
Abstract
In the work the risk of CO2 migration in deep wells, caused by integrity loss on cement–rock interface and thow wellbore integrity correlated with the formation rock lithology were determined. 19 composed samples of rock and wellbore cement were exposed to CO2-saturated brine, in the autoclave reactor, under the formation conditions (50 °C and 10 MPa). Mineralogical and textural changes in the cement–rock interface in the case of selected rocks (sandstones, shale, limestones, dolomites and anhydrites) were characterised. The performed examination indicates that both cement and formation rocks react with CO2 saturated brine under the experimental conditions. The cement alteration is characterised by carbonation process in the outer rim, but it is enhanced on the interface with formation rock. It was stated that the performance of the cement–rock interface is essentially dependent on the rock lithology, including mineral composition and rock structure. Some minerals are very easily dissolving, e.g., anhydrite, gypsum, calcite and feldspars, what is contributing to an increase in the porosity and permeability in cement–rock contact zone. Primary dissolution of certain minerals in the first stages of the experiment results in the secondary precipitation after the last stage of reaction contributing to a secondary reduction of pore space.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Wellbore cement
Wellbore cement is used as a sealant to secure and support casing inside the well and prevent fluid communication between the underground fluid-containing layers. It has been used as the primary sealant in oil and gas wells throughout the world and is manufactured to meet specific chemical and physical standards. Sour natural gas contains undesirable components: hydrogen sulfide (H2S) and carbon dioxide (CO2). The petroleum industry has to develop alternative ways for acid gas elimination. Reinjection into geological reservoirs (i.e. deep saline aquifers in western Canada and depleted oil and gas reservoirs) seems to be one of the most often considered solutions for acid gas release mitigation (Chakma 1997; Connock 2001). During geological sequestration CO2 is likely to react with formation fluids, reservoir rocks and wellbore cement in the injection zone. The acidification of the fluids may result in the dissolution of solid phases and transition into new solid phases which may cause changes in rock and cement composition.
Wellbore cement degradation forms a potential risk pathway for leakage of CO2 from the storage reservoir to water aquifers and even back to the atmosphere. Cement carbonation is the result of salvation of Ca ions from the solid phase by diffused CO2 and precipitation of CaCO3 minerals, what can affect both the chemical and physical properties of the cement. The course of the process of cement degradation depends on the pH of the CO2-saturated formation fluids, which is affected by aquifer types, connected to the formation rock lithology (e.g. sandstone or limestone formation) (Duguid and Scherer 2010).
Research on cement carbonation has been conducted on laboratory and field scales, and using computer models. Carbonation research can be divided into two broad categories. So-called “dry carbonation” takes place on cement that is not submerged in water. In this case the carbonation process generally stops after the creation of calcium carbonate and acts to increase the strength of the cement and decrease the permeability (Kutchko et al. 2007, 2009; Rimmelé et al. 2008). Wet carbonation occurs on cements that are submerged in liquid (water or brine). This category is similar to the in situ conditions of the cement in an abandoned well. In this case the cement in the annulus of the well will be saturated with brine, under the right conditions; this allows the dissolution of the calcium carbonate reaction product (Duguid and Scherer 2010). In contrast, dry carbonation (carbonation under unsaturated condition) is much less likely to cause calcium carbonate dissolution. The experimental results obtained by Jung and Um (2013) showed that slow and minor cement carbonation under dry supercritical exposure of CO2 was formed after 15 months. In the case of wet gaseous CO2 exposure the extensive calcite coating was formed on the outside surface of a cement sample after exposure of 1–3 months. As a result, the pore volume of the cement was reduced by a factor of 3–6. Most of the researchers suggest that the precipitation of calcium carbonate increases the strength of cement. However, excessive carbonate formation may cause mechanical failure (Fabbri et al. 2009; Gherardi et al. 2012).
Though reduction of the pore volume in the outer zone of cement samples is observed in many experimental works, the formation of a thin zone with high porosity and high permeability at the outer boundary of the carbonate layer is observed in some studies (Kutchko et al. 2007; Walsh et al. 2013). This thin zone (commonly referred as amorphous SiO2 zone) may serve as a potential brine and CO2 leakage pathway.
Field studies of well bores exposed to CO2 for more than 30 years have indicated that alteration of hydrated cement by CO2 under geologic sequestration conditions is limited. The long-term performance of wellbore cement in a CO2-enhanced oil recovery (EOR) field was good, providing an effective barrier to significant fluid flow (Carey et al. 2007).
Experimental approaches to study reactivity of host-rocks minerals were presented by Pearce et al. (1996), Gunter et al. (1997), Kaszuba et al. (2003), Fischer et al. (2013) and others. Well cements durability in presence of CO2 at high P–T has been studied extensively by Onan (1984), Krilov et al. (2000), Kutchko et al. (2007, 2009, 2011), Carey et al. (2007), Barlet-Gouédard et al. (2009), Um et al. (2011) and others. Nonetheless there is still limited information on the physical and chemical characteristics of wellbores environment exposed to acid-gas under geologic sequestration conditions.
Kutchko et al. (2011) found the cement deterioration mechanism is connected to secondary ettringite formation, as well as oxidation of pyrite. There is significant experimental variability in the interpretation of cement durability in the wellbore environment. Experiments of Duguid et al. (2005) suggest rapid carbonation, while those of Kutchko et al. (2007) and Carey et al. (2007) suggest more limited rates of CO2 penetration and reaction. Despite numerous studies reporting the geochemical and mineralogical alteration of well cement by acid gases (mostly CO2) under geologic sequestration conditions, the pore structure and permeability change of cement is still poorly understood.
Carey and Lichtner (2011) by numerical simulation showed that the capillary properties of good-quality cement will prevent flow of CO2 into and through cement. Rather CO2, if present, is likely to be confined to the casing/cement or cement/formation interface. Carey et al. (2007) and Carey and Lichtner (2011) assumed that CO2 migrate along the cement/formation interface and then diffuses into the cement. Crucial to long-term safety and security of CO2 sequestration is the determination of whether zonal isolation deteriorates or improves through chemical reactions on formation rock and cement contact zone.
Deterioration within rock-cement interface
The research works conducted on CO2–rock interaction are mostly based on the assumption that the geochemical reactions occur in an aqueous environment. However in the context of CO2 storage two other non-aqueous types of interactions are possible: chemical interactions between water dissolved in CO2 and rock, and pure CO2–rock interactions in complete absence of water (Gaus 2010).
The experiments performed by Jung and Um (2013) suggest that preferential cement alteration by CO2 and CO2-saturated water can occur along the cement–steel and cement–rock interfaces, which highlights the importance of further investigation of cement degradation along the interfaces to ensure permanent geologic carbon storage.
Some researchers indicate to the necessity of improving cement bonding to the formation rock (Brandl et al. 2011). Newhall (2006) reported that a preflush with a sodium silicate will improve cement bond. An aqueous film of silicates on the surface of the formation and the pipe will form calcium silicate precipitation when in contact with the cement slurry.
Carroll et al. (2011a, b) have performed an experimental study on cement–sandstone/shale–brine–CO2 interactions. They observed marked changes in solution composition when CO2 reacted with cement, sandstone and shale components at reservoir conditions. In the case of cement–sandstone reaction, some iron and amorphous silica precipitate. The alteration of shale by CO2-rich brines was nearly negligible. The research of Carroll et al. (2011a) also shows that relatively simple geochemical models can describe the dominant reactions that are likely to occur in case of CO2 storage. On the contrary, the results obtained by Fischer et al. (2013) indicate that geochemical modeling is not capable of covering the complete range of natural variability.
The different lithologies of formation rock were considered in experimental and modelling studies, i.e., basalt (Jung and Um 2013; Jung et al. 2014), limestone (Duguid and Scherer 2010), shale and sandstone (Carroll et al. 2011a, b) and siltstone (Fischer et al. 2013). It is worth mentioning however, that sandstones are highly differentiated, as a result of various mineral composition and diagenesis level. The specific nature of CO2–rock interactions makes it impossible to provide the description of detailed reaction patterns that are valid for a wide range of host and cap rocks (Gaus 2010). This is the reason why it is necessary to characterise a geochemical baseline of each potential storage site, before starting injection. Most of the researchers (e.g. Chadwick et al. 2007; Fischer et al. 2013; Holloway 1997) suggest that a baseline programme should consist of the analysis of the cap rock and host rock mineralogy, the composition of the pore water, prevailing pressure and temperatures in the CO2 storage environment.
CO2 injected into an aquifer will partially dissolve into the formation brine to form carbonic acid, causing dissolution of some primary minerals of reservoir and cap rocks, or precipitation of secondary minerals, which in turn affect petrophysical properties, such as permeability and porosity of the rock.
Objectives of the research
The objective of presented research was to determine the risk of CO2 migration in deep wells, caused by integrity loss on cement–rock interface. It should be expected that corrosion and dissolution at the rock-cement interface under carbon dioxide sequestration conditions is dependent on the rock lithology. To realize the goals there were analysed several samples of sedimentary rocks, including reservoir and cap rocks, occurring in the Polish hydrocarbons deposits. These rocks were not previously examined in terms of CO2 sequestration.
Materials and methods
Samples description
The rocks which were selected for composite (cement–rock) samples preparation represent host and sealing rocks for hydrocarbons deposits in Poland, and were collected from several deep boreholes. The representatives for reservoir formations are sandstones and carbonate rocks. Middle Cambrian sandstones (samples 1 and 2) are the main oil- and gas bearing strata in the Baltic Sea region. Pennsylvanian and lower Permian red sandstones of Western Pomerania and Fore-Sudetic Monocline are mostly good reservoir rocks for tight gas (samples 3–7). Upper Jurassic limestones originating from Outer Carpathians (samples 9, 10, 12) are host rocks for gas accumulations. Permian carbonates (samples 11, 13–16) occuring at the base of Zechstein cyclotem Stassfurt belong to one of the most important exploration target in the Polish Basin (Karnkowski 2007). The other evaporates (samples 17–19) are the sealing rocks overlying carbonates mentioned before. Shale rock (sample 8) represents the potential source rock for shale gas in Baltic Sea region.
In Table 1 the stratigraphic position, and mineral composition of these rocks is presented. The description has been made on the basis of optical microscopy and XRD analysis. X-ray diffraction results are given in the last column, as the weight fraction of main minerals in the rock. The accessory minerals are usually not detected by this method, and are only mentioned in sample description column, which was based on microscopic image observation.
Samples preparation
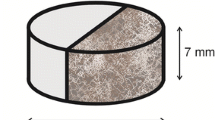
Rock samples were prepared in the form of cylinders with a height of approximately 4 cm and a diameter of 2.5 cm. Rock cores were cut lengthwise in half and supplemented with cement grout. The cement grout was composed of the following materials: tap water, defoamer, fluid loss additive, plasticizer, latex, latex stabilizer, retarder, microcement, swelling additive and the Portland cement CEM I 32.5, at a water-to-cement ratio of 0.52. Grout was prepared in accordance to the procedure laid down in the relevant standards (DIN EN 10426-2). The samples were cured in an autoclave at 50 °C, at a pressure of 17 MPa. From the prepared samples the thin cuts were made. The rest of the sample was sliced and placed in the reactor (Fig. 1).

Example of composed cement–rock cylindrical sample which was divided into three parts for each stage of the experiment
After each stage of the experiment the composed cement–rock samples’ slices were washed with distilled water and dried. For SEM analysis the samples were embedded in epoxy rasin to prevent sample from its disintegration.
Experiment conditions
The conditions of the CO2 treatment in autoclave were corresponding to brine saturated reservoir. Provided the average increase of the temperature in the well of about 3 °C for each 100 m, it can be assumed that the temperature of 50 °C is corresponding to the depth of 1000 m below the ground level in static conditions. It should be mentioned however, that the hydrostatic pressure in the wellbore depends on the amount and specific gravity of the drilling fluid (e.g. at a depth of approximately 3000 m in the case of heavy cement slurry the pressure can be about 60–70 MPa) (Rzepka 2010). The assumed conditions (50 °C and 10 MPa) can be present in the wellbore in case of deep carbon injection and storage depth (Jung and Um 2013).
Prepared samples were exposed to CO2-saturated brine, under static conditions. Gas was injected until the required pressure of 10 MPa and supplemented, if needed, during the experiment. The autoclave experiment was performed in 3 stages of different duration: stage I—30 days. stage II—60 days, stage III—200 days. The reactor which was constructed in the laboratories of VSB Ostrava is divided into two cells (4 dm3 each of them). The general view of the reactor is shown in Fig. 2. The composite cement–rock samples were fully covered with fluid: CO2-saturated brine, pH of which was measured in the beginning of the experiment, and after each, mentioned above, stage.

General view of the autoclave reactor used for the experiment
SEM analysis
Scanning electron microscope (SEM) analysis were performed in the Institute of Clean Technologies (Ostrava, Czech Republic) with FEI Quanta-650 FEG, equipped with the analyzers: energy dispersive analyzer (EDX)—EDAX Galaxy, wave dispersion analyzer (WDA)—EDAX LEXS, cathodoluminescence detector (CL)—Gatan MonoCL4 detector and analyzer of electron backscatter diffraction (EBSD). Microphotographs were taken using backscattered electron detector (EBSD) enabling monitoring of the chemical elements distribution in the samples.
The analysis were conducted without standard, using the correction contents of light elements on the basis of a set of standard materials. Energy dispersive microanalysis under these conditions are considered only semi-quantitative. Microscope working conditions: voltage 15 kV, current of 8–10 nA, beam diameter 5.5 mm, reduced vacuum pressure in the chamber 50 Pa, samples without coating. Identification of the spectral lines was performed using the spectral decomposition using function halographic peak deconvolution.
XRD analysis
XRD analysis was performed at the Institute of Clean Technologies (Ostrava, Czech Republic). Rocks’ and cement samples were ground in McCroneMicronisingMill in isopropanol to the granularity of less than 5 mm and were homogenised by micronising in a vibration mill and then pressed into the cells of depth of 2 mm.
XRD measurements were carried out with use of powder diffractometer Bruke-AXS Advance D8 (Germany) of 2Θ/Θ geometry, equipped with linear semiconductor detector LynxEye and energy-dispersive detector SOL-XE. Measurement conditions: radiation CoKα/Fe filter, voltage 40 kV, current 40 mA, step of 0.014 2 Θ, step time 1.25 s (the sum of five measurements with a step of 0.25 s) and digital processing of the resulting data. For the measurement and calculation there was used software of BrukerDiffracSuite.
For qualitative assessment, the diffraction database PDF-2, version 2011 (International Data Centre for Diffraction, Pennsylvania, USA) was used. For semiquantitative analysis software Bruker Topas version 4.2 was used. Quantification is based on a Rietveld method of structural analysis of the powder diffraction data and consists in modeling diffraction spectrum using the known structural data (lattice parameters, the positions of atoms in the structure, space group, etc.) for each of the minerals and the subsequent comparison of the measured diffraction pattern using multivariate regression. Input structural data were taken from BrukerDiffracPlus Topas structure database.
In XRD analysis of cement samples the internal standard (ZnO, cca 10 %) was used. Quantitative analysis is possible by comparing the integral intensity of the reflections of individual minerals in the sample to the intensity of the standard (ZnO) reflex.To determine the amount of amorphic components (e.g. C–S–H phases) the sum of crystalline components was substracted from 100 %.
Results and discussion
Wellbore cement alteration
Wellbore cement is considered as a very heterogeneous material with discrete particles typically in the size range of about a few hundred nanometers up to a few hundred micrometers, mainly consisting of variable amounts of calcium silicate hydrates (C–S–H), portlandite, calcium aluminates, and minor reactive phases (e.g., hydrocalumite, ettringite, etc.). The phase composition of analysed wellbore cement is given by XRD analysis (Fig. 3). As it was described in "SEM analysis", the internal standard applcation lets determining the quantity of amorphic components (Table 2).

XRD pattern of cement in course of the experiment. B belite (C2S beta), C calcite, E ettringite, H hydrocalumite, P portlandite, V vaterite, ZnO internal standard
The determined components of the initial cured cement sample (Stage 0) are hydration products of Portland cement: amorphic phase (probably calcium-silicate-hydrate C–S–H), portlandite (Ca(OH)2), and C2S-β which is a polymorph of belite (Ca2(SiO4)). As C2S is slow reactive with water, some amount of this component is still not hydrated on this stage. The other mineral components are ettringite (Ca6Al2(SO4)3(OH)12·26H2O) and hydrocalumite \(({\text{Ca}}_{ 2} {\text{Al}}\left( {\text{OH}} \right)_{ 6} \left[ {{\text{Cl}}_{{ 1\subseteq {\text{x}}}} \left( {\text{OH}} \right)_{\text{x}} } \right] \cdot 3 {\text{H}}_{ 2} {\text{O}}\). Ettringite on the early stage of cement hydration is known as “primary ettringite” and is necessary and beneficial component of Portland cement system.
After the I stage of the experiment (30 days) the content of ettringite and hydrocalumite decreases. All portlandite has reacted to form calcium carbonates (calcite and vaterite).
After the III stage of the experiment (200 days) the diffraction pattern shows the occurrence of abundant calcite. The presence of other calcium carbonate—aragonite—is possible, as some of the peaks could overlapping calcite signals. However, the presence of aragonite is uncertain, and vaterite was not detected at all. The absence of vaterite and aragonite suggests the rapid rate of the carbonation reaction.
The SEM observations of rock-cement interface support the results of XRD analysis. In the cement part of the composed samples the precipitation of CaCO3 is noted. The carbonated zone has a thickness of about 1 mm after the III stage of experiment. In the case of composite cement–sandstone samples (sample 4 and 5) the precipitation of some secondary minerals within cement is noted. The crystals visible in Fig. 4 are probably of zeolite. The given elemental composition is slightly disturbed by the surrounding background, but it could be assumed that the precipitated mineral is Ca–zeolite, probably Ca-phillipsite (Ca,K,Na)1–2(Si,Al)8O16·6(H2O), as it was modelled by Gherardi et al. (2012), when predicting geochemical alteration of wellbore cement. These authors stated that zeolites could be thermodynamically stable at the temperature of their simulations (i.e. 75 °C), and that their precipitation may exert some control on aqueous Ca concentration of caprock and cement porewaters. Gherardi et al. (2012), suggest also the phillipsite re-dissolution in long-term view of cement alteration.

Zeolite crystals precipitated in cement part of subarcose sandstone (sample 5) after III stage of experiment—SEM image and EDS elemental composition (analysed point marked with the cross)
Rocks alteration in composite samples
Cement–sandstone interface
Sandstone samples which were taken into the experiments are the most differentiated group of rocks. The observed alteration in course of the reaction permits us to divide these rocks into at least two groups of sandstones: samples 1 and 2 which are quartzitic, fine grained rocks, and samples 3–7, which represent sandstones of more complex mineral composition.
The contact zone of cement–sandstone in the case of quartzitic sandstones (1 and 2) is nearly not altered in course of the experiment. The carbonated zone in cement part of the sample is quite narrow—of about 0.6 mm after the III stage of the experiment. In some cases the texture loosening is noted (Fig. 5b). On the interface of cement and sandstone in sample 2 the opaline efflorescent is visible (Table 3).

Examples of cement–sandstone contact zone. SEM images of polished composite samples: a quartzitic sandstone (sample 1) after I stage (30 days) of the experiment, b quartzitic sandstone (sample 1) after III stage (200 days) of the experiment, c sublithic arenite (sample 4) after II stage (60 days) of the experiment—chlorites on the quartz grain, d gypsum crystal in the centre, calcite crystals in the background, precipitated on the sandstone grains (sample 6)
Within sandstones which are weakly consolidated (samples 3–6), the primary porosity is dominant. In course of the experiment, just after the I stage, the secondary porosity is occurring, as a result of rock cement dissolution, and K-feldspar dissolution. The main components of rock cement which are subject to dissolution are carbonates: calcite, siderite and dolomite; or sulphates: anhydrite. Plagioclase and K-feldspar grains, as well as lithic clasts show intense alteration. In the case of sample 4 the sandstone originally contains diagenetic minerals such as chlorite and authigenic quartz filling pore space. After the reaction, especially after the III stage, sandstone porosity is increased, but the dissolved minerals exhibit secondary precipitation on the quartz grains (Fig. 5c). For the samples originally containing evaporate minerals or carbonates (e.g. sandstone 5 and 6) the secondary precipitation of these minerals is observed. In Fig. 5d we can see secondary gypsum, which is formed as a result of anhydrite dissolution. The small calcite crystals in the background also precipitate on the quartz grains. Newly precipitated clay minerals (chlorites), carbonates (mainly calcite) and sulphates occlude pore space.
Cement–shale interface
The cement–shale interface was examined on the example of one sample (8). It was observed that the slate structure of the rock facilitates its fracturing, which enables fluid migration within the shale and in the space between rock and cement. Just after I stage of the experiment shale was splitted, and the process progressed with duration of the reaction. After III stage of the experiment, cement and rock became separated, and in the resulting aperture, the crystallization of CaCO3 occurs (Fig. 6). An interesting phenomenon is the emergence of a zone enriched in calcite, in the rock part of the sample. This zone is of 50–100 µm thick, and finely laminated, maintaining slate structure of shale. Beneath this carbonated stripe the rock is splitted and of a high porosity.

SEM image of cement–shale interface after III stage (200 days) of the experiment. Cement became separated from the rock. On the border with cement shale is compressed and enriched with calcite, below—splitted
Cement–limestone/dolomite interface
The alteration on cement–limestone and cement–dolomite interface is not as significant as in the case on the other rocks. Usually, the cement–rock contact is very strict, and within the carbonate rock the changes are restricted to local dissolution of CaCO3, causing the enlargement of existing caverns. Within rock samples containing some amount of anhydrite (Table 1), which are sample 11 (limestone) and samples 14 and 15 (dolomites), the dissolution of anhydrite is visible. The zone in which the dissolution takes place develops in contact with the cement, and has a thickness of up to 1 mm (Fig. 7).

SEM image of cement–anhydrite interface. a mineral anhydrite (A: light gray colour) is dissolving; the undissolved mineral is dolomite (D: dark gray), b calcite (C) is precipitating filling the pores
The carbonated zone in cement part of the composed cement–carbonates samples is forming, as in the case of other types of rocks. After the III stage of experiment one of the samples (9) has been splitted on the contact zone of cement–dolomite. In this particular case the precipitation of new forming mineral is visible (Fig. 8). The elongated crystals represent vaterite, which is a polymorph of calcium carbonate CaCO3.

SEM image of the polished composite cement–dolomite sample (sample 9): a the fissure at the interface which was formed after the III stage of the reaction, b hexagonal vaterite crystallizing in the void
Cement–evaporate rock interface
The rock part in the composed samples 17–19 represent evaporates, containing minerals such as anhydrite, magnesite and gypsum (Table 1). Anhydrite and gypsum are minerals which easily dissolve in CO2 saturated brine under experiment conditions. In the case of the mentioned samples the significant increase of porosity in outer zone of the sample and on the cement–rock interface is visible. The alteration zone is increasing with progressing run time (Labus and Lorek 2015). The particularly vulnerable mineral to dissolution is anhydrite, and to a lesser extent—gypsum. The remaining (not dissolved) mineral is magnesite. Moreover, the secondary precipitation of calcite on the interface of cement–rock is noted.
Discussion
The wellbore cement alteration under sequestration conditions is a widely studied issue, and the degradation processes are relatively well understood. As it was stated in several works, the wellbore cement exposure to different phases of CO2 during geologic carbon sequestration, is unlikely do damage the wellbore integrity. The preferential cement alteration occurs along cement–steel and cement–rock interface. The performed in this study experiments confirm the validity of this statement, with regard to the zone of cement contact with the formation rock.
The most characteristic, and well known cement alteration form is the carbonation front, sealing the outer part of the sample. The next stage of carbonated zone dissolution was not observed in this study, in contrary to the observations by Barlet-Gouédard et al. (2009), who noted the dissolution process even earlier in CO2 saturated water than in wet supercritical CO2. In the mentioned investigation the authors emphasize that the high salinity of a brine slows down the alteration process.
The depth of the observed cement carbonation is rather small—it reaches about 1 mm after 6 months exposure to CO2 saturated brine. Jung and Um (2013) have reported 3.17 mm alteration depth after 8 months (w/c = 0.38), and 2.98 mm after 5 months (w/c = 0.33) under exposure to CO2 saturated groundwater. In the case of our study (w/c = 0.52) the alteration depth is smaller, probably due to the cement curing conditions, which were: 48 h, 50 °C, 17 MPa. Jung and Um (2013) have performed their experiments on cement samples which were cured under ambient conditions. As it was reported by Kutchko et al. (2007) the cement alteration depth is shallower by a factor 2–3 for the cement cured at high P–T conditions than the cement samples cured at ambient conditions.
The carbonation process is considered to be a phenomenon improving wellbore cement integrity, by reducing its permeability and sealing fractures. We can observe that although some secondary calcium carbonate minerals precipitate on the cement–rock interface (as it was observed here on the example of vaterite), and mineralise the fracture, they do not seal the fracture completely. It is worth mentioning however that occurring of such severe, wide fractures on the cement–rock interface is possible only on laboratory scale and is not expected under real borehole conditions (Carey et al. 2007). The downhole pressure of surrounding formation prevents fracture widening.
The alteration of the cement–rock interface is strongly dependent on the rock lithology, which includes rock texture (mainly porosity) and mineral composition. The degradation processes occur with the lowest intensity in the case of carbonate rocks—limestones and dolomites, as the chemical composition of cement and rock is similar. Some of the examined carbonate rocks (Permian limestones and dolomites of Fore-Sudetic Monocline) are of evaporate origin, and hence include certain amounts of anhydrite or sylvite. In such cases the dissolution of anhydrite is the most important degradation process. Sylvite is so highly soluble that its dissolution is even not detectable by SEM observation. Caprocks which are mainly composed of anhydrite or anhydrite and magnesite are the most vulnerable to dissolution during the experiment. The essential for this process is the interaction of fluid—in this case brine, simulating formation water. Rochelle et al. (2004) report that the effect of CO2 on anhydrite dissolution is rather small, whereas the increasing solubility of anhydrite is observed with increasing salinity of pore water. Czernichowski-Lauriol et al. (1996) found caprock (mudstone and anhydrite) unaltered in their experiment with supercritical CO2. On the other hand, significant reactions were observed in wet conditions experiment (CO2 saturated brine); the most important was rapid dissolution of anhydrite.
In the case of sandstone samples, which represent host rock, the performance under CO2 sequestration conditions is differentiated, regarding their mineral composition. Quartzitic sandstones on the interface with wellbore cement are relatively low altered. The secondary precipitation of amorphous silica is noted, as identified in cement/sandstone system by Carroll et al. (2011a). The alteration of sandstone of more complex mineral composition is a process of rather complicated nature. The alteration is enhanced by high porosity of these rocks, which enables more rapid fluid flow. In the course of the experiment the porosity initially increases, due to dissolution of some components. Out of the skeletal grains, the most susceptible to dissolution are plagioclase and K-feldspar, as well as lithic clasts. The most soluble components of rock cement are: calcite and anhydrite. Primary dissolution of certain minerals results in the secondary precipitation of calcite, gypsum, chlorites and authigenic quartz, causing the secondary reduction of pore space. There could be also expected the precipitation of kaolinite in sandstone pores, as it was reported by Fu et al. (2009), but in our experiments secondary kaolinite was not noted in any of sandstone samples.
Also in the case of shale rock, the secondary precipitation of chlorites and authigenic quartz was observed, but to a much lesser extent than for sandstones. It seems that a critical feature in shale, accelerating the destruction of a cement–shale interface, is its slate structure, which contributes parallel fracturing of the rock. This process facilitates fluid migration within the rock itself and in the fractures between rock and cement, ac celerating the geochemical alteration.
Conclusions
The performed examination indicates that both cement and formation rock react with CO2 saturated brine under the experiment conditions. The cement alteration is characterised by carbonation process in the outer rim, but it is enhanced on the interface with formation rock.
Most of the examined cement–rock samples provide evidence for dissolution and corrosion processes. With progressing run time the corrosion and dissolution intensify. The most vulnerable minerals to dissolution are anhydrite, gypsum and calcite. Formation of secondary pore spaces within sandstones is also resulting from K-feldspar dissolution. As the processes of alteration and dissolution are coming more intensely on cement–rock interface, this zone is a potential pathway for flowing pore waters rich in CO2 to come into contact with the wellbore cement.
The performance of the cement–rock interface is essentially dependent on the rock lithology, which means mineral composition and structure. Thus, to predict processes that can take place in boreholes under acid gas injection, there are necessary experimental tests, taking into account the different types of formation rocks. The geochemical modeling to simulate the reactions between wellbore cement and different formation rocks under specific P–T conditions is needed. The simulation however could not provide the complete idea of occurring changes, due to the complicated nature of rocks. The mineral composition and porosity of rocks could be implemented to the model in a relatively easy way, but specific features, like slate structure, or differentiated grain sizes are more challenging.
When regarding the geological storage, the consideration of the reactions between wellbore cement and different formation rocks is required. The full lithological profile of the borehole must be taken into consideration, regarding the wellbore cement–rock interface behavior under sequestration conditions.
References
Barlet-Gouédard V, Rimmelé G, Porcherie O, Quisel N, Desroches J (2009) A solution against well cement degradation under CO2 geological storage environment. Int J Greenh Gas Control 3:206–216
Brandl A, Cutler J, Seholm A, Sansil M, Braun G (2011) Cementing solutions for corrosive well environments. SPE Drill Complet 26(2):208–219
Carey JW, Lichtner PC (2011) Computational studies of two-phase cement/CO2/brine interaction in wellbore environments. SPE J 16(4):940–948
Carey JW, Wigand M, Chipera SJ, Woldegabriel G, Pawar R, Lichtner PC, Wehner SC, Raines MA, Guthrie GD Jr (2007) Analysis and performance of oil well cement with 30 years of CO2 exposure from the SACROC Unit, West Texas USA; Int. J Greenh Gas Control 1:75–85
Carroll SA, McNab WW, Torres SC (2011a) Experimental study on cement-sandstone/shale-brine-CO2 interactions. Geoch Trans 12:9
Carroll SA, McNab WW, Torres SC, Singleton M, Zhao P (2011b) Wellbore integrity in carbon sequestration environments 1: experimental study on cement-sandstone/shale-brine-CO2. Energy Procedia 4:5186–5194
Chadwick A, Arts R, Bernstone C, May F, Thibeau S, Zweigel P (2007) Best practice for the storage of CO2 in saline aquifers—observations and guidelines from the SACS and CO2STORE projects. British Geological Survey, Nottingham. p 267
Chakma A (1997) Acid gas re-injection—A practical way to eliminate CO2 emissions from gas processing plants. Energy Convers Manag 38(supplt):S205–S209
Connock I (2001) Acid gas re-injection reduces sulphur burden. Sulphur 272:35–41
Czernichowski-Lauriol I, Sanjuan B, Rochelle C, Bateman K, Pearce J, Blackwell P (1996) Analysis of the geochemical aspects of the underground disposal of CO2: scientific and engineering aspects. In: Apps JA, Tsang CF (eds) Deep injection disposal of hazardous and industrial waste. Academic Press, Cambridge, pp 565–583
DIN EN ISO 10426-2: Petroleum and natural gas industries—Cements and materials for well cementing—Part 2: testing of well cements
Duguid A, Scherer G (2010) Degradation of oilwell cement due to exposure to carbonated brine. Int J Greenh Gas Contr 4:546–560
Duguid A, Radonjic M, Scherer G (2005) Degradation of well cements exposed to carbonated brine. In: Fourth Annual Conference on Carbon Capture and Sequestration, May 2005. Alexandria
Fabbri A, Corvisier J, Schubnel A, Brunet F, Goffé B, Rimmele G, Barlet-Gouédard V (2009) Effect of carbonation on the hydro-mechanical properties of Portland cements. Cem Concr Res 39:1156–1163
Fischer S, Liebscher A, De Lucia M, Hecht L, The Ketzin Team (2013) Reactivity of sandstone and siltstone samples from the Ketzin pilot CO2 storage site-Laboratory experiments and reactive geochemical modelling. Environ Earth Sci 70:3687–3708
Fu Q, Lu P, Konishi H, Dilmore R, Xu H, Seyfried WE Jr, Zhu Ch (2009) Coupled alkali-feldspar dissolution and secondary mineral precipitation in batch systems: 1. New experiments at 200 and 300 °C bars. Chem Geol 258:125–135
Gaus I (2010) Role and impact of CO2-rock interactions during CO2 storage in sedimentary rocks. Int J Greenh Gas Contr 4:73–89
Gherardi F, Audigane P, Gaucher EC (2012) Predicting long-term geochemical alteration of wellbore cement in a generic geological CO2 confinement site: tackling a difficult reactive transport modelling challenge. J Hydrol 420–421:340–359
Gunter WD, Wiwchar B, Perkins H (1997) Aquifer disposal of CO2-rich greenhouse gases: extension of the time scale of experiment for CO2-sequestring reactions by geochemical modelling. Mineral Petrol 59:121–140
Holloway S (1997) An overview of the underground disposal of carbon dioxide. Energy Convers Manag 38:193–198
Jung HB, Um W (2013) Experimental study of potential wellbore cement carbonation by various phases of carbon dioxide during geologic carbon sequestration. Appl Geochem 35:161–172
Jung HB, Kabilan S, Carson JP, Kuprat AP, Um W, Martin P, Dahl M, Kafentzis T, Varga T, Stephens S, Arey B, Carrol KC, Bonneville A, Fernandez CA (2014) Wellbore cement fracture evolution at the cement-basalt caprock interface during geologic carbon sequestration. Appl Geochem 47:1–16
Karnkowski PH (2007) Permian Basin as a main exploration target in Poland. Przegl Geol 55:1003–1015
Kaszuba JP, Janecky DR, Snow MG (2003) Carbon dioxide reaction processes in a model brine aquifer at 200 °C and 200 bars: implications for geologic sequestration of carbon. Appl Geochem 18:1065–1080
Krilov Z, Loncaric B, Miksa Z (2000) Investigation of a long-term cement deterioration under a high-temperature, sour gas downhole environment, SPE Int. Symp. Formation Damage Control. Lafayette
Kutchko BG, Strazisar BR, Dzombak DA, Lowry GV, Thaulow N (2007) Degradation of well cement by CO2 under geologic sequestration conditions. Env Sci Technol 41:4787–4792
Kutchko BG, Strazisar BR, Huerta N, Lowry GV, Dzombak DA, Thaulow N (2009) CO2 reaction with hydrated Class H well cement under geologic sequestration conditions: effects of flyash admixtures. Environ Sci Technol 43:3947–3952
Kutchko BG, Strazisar BR, Hawthorne SB, Lopano CL, Miller DJ, Hakala JA, Guthrie GD (2011) H2S-CO2 reaction with hydrated class H well cement: acid gas injection and CO2-co-sequetration. Int J Greenh Gas Control 5(4):880–888
Labus M and Lorek A (2015) CO2-rock interaction on the example of Permian sedimentary rocks, 77th EAGE Conference and Exhibition in Madrid, Spain
Lorek A and Labus M (2015) Carbonated brine effect on wellbore cement degradation in contact zone with formation rock, 3rd Sustainable Earth Science Conference and Exhibition, Celle, Germany
Newhall C (2006) Improving cement bond in the Appalachian Basin with adjustments to preflush and spacer design. Paper SPE 104576 presented at the SPE Eastern Regional Meeting, Canton
Onan DD (1984) Effects of supercritical carbon dioxide on well cements, SPE Journal, Permian Basin Oil and Gas Recovery Conference, Midland
Pearce JM, Holloway S, Wacker H, Nelis MK, Rochelle C, Bateman K (1996) Natural occurences as analogues for the geological disposal of carbon dioxide. Energy Convers Manag 37(6–8):1123–1128
Rimmelé G, Barlet-Gouédard V, Porcherie O, Goffé B, Brunet F (2008) Heterogeneous porosity distribution in Portland cement exposed to CO2-rich fluids. Cem Concr Res 38:1038–1048
Rochelle CA, Czernichowski-Lauriol I, Milodowski AE (2004) The impact of chemical reactions on CO2 storage in geological formations: a brief review. In: Baines S, Worden RH (eds) Geological storage of carbon dioxide, vol 233. Geological Society, Special Publication, London, pp 87–106
Rzepka M (2010) Zaczyny cementowe do uszczelniania kolumn rur okładzinowych w głębokich otworach wiertniczych, w temperaturze dynamicznej do ok. 120 °C, (Cement slurry used for sealing facing pipes in deep drilled holes in dynamic temperature of 120 °C) Nafta-Gaz 4: 274–279
Um W, Jung HB, Martin PF, Mcgrail BP (2011) Effective permeability change in wellbore cement with carbon dioxide reaction, PNNL-20843. Pacific Northwest National Laboratory, Richland
Walsh SDC, Du Frane WL, Mason HE, Carroll SA (2013) Permeability of wellbore-cement fractures following degradation by carbonated brine. Rock Mech Rock Eng 46(3):455–464
Acknowledgments
The research leading to these results has received funding from the Polish-Norwegian Research Programme operated by the National Centre for Research and Development under the Norwegian Financial Mechanism 2009–2014 in the frame of Project Contract No Pol-Nor/207125/66/2013.
The results of the research presented in this paper are a part of one of the authors (Agata Lorek) PhD thesis, which are being prepared.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lorek, A., Labus, M. & Bujok, P. Wellbore cement degradation in contact zone with formation rock. Environ Earth Sci 75, 499 (2016). https://doi.org/10.1007/s12665-015-5114-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12665-015-5114-z