-CO2. Energy Proc 4:5186–5194
Czernichowski-Lauriol I, Rochelle C, Gaus I, Azaroual M, Pearce J, Durst P (2004) Geochemical interactions between CO2 pore-waters and reservoir rocks—Lessons learned from laboratory experiments, field studies and computer simulations. In: S.A.L.K.B.S.E. Lombardo (ed.) NATO Advanced Research Workshop on Advances in CO2 geological sequestration in eastern and western European countries. Springer, Tomsk Russia 157–174
DIN EN ISO 10426: Petroleum and natural gas industries—Cements and materials for well cementing
Duguid A, Scherer G (2010) Degradation of oilwell cement due to exposure to carbonated brine Int. J Greenh Gas Control 4:546–560
Erickson KP, Lempp Ch, Pöllmann H (2015) Geochemical and geomechanical effects of scCO2 and associated impurities on physical and petrophysical properties of Permotriassic Sandstones (Germany): an experimental approach. Environ Earth Sci 74:4719–4743
Fischer S, Liebscher A, Wandrey M, Grp CS (2010) CO2-brine-rock interaction—First results of long-term exposure experiments at in situ P–T conditions of the Ketzin CO2 reservoir. Chemie Der Erde-Geochemistry 70:155–164
Fischer S, Liebscher A, De Lucia M, Hecht L, The Ketzin Team (2013) Reactivity of sandstone and siltstone samples from the Ketzin pilot CO2 storage site-Laboratory experiments and reactive geochemical modelling. Environ Earth Sci 70:3687–3708
Galí S, Ayora C, Alfonso P, Tauler E, Labrador M (2001) Kinetics of dolomite–Portlandite reaction: application to Portland cement concrete. Cem Concr Res 31(6):933–939
Gaucher EC, Blanc P (2006) Cement/clay interactions—a review: experiments, natural analogues, and modeling. Waste Manag 26:776–788
Gherardi F, Audigane P (2013) Modeling geochemical reactions in wellbore cement: assessing preinjection integrity in a site for CO2 geological storage. Greenh Gas Sci Technol 3:447–474
Gherardi F, Audigane P, Gaucher EC (2012) Predicting long-term geochemical alteration of wellbore cement in a generic geological CO2 confinement site: tackling a difficult reactive transport modelling challenge. J Hydrol 420–421:340–359
Huet BM, Prevost JH, Scherer GW (2010) Quantitative reactive transport modelling of Portland cement in CO2-saturated water. Int J Greenh Gas Control 4:561–574
Huq F, Haderlein SB, Cirpka OA, Nowak M, Blum P, Grathwohl P (2015) Flow through experiment on CO2-brine-rock interaction in a sandstone from the Altmark gas reservoir 77th EAGE Conference and Exhibition Madrid Spain
Jung HB, Um W (2013) Experimental study of potential wellbore cement carbonation by various phases of carbon dioxide during geologic carbon sequestration. Appl Geochem 35:161–172
Jung HB, Kabilan S, Carson JP, Kuprat AP, Um W, Martin P, Dahl M, Kafentzis T, Varga T, Stephens S, Arey B, Carrol KC, Bonneville A, Fernandez CA (2014) Wellbore cement fracture evolution at the cement-basalt caprock interface during geologic carbon sequestration. Appl Geochem 47:1–16
Kaszuba J, Yardley B, Andreani M (2013) Experimental perspectives of mineral dissolution and precipitation due to carbon dioxide-water-rock interactions. Rev Miner Geochem 77(1):153–188
Kutchko BG, Strazisar BR, Dzombak DA, Lowry GV, Thaulow N (2007) Degradation of well cement by CO2 under geologic sequestration conditions. Environ Sci Technol 41:4787–4792
Kutchko BG, Strazisar BR, Huerta N, Lowry GV, Dzombak DA, Thaulow N (2009) CO2 reaction with hydrated Class H well cement under geologic sequestration conditions: effects of flyash admixtures. Environ Sci Technol 43:3947–3952
Labus M, Lorek A (2015) CO2-rock interaction on the example of Permian sedimentary rocks, 77th EAGE Conference and Exhibition 2015 in Madrid Spain
Labus M, Such P (2016) Microstructural characteristics of wellbore cement and formation rocks under sequestration conditions. J Petrol Sci Eng 138:77–87
Lasaga AC (1981) Rate laws in chemical reactions. In: Lasaga AC, Kirkpatrick RJ (eds) Kinetics of Geochemical Processes Reviews in Mineralogy, vol 8. pp 135–169
Liu FY, Lu P, Griffith C, Hedges SW, Soong Y, Hellevang H, Zhu C (2012) CO2-brine-caprock interaction: reactivity experiments on Eau Claire shale and a review of relevant literature. Int J Greenh Gas Control 7:153–167
Lorek A, Labus M (2015) Carbonated brine effect on wellbore cement degradation in contact zone with formation rock, 3rd Sustainable Earth Science Conference & Exhibition, Celle, Germany
Lorek A, Labus M, Bujok P (2016) Wellbore cement degradation in contact zone with formation rock. Environ Earth Sci 75:499–514
Palandri JL, Kharaka YK (2004) A compilation of rate parameters of water–mineral interaction kinetics for application to geochemical modelling. U.S. Geological Survey Report 2004-1068
Schweizer Ch R (1999) Calciumsilikathydrat-Mineralien. Lösungskinetik und ihr Einfluss auf das Auswaschverhalten von Substanzen aus einer Ablagerung mit Rückständen aus Müllverbrennungsanlagen. PhD thesis, dipl. Chem. Universität Basel Zürich und Dübendorf
Soler JM, Mäder UK (2010) Cement-rock interaction: infiltration of high-pH solution into a fractured granite core. Geol Acta 8:221–233
Stegemann JA, Russell MI, Zhou Q, Bai Y, Abdul Rani L, Maduka I (2012) Acid resistance of Ettringite-based material WASCON 2012—Towards effective, durable and sustainable production and use of alternative materials in construction
Um W, Jung HB, Martin PF, McGrail BP (2011) Effective permeability change in wellbore cement with carbon dioxide reaction, PNNL-20843, Pacific Northwest National Laboratory, Richland, WA
Wertz F, Gherardi F, Blanc P, Bader A, Fabbri A (2013) Cement CO2-alteration propagation at the well-caprock-reservoir interface and influence of diffusion. Int J Greenh Gas Control 12:9–17
Wertz F, Vrbova V, Havlova V (2014) Numerical simulations of supercritical injection of CO2 in Dunajovice Sandstone. Fuels 6:108–115
Xu H, Sonnenthal EL, Spycher N, Pruess K (2004) TOUGHREACT user’s guide: a simulation program for nonisothermal multiphase reactive geochemical transport in variably saturated geologic media. Lawrence Berkeley National Laboratory Berkeley California Report LBNL-55460
Acknowledgements
The research leading to these results has received funding from the Polish-Norwegian Research Programme operated by the National Centre for Research and Development under the Norwegian Financial Mechanism 2009–2014 in the frame of Project Contract No Pol-Nor/207125/66/2013.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Labus, M., Wertz, F. Identifying geochemical reactions on wellbore cement/caprock interface under sequestration conditions. Environ Earth Sci 76, 443 (2017). https://doi.org/10.1007/s12665-017-6771-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12665-017-6771-x