
Abstract
Bone waste is a problematic slaughterhouse waste typically disposed of in landfills. The pyrolyzed product of this waste shows strong potential in mine and industrial waste water remediation and work is needed to identify chemical and structural parameters which drive performance. Diffuse Reflectance Fourier Transform Spectroscopy (DRIFTS) was used to probe carbonate (CO32−), phosphate (PO43−) and hydroxyl (OH−) environments of mineral phases and functional group chemistry in carbonaceous phase, revealing a potentially synergistic functionality between the two in bone char. CO32− and water substitutions in the mineral lattice were found to persist after pyrolysis to 750 °C, and more soluble non-apatite calcium phosphate phases were observed using second derivative analysis of the v3 PO43− band. Nitrogen-rich functional groups were found in the carbonaceous phase which are associated with complexation of aqueous metals, and ordered aromatic clusters identified by Raman spectroscopy indicate a porous carbon skeletal structure to promote metals adsorption and complexation. These results point to unique chemical and structural features of bone char which are not easily replicated by synthetic carbonated apatite or activated carbon and which contribute to the excellent aqueous metals removal power of bone char.
Graphical Abstract

Similar content being viewed by others

Avoid common mistakes on your manuscript.
Statement of Novelty
Despite the strong performance of bone char compared to other porous materials in removing toxic metals dissolved in water, thorough characterization of the chemical and structural diversity which drive this behavior has been limited. In this work we present a rigorous analysis of the chemistry and structure of bone char using vibrational spectroscopy, X-ray diffraction, and thermal gravimetry. Two materials which can be considered comparative to the mineral and carbonaceous phases of bone char have been characterized in parallel, and the resulting information provides new guidance on why bone char is a superior material for heavy metals removal.
Introduction
Beginning in the early nineteenth century, bone char, made of pyrolyzed animal bone waste, was valued for its unique capacity in simultaneously removing colorants and ash from sugar liquor, with performance in this application attributed solely to the mineral phase—a biogenic, highly defected, carbonated apatite [1,2,3]. Shipping restrictions during World War II resulted in a shortage of foreign-supplied raw bone feedstock to supply the booming U.S. sugar industry, fueling a search for promising replacement materials [1]. Synthetic calcium phosphate hydroxyapatite, Ca10(PO4)6(OH)2, is structurally and chemically similar to bone mineral, and was investigated as a high performing, affordable replacement. In this early research, direct comparative studies showed that bone char removed more ash and colorants when normalized for surface area and imparted a slightly higher pH to liquors than synthetic hydroxyapatite indicating differences in surface chemistry and reactivity between the two [1].
The adsorptive properties of bone char have been tested in a range of other, primarily aqueous, contaminant removal applications, including municipal water treatment—although in this case the taste imparted into the water was unpalatable resulting in bone char being abandoned in favor of other methods [4]. More recently, bone char has demonstrated high potential as an inexpensive, effective adsorbent for treating mine, industrial and municipal waste water laden with heavy metals or fluoride [5,6,7,8,9,10,11]. Structurally and chemically similar synthetic hydroxyapatite has also been applied in toxic metals and fluoride remediation, but comparative studies of the relative performance of bone mineral and hydroxyapatite in these applications are largely absent from recent literature [12,13,14,15,16,17].
One exception is a study by Hashimoto, et al. in which the effects of crystallinity on lead adsorption were investigated [14]. In that study, poorly crystalline, gypsum-derived hydroxyapatite and poultry bone mineral were more effective in removing lead from water than highly crystalline hydroxyapatite over a wide pH range. These findings support the early results of Barrett, Joyner, and Halenda regarding the superior performance of bone char in sugar purification and seem to indicate that the inherent disorder in natural bone mineral is beneficial for aqueous metals remediation applications [1, 2, 18]. In another notable study supporting similar conclusions, Betts, et al. briefly discuss carbonate content in bone apatite as a source of disorder which enhances dissolution of bone mineral in Zn2+ solution, resulting in high Zn2+ removal by precipitation of less soluble zinc phosphate [5].
These past studies as well as in-house investigations into Cu2+ uptake from Cu(NO3)2 aqueous solution raised questions regarding (1) the impacts of the chemistry and nanostructure of pyrolyzed bone mineral compared to those of synthetic carbonated hydroxyapatite, and (2) the role of the residual nitrogen-rich carbonaceous phase on Cu2+ removal [19]. It is clear that a more detailed understanding of bone char and comparative carbonated hydroxyapatite used in aqueous metals removal applications is needed in order to optimize their performance for toxic metals remediation. The specific aims of this present study were to identify features unique to bone char which could be related to metals removal processes like cation exchange, coprecipitation, and dissolution/precipitation. Particular attention was paid to factors which are known to play a strong role in disorder and enhanced surface activity of the mineral phase like polyanion substitutional type and level (particularly carbonate substitutions) which may enhance all mechanisms of Cu2+ uptake. The formation of non-apatite phases may primarily enhance dissolution/precipitation and cation exchange mechanisms. Within the carbonaceous phase, functional groups which are known to have a particularly high affinity for Cu2+ in physisorption may indicate a relatively strong contribution by that phase.
Materials and Methods
Sample Preparation
Samples used in this present study are summarized in Table 1. The preparation of pyrolyzed bone was reported elsewhere, but briefly, bovine femurs were boiled to remove residual soft matter and fats, broken into 1-inch pieces, granulated, dried at 90 °C for > 48 h before further grinding into powder containing heterogeneous size fractions [19]. This sample is hereafter referred to as RGB. The as-prepared RGB was heated at \(\sim\) 15 °C/min to 750 °C (30 min dwell) in a tube furnace (MTI, OTF-1200X) with 60 mm × 60 cm quartz tube under flowing argon (3.0 L/min). This pyrolyzed sample is hereafter referred to as BC750. The pyrolysis temperature was chosen as it falls within a range of temperatures most likely to produce high surface area for biochars [20]. Synthetic carbonated hydroxyapatite (Sigma Aldrich), hereafter called CHAp, was used as a comparative material for the mineral phase of BC750. This material was observed in unpublished studies to have lower total Cu2+ uptake compared to BC750. It was used as-received without further processing.
Three additional samples were prepared to provide insight into the substitutional polyanion environments and the effects of pyrolysis on the mineral and organic components of bone. First, a small amount of CHAp was pyrolyzed according to the same method described above for BC750. This sample is labelled CHAp750. The purpose of this sample was to investigate changes in the anion environment with heating to compare and contrast with those observed for RGB and BC750. To isolate the carbonaceous component of BC750, a small amount was demineralized in 5 M HCl, followed by copious rinsing with deionized (DI) water and drying at 75 °C for 12 h. A slight tint to the filtrate suggested that some soluble carbon was lost during the leaching process. However, since this carbon represented a trace amount of the total mass of solids, the remaining powder was assumed to represent the carbonaceous phase for the intended purpose. This sample is labelled D-BC750. The third sample is a wood-based biochar prepared from ground black cottonwood pyrolyzed in argon to 700 °C. This sample is referred to as biochar. The highest heat treatment temperature (HTT) difference (700 °C vs. 750 °C) was considered negligible compared to the effects of feedstock differences between bone char and biochar. This biochar was used to compare general functional group chemistries and carbon hybridization states between bone char and biochar. Comparison between pyrolyzed and un-pyrolyzed samples is denoted in this work as X \(\to\) Y, with the arrow indicating pyrolysis.
Characterization Methods
Secondary electron micrographs of all samples were taken on a Mira 3 Tescan field emission scanning electron microscope.
Powder x-ray diffraction was performed for mineralogical phase determination and to investigate the effects of heating on lattice parameters and crystallinity. Measurements were made for powdered samples without further preparation on a Rigaku Ultima IV X-ray Diffractometer using Cu k-alpha 1 radiation (1.54059 Å) at − 40 kV and 40 mA with a Ni k-beta filter. Scans were taken in Bragg–Brentano geometry between 5° and 70° 2θ with step size of 0.02°.
Specific surface area (SSA) and pore size distribution (PSD) analysis were outsourced to Dynalene Laboratory Services. SSA and PSD were extracted from N2 adsorption–desorption isotherms collected at 77 K using a Micromeritics ASAP 2020 apparatus. SSA for each sample were calculated from the adsorption branch using the Brunauer–Emmett–Teller (BET) equation and pore size distribution from the desorption branch using Density Function Theory (DFT) methods [21]. Prior to measurement, all samples were degassed at 0.667 Pa and 200 °C for at least 8 h.
Diffuse Reflectance Fourier Transform Infrared spectra (DRIFTS) were collected to characterize mineral and carbonaceous phases. All data were taken on a Shimadzu IR Tracer100 with Harrick Praying Mantis DRIFTS attachment and DLTAGS detector under ambient conditions with atmospheric correction applied. Samples were prepared for DRIFTS by grinding individual powdered samples (\(\sim\) 7% wt/wt) in KBr powder using a mortar and pestle. References collected from pure KBr were used to remove backgrounds from all sample spectra. Spectra were collected in the 4000–400 cm−1 range at 2 cm−1 resolution with 120 accumulations. At least three samples from each material were tested. Second derivatives of each raw spectrum were mathematically calculated using Origin Pro, and were used to identify peak locations and therefore guide peak fitting with the appropriate number of Gaussian curves in the v2 CO32− band between 820 and 920 cm−1. Spectra were cropped between 820 and 920 cm−1, and a linear background was applied and subtracted prior to fitting.
Raman measurements of undiluted, powdered samples were made on a Renishaw inVia Spectrometer using a 20 × LWD objective. Because strong photoluminescence of the mineral phase was observed with longer wavelengths, the 457 nm emission line from an argon ion laser was chosen with a power of 30 μW power and 1–5 µm spot size. Scans were taken between 500 and 2300 cm−1 with 3–5 s accumulation time and 5–20 repetitions in order to achieve sufficient signal/noise. Low power density is often necessary to prevent thermal damage to opaque samples such as amorphous carbon. For each sample, 3–5 randomly chosen spots were analyzed to ensure uniformity. Reported fit parameters are the arithmetic mean for these scans with reported standard error.
Thermal analysis of all materials was accomplished using thermal gravimetry coupled with mass spectrometry (TGA-MS) for semi-quantitative assessment of the composition and thermal decomposition products for each material. As-prepared samples were heated in ultra-high purity argon or oxygen in the evolved gas analysis furnace (EGA) furnace on a TA Instruments Q500 thermal analyzer. Ion current for m/z of particular ionized species were measured using a Pfeiffer ThermoStar electron impact quadrupole mass spectrometer. For these measurements, the MS probe was coupled with the EGA furnace via special attachment (TA Instruments, Inc.) so that the quartz capillary sampled evolved gases directly above the Pt sample pan inside the TGA furnace. For this set of measurements, all samples were initially heated to 35 °C and held at that temperature for 1 h to stabilize the ion current signal, and then heated at 2 °C/min from 35 to 1000 °C. Water, hydrogen, carbon dioxide and a characteristic polymer fragment were tracked from all materials, as applicable. All materials were heated in dry argon to measure water content and thermal decomposition products. Ash content (mineral phase relative composition) was determined as the residual mass at 600 °C in 90% O2 (bal. Ar). Integration of the smoothed differential weight loss curves was used to calculate water (weight loss between 35 and 150 °C), thermal decomposition (weight loss between \(\sim\) 150 and 800 °C) and thermally stable carbon or ash, which was characterized as the remaining mass at 800 °C for runs in argon (D-BC750) and 600 °C for runs in oxygen, respectively.
Results and Discussion
Characterization of Whole Particles
Scanning Electron Microscopy
The morphology of BC750 and CHAp particles were investigated using field emission scanning electron microscopy (FE-SEM). Micrographs of BC750 and CHAp at different magnification are shown in Fig. 1a–d. Both were found to consist of polydisperse particle size fractions. CHAp particles were spherical in general with larger particles present as hollow spheres, consistent with synthetic hydroxyapatites prepared by aerosol spray method [22]. BC750 particles are oblong and have a heterogeneous size distribution, with the largest typically being < 1 mm in the longest dimension. In BC750, mineral platelets are observed in fractured particles (Fig. 1b).

FE-SEM micrographs of a BC750 particles have elongated, irregular shape and heterogeneous size distribution; b Broken BC750 particle reveals the interior consisting of carbonaceous residue and plate-like mineral components; c CHAp particles are polycrystalline spherical particles as is typical for aerosol-formed synthetic hydroxyapatite (Vallet-Regi 2004), and have overall a smaller average size than BC750 particles; d CHAp surface showing fine grained, mesoporous structure; it is difficult to determine whether CHAp crystals have columnar or plate-like growth habit
Nitrogen Adsorption
BC750 and CHAp showed similar surface areas calculated using the BET method—CHAp: 113 m2/g, BC750: 112 m2/g. Graphical results are summarized in Supplementary material Section S.3. Both CHAp and BC750 exhibited IUPAC Type IVa N2 adsorption/desorption isotherms (Fig. S6) [21]. The incremental surface areas for CHAp and BC750 are presented in Fig. S7a,b and Table S2. Both CHAp and BC750 are primarily macroporous, meaning most of the specific surface area is associated with pores > 500 Å, or with outer surfaces. CHAp had only 5% of specific surface area associated with micro- or mesopores and BC750 had 15%. BC750 has five times greater specific surface area in micropores (< 20 Å) than CHAp and nearly three times greater specific surface area in mesopores (20–500 Å). The impact of the carbonaceous phase on the specific surface area was not quantified because of limitation in availability of D-BC750, however others have observed that this phase in pyrolyzed bone is primarily microporous [23]. Thus, it is possible that the presence of the carbonaceous phase in BC750 enhances the micropore content. Microporosity in CHAp and BC750 was also observed to have different size distributions. CHAp has microporosity of \(\sim\) 7 Å, while BC750 microporosity is slightly larger at \(\sim\) 15 Å.
Thermal Analysis and Mass Spectrometry
Thermal degradation products correlated to a particular temperature or range of temperatures can provide clues about composition and bonding in complex materials like bone char. Using TGA-MS, thermally-induced chemical changes can then be correlated to the temperature range over which they occurred and related to the weight lost over that range. In this current work, TGA-MS was used to investigate bulk compositional components like surface adsorbed water and relative amounts of carbonaceous and mineral phases. Mass spectra also provided some sample-specific information, particularly giving insight into bonding in the amorphous carbon of bone char and demineralized bone char as well as the high-temperature decomposition behavior of CHAp. Evolution of water, carbon dioxide, hydrogen, and a characteristic polymer fragment, C3H6, associated with volatilization of light hydrocarbons, were tracked to investigate the products of thermal decomposition (argon) and combustion (90% O2/bal. argon). TGA curves for BC750, CHAp, and D-BC750 are presented in Fig. S9a,b and DTG curves are presented in Fig. S10a,b. All samples showed between 2 and 5% by weight physisorbed water loss between 80 and 150 °C with residual water and CO2 evolving at varying quantities up to \(\sim\) 450 °C, especially for CHAp.
Using combustion analysis (in 90% O2/bal. argon), BC750 was found to consist of 2.5% water, 86% ash (mineral phase), and 11.5% carbonaceous phase by weight. In argon, D-BC750 consisted of 1.4% surface adsorbed water, < 0.5% ash, and 81% thermally stable carbon while 17% mass loss occurred to thermal decomposition between 80 and 150 °C.
For all samples in Ar, water and carbon dioxide generated between 80 and 250 °C are attributed to surface adsorbed species [24]. Between 250 and 450 °C evolution of these species is attributed to those more strongly adsorbed in pores (i.e., in CHAp) or due to volatile carbon (i.e., in BC750 and D-BC750). For BC750 and D-BC750, evolution of CO2 beginning at \(\sim\) 200 °C for BC750 and \(\sim\) 400 °C in D-BC750 may be indicative of the catalytic action of the mineral phase, present in BC750, lowering the activation energy for CO2-forming reactions. Above 650 °C significant effusion of water and CO2 was observed for CHAp (Fig. 2, top). This is indicative of the onset of thermal degradation of some carbonate and hydroxyl species, as observed by others [24, 25]. High temperature decomposition of bone mineral was not directly observed in this study because effusion of CO2 and water associated with pyrolysis of the carbonaceous phase obscured the onset of bone mineral decomposition.

Effusing gases from CHAp (top), BC750 (middle), and D-BC750 (bottom). All signals were normalized to the argon signal and by their respective min–max., H2 and H2O curves for BC750 and D-BC750 were multiplied by 5 to improve visibility, and all were vertically offset. The intense background at the start of the H2O curve for BC750 is attributed to the effects of normalization
Thermal decomposition (in Ar) of amorphous carbon in D-BC750 (Fig. 2, bottom) resulted in effusion of water, CO2, and C3H6 between 150 and 400 °C attributed to volatile, surface adsorbed species, while formation of CO2 and polymeric fragments between 450 and 800 °C are attributed to thermal decomposition of the carbonaceous phase. CO2 effusion above 450 °C is attributed to decomposition of carboxylic anhydride, lactone, and pyrone groups [26]. Water effusion above 600 °C is attributed to result from break-down of phenolic alcohols [26]. Hydrogen effusion begins at approximately 600 °C, behavior which was previously observed in acetylene-based amorphous carbon films [27]. Hydrogen evolution above 900 °C is indicative of large-scale carbonization.
It is worth noting that the temperature profiles for evolved gases from D-BC750 decomposition are consistent with those observed by others [27, 28]. Hydrogen evolution has been reported as an indicator of sp3 structure in amorphous carbon, with H2 effusion resulting from carbonization processes [27]. Hydrogen, water, and polymer fragment effusion during annealing of amorphous carbon films was studied by Conway, et al. [27], with differences in the onset of H2 effusion attributed to the formation of different hydrogen environments within the amorphous carbon from either methane or acetylene precursors. Conway, et al. found that onset of effusion at high temperatures was indicative of a dense carbon skeleton of highly cross-linked sp3 carbon having a lower overall hydrogen content compared to polymeric amorphous carbon. Fragmentation of polymeric structures was observed to occur just before hydrogen effusion for acetylene-based carbon films, and at the same time as hydrogen effusion from methane-based films. The former was observed in D-BC750, providing some evidence for the presence of dense, mechanically robust and chemically inert cross-linked sp3 carbon which may have impact on adsorption of hydrated metal cations, but which give hardness and mechanical stability to the carbonaceous phase. Further study is needed to validate this observation, and to determine whether it is inhibitive to metals removal.
Characterization of Mineral Phases
For the remainder of this current work, results and discussion pertaining exclusively to the carbonaceous and mineral phases of each material is presented separately, with characterization of bone mineral and CHAp presented first and characterization of the carbonaceous phase and biochar last.
Review of the Chemistry and Structure of Bone Mineral
The inorganic phase in bone is structurally similar to calcium phosphate hydroxyapatite (Ca10(PO4)6(OH)2) sharing the same hexagonal unit cell, with P6 3/m space group symmetry, lattice parameters a and b being coplanar and c being normal to this plane [29]. In bone mineral, plate-like nanocrystallinity (⁓50 nm × 25 nm × 4–6 nm) of primary crystallites implies that, geometrically, a very high percentage of ionic species are on the surface. In general, surface species are less likely to be charge compensated, and therefore are more likely to be chemically active in processes of metals removal. In addition to surface effects, the presence of substitutional defects in the bulk of bone mineral is likely to extend this solubility behavior in aqueous environments to the whole crystallite [22, 30].
Primary crystallites in bone exist as part of a larger mineral macrostructure rather than as isolated crystallites in a bulk collagen matrix. Within this larger macrostructure, individual nanocrystalline apatite platelets are joined by hydrated, interfacial surface structures consisting of amorphous phases of Ca2+, HPO42−, HCO3−, Mg2+, water, and organic polyanions such as citrate [31,32,33]. Water in these layers is believed to take on a structural role, orienting the organic polyanions into a distinct, non-apatite phase [34]. The presence of structural water in these phases is also linked to the arrangement of bone crystallites in an oriented, stacked configuration [34]. The close proximity between layers of ordered and amorphous phases implies some interaction may occur during thermal treatment, for instance precipitation of amorphous calcium phosphate or other non-apatite phase on the surface of primary crystallites or possible incorporation of interlaminar species into primary crystallites. The fate of interlaminar species during pyrolysis has not been well-characterized, but it can be reasoned that these regions are particularly affected by pyrolytic processes including dehydration and thermally-driven, secondary reactions between constituent species. Phase transformation within interlaminar layers is thus possible during heat treatment, with nucleation of new apatite or non-apatite species—probably on the surface of existing primary crystallites.
While often referred to as “hydroxyapatite”, bone mineral is known to be chemically quite different from pure hydroxyapatite [30, 35]. First, bone mineral has very little hydroxyl ion content, with so-called structural water occupying most of the hydroxyl sites and some other lattice positions via substitution, and OH− being found extensively only in mature crystallites [30, 36]. Low hydroxyl content in bone mineral has been linked to high susceptibility for dissolution and low acid-buffering capability which enables resorption by osteoclasts in vivo [29]. Other differences include ionic substitution and vacancies. Important cation substitutions occur in bone mineral in which primarily Mg2+ and Na+ replace Ca2+. Na+ substitutions are known to be extensive in biogenic apatite and to occur in conjunction with CO32− substitutions in the OH− position in bone mineral [38]. Cation substitutions impact the overall crystallinity in bone mineral. They may enhance cation exchange with heavy metals species, including Cu2+, as the ionic radii of Mg2+ and Na+ are smaller than Ca2+, making it easier to exchange with the even smaller Cu2+.
Another substitutional species, carbonate (CO32−), is typically present in bone mineral at about 4–8% wt, and is critical for biocompatibility in synthetic hydroxyapatites for bone implants [35, 39]. Carbonate can be present in different forms in both synthetic and biogenic apatite, and within the hydroxyapatite lattice can occupy two primary positions: the OH− site (also called the channel site; the channel is oriented parallel to the c-axis), which is known as A-type substitution, and the PO43− site, known as B-type substitution [37, 40, 41]. In A-type substitutions the planar carbonate ion orients normal to the channel axis, having the effect of contracting the c-lattice parameter. In B-type substitutions, the planar CO32− ion is believed to primarily occupy the position of one of the faces of the PO43− tetrahedra roughly parallel to the c-axis, and has the general effect of contracting the a-axis [39]. Bone mineral is AB-type, containing both types of substitutions, with the degree of A-type substitution observed to depend on the age and species of the bone tissue [40]. Increased CO32− content in bone apatite has been linked to reduction in crystallinity and to the plate-like habit and nanometer-scale of primary crystallites, which impart high biological activity, including ease of resorption in vivo [30, 35, 41,42,43]. Importantly for the current work, A-type substitutions have been observed to reduce the crystallinity of apatite more than B-type [39]. Both bone mineral and hydroxyapatite can also contain a labile (L-type) carbonate species and HCO3−, which are typically present in amorphous phases and on the surface of crystallites [38]. Rey, et al. indicated that this species may be associated with early stages of apatite crystal formation and with disordered domains [40]. Fleet, et al. also referred to L-type CO32− as “stuffed channel” CO32−, suspecting that it occupies a surface position of the channel compared to A-type carbonate, which is within the bulk [44]. Those authors found that in Na-bearing apatite such as bone mineral, L-type carbonate largely disappears with heat treatment above 600 °C.
Acid phosphate (HPO42−) is another polyanion impurity which is frequently found in both biogenic apatite and synthetic hydroxyapatite. Most HPO42− in bone mineral is found within the interlaminar domains, outside the primary crystallites [45, 46]. However, HPO42− has been observed within the primary crystal lattice in the phosphate position in smaller amounts and its presence there is believed to relate to charge neutrality, particularly in newly forming crystals [33]. This species is present in synthetic hydroxyapatite as a result of incomplete reaction synthesis. Alternate phases found in thermally treated bone and hydroxyapatite can also contain HPO42−, especially octacalcium phosphate (OCP) and brushite [24, 47, 48].
Diffuse Reflectance Fourier Transform Infrared Spectroscopy
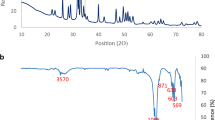
Collagen, amorphous carbon, bone mineral and hydroxyapatite all have components with strong IR activity, making infrared spectroscopy a powerful and well-used tool for characterizing bone, bone char and hydroxyapatite. In this present work DRIFTS was utilized because of the ease of sample preparation and sensitivity to surface species. The assignment of IR vibrational modes for biological and synthetic apatite have been reviewed extensively elsewhere, and many are labelled in Fig. 3a and b [38,39,40, 44, 49,50,51]

Raw, vertically offset DRIFTS spectra for RGB, BC750, CHAp, and CHAp750. a OH, NH, and CH stretch region; b v1,v3 PO43−, v2 CO32− and v4 PO43− regions with OH− libration mode. Important vibrational modes are annotated. Overall intensity for mineral peaks in synthetic CHAp and CHAp750 was higher than for RGB and BC750, which may be due to the impact of carbonaceous char and collagen on reflectance and is not a direct function of concentration. All species were mixed with KBr at \(\sim\) 7% wt
The vibrational modes of phosphate, carbonate, and hydroxyl ions are highly sensitive to the local environment, including protonation, making small changes in lattice position or bonding easily detectable with IR spectroscopy. In the present work, OH− stretch (4000–2700 cm−1), v2 CO32− (\(\sim\) 875 cm−1) and v3 PO43− (1200–900 cm−1) modes were used to track differences in anion content which may be relevant to aqueous metals removal application. Representative spectra from each material are presented in Fig. 3a and b.
In materials such as bone mineral, which can contain multiple substitutional species like CO32−, HPO42−, and so-called structural water, the challenges associated with identifying structural effects due to a single species are non-trivial. IR spectroscopy can provide clues about the fate of substitutional species by probing the degree of protonation, substitutional position, and the formation of alternate phases. It is worth noting that the quantitative capability afforded by the Beer–Lambert law, used for interpretation of transmission IR spectra, does not directly apply to DRIFTS spectra because the optical path and processes involving multiple reflections through the sample are difficult to reproduce. For this reason, DRIFTS results were interpreted qualitatively.
OH− and Water Bands
Key differences in OH−—and water-related bands are visible in the O–H stretch region between 2700 and 4000 cm−1 (Fig. 3a) and in the O–H libration mode at 630 cm−1 (Fig. 3b) [52]. The libration mode is associated with lattice species and not metal hydroxides or water. CHAp and CHAp750 have an intense lattice O–H stretch peak at 3570 cm−1 and libration mode peak at 630 cm−1, while RGB and BC750 do not show a libration mode and very low to no intensity for the lattice OH− stretch. This result agrees with the very low lattice OH− content in biogenic apatite. With pyrolysis, lattice OH− modes do not appear for BC750, indicating very little to no new lattice OH− is created by pyrolytic processes. A broad, intense band associated with surface adsorption of atmospheric water is observed for all materials, however heated samples show lower intensity for this band compared to un-heated precursors. This is attributed to the effects of pyrolysis where (1) at least partial coating of bone mineral by the more hydrophobic carbonaceous product of collagen pyrolysis was formed, (2) excess surface water was lost, and (3) enhanced crystallinity (including potential loss of specific surface area due to growth of primary crystallites), which has been observed in bone mineral to inhibit atmospheric water binding [34].
Carbonate v2 Band
There are two primary carbonate bands which show up in the IR spectra of bone mineral and hydroxyapatite: the v2 out of plane bend (Fig. 4) and the v3 asymmetric stretch modes [38]. The v3 CO32− mode (1500–1400 cm−1) is often used to characterize CO32− in carbonated hydroxyapatite, but in BC750 this band overlaps with intense bands which are associated with the carbonaceous phase (see Fig. S5), so was not used in analysis [39, 40, 53]. The v2 CO32− mode at approximately 875 cm−1, on the other hand, is largely unobscured by amorphous carbon bands of bone char because the carbonaceous phase of bone char does not show the intense peaks associated with aromatic ring C-H out-of-plane wag modes (see Fig. 8 and SI Fig. S5). Like the v3 mode, the carbonate v2 mode is highly sensitive to substitutional position, protonation and local environment. The frequency for A-type substitutions is observed at \(\sim\) 880 cm−1. B-type substitutions are observed over the range 871–875 cm−1 with the exact frequency depending on local environment and coupling with A-type carbonate modes [37]. The effect of structural water in channel sites on the frequency of B-type CO32− has not been well characterized, but some effect is possible. Other forms of CO32− found in biogenic and synthetic hydroxyapatite also have vibrational modes nearby: labile carbonate (L-type) at \(\sim\) 866 cm−1 and HCO3− at \(\sim\) 843 cm−1 [40, 44]. There may also be some weak contribution from P-(OH) or H2CO3 modes at \(\sim\) 890–900 cm−1 [40, 46, 54].

The v2 CO32− band of representative spectra for each material. With heating, the total peak area for CHAp → CHAp750 and RGB → BC750 remaining constant with heating which indicates little to no total CO32− was lost during heating. The discrepancy between peak intensities for bone-based versus synthetic materials does not necessarily indicate that CHAp and CHAp750 contain more CO32− than RGB and BC750. See Sect. 3.2.2. for further discussion
According to the method of Rey, et al. comparison of the area of peaks within the v2 CO32− band is indicative of the relative content of each substitutional species [38,39,40]. In this current work relative change of each type due to pyrolysis was the primary focus. In addition, using this method enabled investigation into the effects of pyrolysis on CO32− substitutions in synthetic vs. biogenic materials, and provided insight into thermal recalcitrance of defects in bone mineral which are not observed in synthetic carbonated hydroxyapatite.
The HPO42− peak area was subtracted prior to calculation of percent areas so that relative differences in CO32− band areas only were considered. It is of note that with heating, the HPO42− peak area for biogenic and synthetic materials showed a significant decrease. Results of peak fitting for all four materials in this present study (Table 2) revealed significant differences in the material response to pyrolysis for synthetic hydroxyapatite compared to bone mineral. Differences in the relative contributions of all four carbonate types to the total band area were observed for BC750 and CHAp which are likely to impact solubility, acid/base buffering capacity, and mechanisms of aqueous metals removal.
With heating to 750 °C, no significant change in total v2 CO32− band area was observed for either CHAp \(\to\) CHAp750 or RGB \(\to\) BC750 (Fig. 5a,b). Both synthetic hydroxyapatite and bone mineral showed a decrease in L-type and HCO3− with heating, in agreement with trends observed by others [37, 40]. The percentage of L-type lost with heating was greater for RGB than for CHAp. An increase in B-type CO32− was observed for RGB \(\to\) BC750, however for CHAp \(\to\) CHAp750 a decrease was observed. A-type CO32− increased with heating for both bone mineral and synthetic apatite.

Carbonate v2 band for a RGB and b BC750; with raw spectrum (blue), fitted total band (solid black), and subpeaks.
CHAp contained primarily B- and L-type CO32− substitutions with only minor contributions from HCO3− and A-type. Following pyrolysis, CHAp750 lost nearly all HCO3−, and saw approximately 29.5% reduction in L-type CO32−. More than four-fold increase in A-type CO32− contribution was observed following pyrolysis. While this may initially seem exceptional, it is likely explained by conversion of other forms of CO32−, particularly L-type, HCO3− or even B-type, into A-type as has been proposed by others, this conversion being enabled by the loss of channel OH− at temperatures above 600 °C [39, 40, 55]. Conversion of B-type to A-type typically been attributed to migration of CO32− from the phosphate position into the neighboring channel as OH− vacancies are created by thermal effusion of this species (and subsequent water and oxygen formation) during heating or reaction with other protonated species [39]. De-hydroxylation has been observed in hydroxyapatite having high initial OH− content [25]. Significant water effusion (Fig. 2, top) observed above 600 °C for CHAp supports this mechanism.
RGB contained comparable levels of A- and B-type CO32−, typical for bone mineral, with nearly half the total CO32− present as L-type. With pyrolysis, the relative contributions from L-type and HCO3− decreased significantly. The HCO3− contribution reduced by more than half and L-type contribution reduced by approximately a third. B-type CO32− increased by 37% with heating, in contrast to the behavior of this species in CHAp \(\to\) CHAp750, while A-type CO32− increased with pyrolysis by 77%. Effects of pyrolytic processes such as dehydration of primary bone mineral and interlaminar regions, secondary reactions between water and HPO42−, HCO3− and OH−, and effects of gaseous products (i.e. CO2 and water) from collagen pyrolysis on carbonate substitutions are likely during bone char formation and have not been well-characterized previously. From v2 CO32− analysis, formation of A-type and B-type CO32− from L-type and HCO32− appears likely and is possibly driven by the loss of excess water, presence of CO2 in evolving pyrolysis products, or reactions at elevated temperature involving water and HPO42−, HCO3− and OH−. These phenomena would explain the observed reduction of HPO42− and HCO3− with heating [30, 39, 45].
Carbonate present in A-sites, B-sites and stuffed channel sites in bone char would have strong impacts on the mechanisms of Cu2+ uptake. In cation exchange, Ca2+ in the apatite lattice is exchanged with a different metal cation from the solution. Ionic interactions between carbonate and calcium are weaker than for phosphate and calcium, therefore high carbonate content in both A- and B-type sites likely enhances ion exchange as a mechanism for Cu2+ uptake. For dissolution/precipitation mechanisms, carbonate substitutions are known to enhance the solubility of biogenic apatite, therefore high carbonate content in bone char would also have the effect of enhancing this mechanism. Labile carbonate, being a surface species, is more likely to enhance surface interactions, including complexation and cation exchange with surface cation species.
Phosphate v3 Band
The phosphate v3 mode, between 1200 and 900 cm−1 is another region rich with structural information, made up of multiple component peaks which are related to the tetrahedral modes of PO43−. For an isolated PO43− tetrahedra, the P–O stretch is triply degenerate. However, inclusion in the apatite lattice, on the surface, or in a non-apatite phases result in distortion of the tetrahedra, observed in the IR spectrum as splitting of these vibrational frequencies. Multiple P–O modes are often observed in bone and hydroxyapatite between 1100 and 900 cm−1, and this has been well-known and exploited in bone research [24, 47, 56, 57]. In addition to tetrahedral PO43−, HPO43− is known to exist within the primary crystallites (believed to persist due to charge balance requirements) and especially in amorphous or non-apatite species in both bone mineral and hydroxyapatite, with more than half found in the hydrated interlaminar regions of bone mineral [45]. Monohydrogen phosphate, or acid phosphate (HPO42−), has (OH)P=O stretch modes between 1300 and 1100 cm−1, with a PO–H bend mode at 989 cm−1, and possible other modes (discussed in Sect. 3.2.4) related to resonance with the OH− stretch [45, 49, 54, 58]. The v1 PO43− mode is visible at 960 cm−1, shown in Fig. 3b [47].
Second derivative IR spectroscopy has been used extensively by bone researchers to track the structural relevance of peaks in the broad, overlapping v3 PO43− band of bone and hydroxyapatite spectra [47, 57, 59]. Through comparison of peak positions in the spectra of a range of calcium phosphates, including OCP, brushite, monetite, amorphous calcium phosphate, and synthetic hydroxyapatites with carefully synthesized crystallinity and substitutional content, some characteristic phosphate modes in bone mineral have been determined [47, 56, 57]. In the present work, the complexity of the v3 PO43− band (Fig. 3b) in all four materials confounded the identification of specific bands in the zero-order absorbance spectra, so second derivative analysis was used instead to identify new peaks and those which showed the most change with pyrolysis. These observed changes point to differences in phosphate environments for BC750 compared to CHAp, and indicate that the phosphate environments in synthetic hydroxyapatite are more thermally stable than for bone mineral under the pyrolysis conditions in this study.
In the zero-order absorption spectrum of BC750, regions with strong intensity are observed which are either not observed directly in RGB or CHAp or are much weaker in those spectra. These regions appear to be PO43− modes associated with hydroxyapatite, poorly-crystalline freshly-deposited apatite, and non-apatite phases for BC750. The primary sub-peaks in the v3 PO43− band for CHAp were found to be related to PO43− in mature hydroxyapatite, with some contributions from mature, poorly crystalline hydroxyapatite containing HPO42− [47]. No significant change is observed in this band structure with heating, as evidenced by the similarity between second derivative spectra of CHAp and CHAp750 in Fig. 6.

Second derivative IR spectra of v1,v3 PO43− band were used to identify peaks which showed enhanced intensity in the spectrum of BC750 compared to RGB and CHAp. Important frequencies are denoted by symbols. CHAp750 did not show significant change compared to CHAp, indicating phosphate environments are largely thermally stable
Increased intensity of second derivative minima at 1033 cm−1, 1055 cm−1, and 1074 cm−1 was observed in BC750. These peaks have previously been associated with stoichiometric hydroxyapatite [47]. Enhancement of these bands in BC750 compared to RGB can therefore be deemed as further evidence of an increase in crystallinity of existing primary crystallites, also evidenced by narrowing of XRD peaks discussed in Sect. 3.2.6.
By tracking changes in the HPO42− vibrational modes for RGB \(\to\) BC750 at 1112 cm−1, 1109 cm−1 and 1127 cm−1, it is hypothesized that some HPO42− was integrated into newly formed apatite and some was present in non-apatite phases. The 1020 cm−1, 1038 cm−1, 1112 cm−1, and 1127 cm−1 (here 1124 cm−1) have been previously assigned to non-apatite PO43− and/or acid phosphate in poorly crystallized hydroxyapatite [47, 58]. A peak at 1116 cm−1 observed in BC750 and CHAp but not in RGB has previously been associated with HPO43− in mature, poorly crystalline hydroxyapatite, which can be attributed to HPO42− remaining in existing primary crystallites [47]. An enhanced minimum at 1112 cm−1, not present in RGB and difficult to determine in CHAp, is observed for BC750. A peak at this frequency has previously been attributed to HPO42− in newly formed, poorly crystalline apatite, and which we propose can probably be attributed here to apatite precipitated from HPO42− originally in the interlaminar regions on the surfaces of existing crystallites. Peaks at 1026 cm−1 and 1100 cm−1 have been assigned to calcium pyrophosphate (β-CaP2O7), at 1038 cm−1 to non-apatite phases and OCP (Ca8H2(PO4)6(H2O)6.5), and at 1060 cm−1 and 1124 cm−1 to brushite (CaHPO4(H2O)2) [24, 47, 58, 60].
While only hydroxyapatite peaks were detected by x-ray diffraction (Sect. 3.2.6), second derivative analysis of the v3 PO43− band provides evidence for non-apatite environments of PO43− and HPO43− which have been previously associated with other phases. It has been well-established that bone mineral phosphate is primarily present as PO43− but can also take the acid form (HPO42−) to compensate charge, for instance in calcium deficient local environments or at surfaces [35, 47, 58, 60]. In raw bone, primary crystallites are surrounded by a hydrated interlaminar phase containing, among other species, HCO3− and HPO42− [31, 61]. Previously identified as having an amorphous structure, the fate of this amorphous surface layer with heat treatment is, to the best of the present authors’ knowledge, not characterized [34, 45]. When water is driven rapidly from these surface layers during heating, nucleation of amorphous or non-apatite phases would be likely, as would secondary reactions between constituent species. Fowler, et al. noted that acid calcium phosphates (specifically OCP), in the presence of hydroxyapatite, form pyrophosphates and brushite plus apatite and water with heat treatment [24]. Young, et al. observed the formation of pyrophosphate with heating to 500 °C as a very weak peak at 730 cm−1 for non-carbonated hydroxyapatite and sheep bone. Those authors attributed this to thermally-driven reaction between HPO42− groups, releasing water and forming pyrophosphate in solid solution with biological apatite [45]. While significant water effusion was observed for CHAp heated above 650 °C, no peaks at 730 cm−1 were observed for BC750 or CHAp750, indicating the formation of pyrophosphate may have been inhibited during pyrolysis. It is not known whether the 730 cm−1 peak or the peaks at 1026 cm−1 and 1100 cm−1 are most indicative of pyrophosphate formation. Since D-BC750 does have some intensity associated with N–H wag in the area of 730 cm−1 (Fig. 8) it is possible that in BC750 these modes obscure this weak pyrophosphate mode.
Non-apatite phases, including those containing HPO42− rather than PO42−, can enhance heavy metals removal processes, and may help explain the strong performance of BC750 in Cu2+ removal compared to CHAp. The presence of more soluble non-apatite phases on the surface of primary apatite crystallites is likely to enhance coprecipitation and dissolution/precipitation mechanisms of heavy metals removal [15]. In particular, amorphous calcium phosphate and pyrophosphate are more soluble than apatite in vitro, so the presence of these phases on the surface is very likely to enhance dissolution/precipitation mechanisms for Cu2+ adsorption [62]. The specific role of hydrogen phosphate in brushite and OCP in BC750 may be linked to enhanced dissolution/precipitation as HPO42− has been observed to readily form less-soluble amorphous precipitates with Cu2+ from copper nitrate solution compared to dissolved PO43− [63].
X-Ray Diffraction of Bone Mineral and Hydroxyapatite
X-ray diffraction was used to investigate mineral phases present in all samples, but not for characterization of carbonaceous phases. Normalized, cropped, vertically offset diffraction patterns for CHAp, CHAp750, BC750, and RGB are shown in Fig. 7 with some primary reflections annotated. Complete patterns for all materials are found in Fig. S2. All materials were found to have a hydroxyapatite crystal structure [64]. While it could be assumed that this means only hydroxyapatite is present, it is possible for other phases have formed in precipitates which are too small or too disordered for Bragg diffraction.

XRD patterns for CHAp750 (top), CHAp (second from top), BC750 (second from bottom), and RGB (bottom). The indices associated with the lattice planes for some reflections are included. All materials exhibit peaks consistent with hydroxyapatite crystal structure with varying degrees of crystallinity
Peak fitting of the (002) and (300) reflections was used to find hexagonal unit cell parameters for hydroxyapatite and bone mineral, reported in Table 3 with standard error, percent change, and propagated error associated with pyrolysis. Raw data was fit with an appropriate number of Lorentzian curves plus a linear background for the (002) and (300) peaks. Calculations and graphical representations of fitted curves are summarized in Supplementary material Section S.1.
The effects of pyrolysis on unit cell parameters of bone mineral and hydroxyapatite in the present study can be approximated by comparing RGB/CHAp (no heating) to BC750/CHAp750 (rapid heating to 750 °C). In the present study, for CHAp \(\to\) CHAp750, the a-lattice parameter decreased by 0.14 ± 0.04% and the c-lattice parameter decreased by 0.13 ± 0.02%. For RGB \(\to\) BC750 the increase in a was negligible, however c decreased by 0.22 ± 0.02%. Changes in lattice parameters of primary crystallites of bone mineral and hydroxyapatite for RGB \(\to\) BC750 and CHAp \(\to\) CHAp750 can result from pyrolytic processes which enhance ordering within the crystallites, including dehydration, potential reactions between species, and changes in substitutional type and content. Additionally, at higher temperature, increased mobility of species may also impact ordering within primary crystallites. Enhanced ordering within primary crystallites, as well as growth of crystallites in the a- and c-directions, impacts lattice parameters in that local lattice strain at defects and surfaces becomes less impactful on the average. Changes observed here are considered as the cumulative effects of all phenomena.
The effect of carbonate ion substitutions on hydroxyapatite unit cell parameters compared to pure hydroxyapatite have been well-studied and reported by others [39]. In perfect hydroxyapatite, when CO32− substitutes for OH− in the channel site (or A-site), the planar ion orients perpendicular to the c-axis and for charge neutrality, one CO32− ion substitutes for two OH− ions. The result is an overall contraction of the c-axis [22, 39]. In the B-site, one CO32− substitutes for one PO43− with charge neutrality being accounted for by vacancies or water substitution [29, 38, 55]. CO32− in purely B-type hydroxyapatite is believed to occupy the face of the PO43− tetrahedra roughly normal to the a-axis, hence contraction in the a-direction and expansion in the c-direction are typically observed for B-type substitutions [22, 39].
For materials which already contain one or more carbonate species as well as other impurities, the changes in lattice parameter may not follow expected trends. In this case, changes relative to the starting carbonate content must be considered. Bone mineral is known to be AB-type, and both RGB and BC750 were found to be of this type in the present study [38, 39, 55]. The changes in lattice parameters for RGB \(\to\) BC750, it seems, could be accounted for by the increase in both A-type and B-type CO32−, with the dramatic effects of increased A-type dampened by the effects of increase in B-type. Etok and Rogers, et al. observed the same trend with bone mineral derived from combusted meat and bone meal [65]. For CHAp \(\to\) CHAp750, one may consider the case of starting with a purely B-type CO32− and transitioning to AB-type. In this case, the work of Madupalli, et al. implies that an increase in a and no change in c should be observed. For CHAp \(\to\) CHAp750, the decrease in c is consistent with an increase in A-type CO32−, however the decrease in a is not consistent with either reduction in B-type or increase in A-type.
Others have observed strong effects on the temperature response of lattice parameters due to CO32− content at lower temperatures than used in this present study, but this effect at elevated temperatures is not well-known [66, 67]. Young, et al. observed a decrease in a with heating of carbonated hydroxyapatite with heating to 400 °C [68]. Those authors propose a reaction between atmospheric CO2 and channel OH−, producing channel carbonate and water vapor. This mechanism seems unlikely with pyrolysis in Ar, however reaction between B-type CO32− and water, or between OH− and residual HCO3−, coupled with integration of labile species into the A-type channel configuration may be responsible. DRIFTS results support this hypothesis, where with pyrolysis, HCO3− and L-type content decreased significantly for both CHAp \(\to\) CHAp750 and RGB \(\to\) BC750. The effect of these species on lattice parameters is not well-characterized, but it is possible they have a measurable impact in addition to that caused by B-type and A-type substitutions. Other secondary reactions could occur, including conversion of HPO42− to PO43− with the loss of OH− observed by TGA-MS. Young and Holcomb observed that for hydroxyapatite heated to 500 °C HPO42− was converted to pyrophosphate, resulting in an overall increase in a [45]. Pyrophosphate was not observed in CHAp750, however was observed in BC750 using second derivative DRIFTS, consistent with the findings of Young and Holcomb.
Peak broadening, measured by the full width at half the peak maximum (FWHM), is associated with nanometer-scale crystallinity and disorder-induced microstrain and was observed for all materials compared to a perfect hydroxyapatite crystal [22, 35, 69]. FWHM for the (002) and (300) peaks is summarized in Table S1. For BC750 and CHAp750 heating resulted in narrower peaks compared to RGB and CHAp, respectively, consistent with enhanced crystallinity. All materials showed more change in FWHM of (002) than for (300), indicating more ordering in the c-axis direction with pyrolysis, consistent with the findings of others [70]. Rogers presented evidence that microstrain is the dominant mechanism of peak broadening in native bone mineral with the collagen matrix intact [70]. Complete structural refinement according to the Rietveld method would be necessary to separate effects of microstrain and size broadening, and this capability was not available for the present study [39, 69, 70]. Evidence for enhanced ordering in the primary crystallites of BC750 due to pyrolysis was found by DRIFTS second derivative analysis, however, in agreement with the general trends observed in the FWHM of the (002) and (300) peaks and with the findings of Rogers, et al.
Characterization of Carbonaceous Phase
The impacts of the carbonaceous phase of bone char on heavy metals removal has not been well-characterized and further study is certainly warranted to determine the maximum capacity and associated mechanisms by which it adsorbs particularly Cu2+. Insight into the surface chemistry of this phase can help guide any future work. Aromatic carbon, amide, amine, and hydroxyl groups have strong IR intensity associated with characteristic vibrational modes, but these often overlap with vibrational modes associated with the mineral phase of bone and bone char. Therefore, in the present work, functional groups in the carbonaceous phase of BC750 were characterized using demineralized BC750 (D-BC750).
The presence of collagen chains and carbohydrates in raw bone tissue indicates that Maillard-type reactions are likely to occur in the early stages of pyrolysis. Maillard reaction products from glycine/glucose reaction up to 300 °C were observed by others to include nitrogen heterocycles with amine, carbonyl, and methyl functional groups [71]. The carbonaceous phase of pyrolyzed bone has been investigated for electrochemical applications because of properties endowed by the presence of nitrogen-doped aromatic clusters [72, 73]. Nitrogen in amine or amide groups (from collagen) and in aromatic compounds both have important implications for Cu2+ adsorption. For instance, Huang, et al. synthesized novel amine-functionalized Fe3O4 nanoparticles to remove Cu2+ from aqueous solution [74]. More recently, similar synergistic effects were observed where pyrolyzed bone was found to adsorb complexes of tetracycline and Cu2+ from aqueous solution [75]. In addition to amine and amide groups, aromatic nitrogen could also be involved in Cu2+ adsorption as indicated by the strong performance of 1,10-phenanthroline in Cu-binding applications [75,76,77,78]. High nitrogen content is known to favor increased clustering of aromatic rings, which promote the formation of a porous, thermally stable macrostructure [79, 80].
DRIFTS Characterization of Carbonaceous Phase
The DRIFTS spectra of RBG, BC750, and D-BC750 are shown in Fig. 8. The spectrum of RGB is dominated by the amide 1° and 2° bands and biological apatite peaks with some potential amide 3° character around 1220 cm−1. Contribution from the acid phosphate P=O stretch is also likely in that same region [54, 81]. Collagen amide components are apparent in the N–H stretch region between 3300 and 3000 cm−1 and the fingerprint region, with prominent peaks there due to –C=O stretch at \(\sim\) 1730 cm−1, and 1°, 2°, and 3° amide –C–N– stretches centered at \(\sim\) 1640 cm−1, \(\sim\) 1550 cm−1, and \(\sim\) 1220 cm−1, respectively. Raw bones were initially boiled to remove soft tissue, so these combined C-N and N–H modes correlate with observed positions for thermally denatured type I collagen [81].

DRIFTS spectra for RGB, BC750, and D-BC750. Despite pyrolysis, some amide and/or amine character of bone collagen is retained, most apparent in D-BC750. This could have strong implications for Cu(II) removal
Despite high temperature pyrolysis and acid leaching, some collagen amide character is still visible in the spectra of BC750 and D-BC750, respectively, apparent in the N–H stretch modes between 3400 and 2700 cm−1 and amide band at 1700–1500 cm−1. For BC750 the intensity of the broad water band between 3500 and 2700 cm−1 is significantly lower than in RGB indicating limited adsorption of atmospheric H2O. For D-B750 this band is practically absent indicating hydrophobicity. A prominent shoulder is visible for BC750 at 3630 cm−1 which is also apparent in D-BC750. In D-BC750, this band is asymmetric with the maximum at \(\sim\) 3625 cm−1 and residual intensity toward lower wavenumbers. The position above 3600 cm−1 is indicative of a free O–H stretch. The phenolic free O–H stretch, absent of hydrogen bonding or confinement effects, is observed at \(\sim\) 3670 cm−1, so decreased frequency signifies this group is involved in hydrogen bonding [54, 82]. Intensity in the 3400–2700 cm−1 region is primarily attributed to N–H stretches, with broadening attributed to heterogeneity in the chemical environment of these groups. The presence of nitrogen heterocycles containing amine functionalities, particularly 5-membered rings which have been observed in Maillard reactions of amino acids, would extend this band well into the 2800 cm−1 region, which is observed in Fig. 8 for D-BC750 [54, 71].
A small peak at \(\sim\) 2200 cm−1 in the spectra for BC750 may contain components of v3 PO43− peak overtones. Because this band persisted following demineralization with a slight change in shape and position, we attribute some intensity here to v3 PO43− band overtone, and some intensity to –C\(\equiv\)N groups in the carbonaceous phase [83].
The fingerprint region of BC750 contains overlapping bands associated with mineral and carbonaceous phases as well, so D-BC750 is used in the following discussion for that region. Some residual amide character is apparent between 1700 and 1600 cm−1 contributions from the C=O stretch at 1650 cm−1 and NH2 scissor modes at around 1620 cm−1. Contributions to intensity from aromatic carbon are apparent in a major peak at \(\sim\) 1580 cm−1 and between 1500 and 1000 cm−1. At least some contribution to the intensity between 1500 and 1000 cm−1 is attributed to epoxide or ether groups as well, common in lignocellulose-based biochar. Similarity between the spectra of D-BC750 and wood-based biochar in this region indicate similar structures may be present (Fig. S5). Intensity in the 1500 and 1000 cm−1 range from sp2 and sp3 C–H bend modes are also present at \(\sim\) 1400 cm−1 and \(\sim\) 1020 cm−1, respectively [54, 80].
The spectrum for D-BC750 is compared to that of a wood-based biochar in Fig. S5 to highlight the unique functional group chemistry on the bone char related to feedstock. BC750 contains –OH and amine and/or amide functional groups, whereas biochar contains primarily carbonyl and ether groups in addition to C–H attached to aromatic and sp3 carbons.
Raman Spectroscopy of Carbonaceous Phase
In the case of B750, the mineral phase contains many Raman active modes, but these were obscured by strong scattering from carbonaceous phases at the low laser power necessitated by the need to avoid thermally damaging the sample. Therefore, Raman spectroscopy was only used in this study for characterization of the carbonaceous phase.
The Raman spectrum for BC750 and D-BC750 exhibit the characteristic shape for amorphous carbons containing defected sp2 and sp3 hybridized carbon. Two characteristic peaks are present in D-BC750: G and D. The so-called G peak originates from the E2g Raman allowed optical phonon mode at the Brillouin zone center. The G peak is present in all polyconjugated carbons and is related to the bond stretching of sp2 hybridized carbon pairs. The D peak, only observed for aromatic clusters. The D peak is due to ring-breathing mode of the A1g optical phonon at K, which is activated by relaxation of the fundamental selection rule associated with the presence of defected clusters of aromatic rings [84]. Different types of defects can activate the D peak, for instance edges of ordered sp2 clusters, sp3 hybridized species, or heteroatoms [84]. Ferrari and Robertson, et al. proposed a model by which to characterize amorphous carbon films which is appropriate for amorphous pyrolysis chars because it accounts for the well-known non-graphitizing thermal behavior, fine size distribution of sp2 and sp3 domains, the presence of 5-, 6-, 7- membered aromatic rings, linear conjugated chains, hydrogen and heteroatom content of biochars [85,86,87,88,89,90]. According to this model, two peaks can satisfactorily fit the first order region, between 1100 and 1700 cm−1, and the parameters for these fitted peaks are related to the average degree of order and aromaticity in the material. In the following discussion, the biochar from Sect. 3.3.1 was included to highlight the enhanced aromatic ring clustering in bone char, possibly catalyzed by the mineral phase.
The two-peak model includes a Breit–Wigner–Fano (BWF) function (Eq. S.4) for the G peak, a single Lorentzian function for the D peak and a linear background. The BWF function is appropriate for amorphous carbon because the asymmetric shape allows for a better fit of the “valley” region between the D and G peaks without the inclusion of additional peaks [27, 83]. There is evidence that this line-shape has a physical origin as well [91]. Further description of the BWF function is found in Supplementary material Section S.4.
Raw data for the Raman spectra of D-BC750 is shown in Fig. 9, plotted together with the fitted curve, D and G bands, and the linear background. The results of fitting for BC750, D-BC750 and biochar (for comparison) are summarized in Table 4, where reported values represent the arithmetic mean value for at least three measurements taken at different points on the sample, with standard error reported. Raman fit for BC750 is shown in Fig. S8.

Raman fits for D-BC750 and a comparative wood-based biochar using the two-peak model according to Ferrari and Robertson which implements a Lorentzian line shape for the D peak and BWF line shape for the G peak. The benefit of the Ferrari-Robertson model is a reasonable fit, including the high-intensity valley between the D and G peaks, with minimized risk of “over-fitting”
The general picture of the carbonaceous phase of bone char from the Ferrari-Robertson model is a highly defected, amorphous carbon consisting of small ordered clusters of aromatic rings and the presence of conjugated linear chains, evidenced by a G position > 1600 cm−1 [92]. I(D)/I(G) lies between 0.73 and 0.66, which indicates clustering of aromatic rings on the order of 1–2 nm, and the FWHM(G) lies at approximately 89 cm−1, indicating relatively low ordering within all sp2 phases [93].
The Raman spectrum of biochar, in comparison, has a lower I(D)/I(G) at 0.48, slightly lower G peak position at 1601 cm−1, and smaller FWHM(G) at 68.2 cm−1. Comparing the results of the Raman fits for D-BC750 and biochar, it is apparent from I(D)/I(G) that D-BC750 contains larger clusters of aromatic rings but the distance between defects in these rings is shorter, possibly due to higher nitrogen heteroatom content of aromatic rings in bone char [80].
Nitrogen groups, for instance amine and amide functional groups, pyrrolic and pyridinic nitrogen groups, may have strong affinity for Cu2+ through complexation or weak acid/base interaction [94,95,96]. The presence of amine functional groups from Sect. 3.3.1. suggests that the carbonaceous phase of bone char is active in Cu2+ uptake [97]. Further study into the mechanisms and performance for Cu2+ uptake by the carbonaceous phase of bone char is recommended to determine the uptake capacity for this phase.
Conclusions
The results of this study point to several fundamental differences in chemistry and structure between bone char and carbonated hydroxyapatite which are likely to account for higher maximum removal capacity of aqueous metals, and particularly Cu2+ by bone char. Despite both materials having the same hydroxyapatite crystalline structure, bone mineral and synthetic carbonated apatite have different levels of substitutional species, including structural water and carbonate, known to influence crystallinity, solubility, and acid-buffering capacity. The presence of high quantities of A-type carbonate in addition to B-type and L-type, coupled with a distinct lack of structural OH− in bone char, is indicative of persistent lattice disorder even after heat treatment. These results indicate AB-type carbonated apatite with low OH− is desirable for metals removal applications.
Non-stoichiometric apatite and non-apatite phases were observed in bone char after pyrolysis, including calcium pyrophosphate (CaP2O7) and brushite (CaHPO42−). The small size of these alternate phases indicates very localized effects, and potentially a lower degree of charge compensation with higher reactivity. In contrast, there is no indication of pyrophosphate or other secondary phase in CHAp, even after pyrolysis. These phases are likely to enhance Cu2+ uptake by coprecipitation, dissolution/precipitation and cation exchange.
The carbonaceous phase of pyrolyzed bone can be considered “amorphous carbon”, which consists of distinct clusters of sp2 and sp3 hybridized carbon, including aromatic rings, olefinic chains and dense tetrahedrally coordinated sp3 carbon, as well as non-trivial quantities of volatiles and polymeric structures. The presence of amine, amide and oxygenated functional groups and heteroatom nitrogen indicates that the carbonaceous phase is likely to play a role in Cu2+ removal. We propose that further experimentation with aqueous Cu2+ adsorption by demineralized bone char would reveal more about the role of the carbonaceous phase in this application, and that there is potential synergy between carbonaceous and mineral phases of bone char driving enhanced metals removal.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available due to privacy restrictions, but are available from the corresponding author on reasonable request.
References
Barrett, E.B., Brown, J.M., Oleck, S.M.: Some granular carbonaceous adsorbents for sugar refining a study of bone char replacements based on hydroxyapatite. Ind. Eng. Chem. 17, 5 (2021)
Barrett, E.P., Joyner, L.G., Halenda, P.P.: Granular adsorbents for sugar refining. Some factors affecting porosity and activity in service. Ind. Eng. Chem. 44, 1827–1833 (2002). https://doi.org/10.1021/IE50512A034
Wayne, T.B.: Influence of absorption spectra of techinical sugar products on the decolorizing effeciency of bone char. Ind. Eng. Chem. 18, 847–854 (2002). https://doi.org/10.1021/IE50200A028
Bhargava, D.S., Killedar, S.D.J.: Batch studies of water defluoridation using fishbone charcoal. Res. J. Water Pollut. Control Fed. 63, 848–858 (1991)
Betts, A.R., Chen, N., Hamilton, J.G., Peak, D.: Rates and mechanisms of Zn 2+ adsorption on a meat and bonemeal biochar. Environ. Sci. Technol. 47, 14350 (2013). https://doi.org/10.1021/es4032198
Deydier, E., Guilet, R., Sharrock, P.: Beneficial use of meat and bone meal combustion residue: “an efficient low cost material to remove lead from aqueous effluent.” J. Hazard. Mater. 101, 55–64 (2003). https://doi.org/10.1016/S0304-3894(03)00137-7
Chen, Y.-N., Chai, L.-Y., Shu, Y.-D.: Study of arsenic(V) adsorption on bone char from aqueous solution. J. Hazard. Mater. 160, 168–172 (2008). https://doi.org/10.1016/j.jhazmat.2008.02.120
Ko, D.C.K., Cheung, C.W., Choy, K.K.H., Porter, J.F., McKay, G.: Sorption equilibria of metal ions on bone char. Chemosphere 54, 273–281 (2004). https://doi.org/10.1016/j.chemosphere.2003.08.004
Wang, H., Luo, P.: Preparation, kinetics, and adsorption mechanism study of microcrystalline cellulose-modified bone char as an efficient Pb (II) adsorbent. Water Air Soil Pollut. (2020). https://doi.org/10.1007/s11270-020-04687-8
Wang, M., Liu, Y., Yao, Y., Han, L., Liu, X.: Comparative evaluation of bone chars derived from bovine parts: physicochemical properties and copper sorption behavior. Sci. Total Environ. 700, 134470 (2020). https://doi.org/10.1016/J.SCITOTENV.2019.134470
Sellaoui, L., Ileana-Mendoza-Castillo, D., Elizabeth-Reynel-Ávila, H., Bonilla-Petriciolet, A., Ben-Lamine, A.: A new statistical physics model for the ternary adsorption of Cu 2+ , Cd 2+ and Zn 2+ ions on bone char: Experimental investigation and simulations • Development of a new statistical physics model. • Interpretation of ternary adsorption of copper (Cu 2+ ), cadmium (Cd 2+ ) and zinc (Zn 2+ ) on bone char. • Development of new microscopic interpretations of adsorption mechanisms. Chem. Eng. J. 343, 544 (2018). https://doi.org/10.1016/j.cej.2018.03.033
Fernane, F., Mecherri, M.O., Sharrock, P., Fiallo, M., Sipos, R.: Hydroxyapatite interactions with copper complexes. Mater. Sci. Eng. C 30, 1060–1064 (2010). https://doi.org/10.1016/J.MSEC.2010.05.010
Núñez, D., Serrano, J.A., Mancisidor, A., Elgueta, E., Varaprasad, K., Oyarzún, P., Cáceres, R., Ide, W., Rivas, B.L.: Heavy metal removal from aqueous systems using hydroxyapatite nanocrystals derived from clam shells. RSC Adv. 9, 22883–22890 (2019). https://doi.org/10.1039/C9RA04198B
Hashimoto, Y., Sato, T.: Removal of aqueous lead by poorly-crystalline hydroxyapatites. Chemosphere 69, 1775–1782 (2007). https://doi.org/10.1016/J.CHEMOSPHERE.2007.05.055
Nzihou, A., Sharrock, P.: Role of phosphate in the remediation and reuse of heavy metal polluted wastes and sites. Waste Biomass Valoriz. 1, 163–174 (2010). https://doi.org/10.1007/S12649-009-9006-X
Xu, Y., Schwartz, F.W., Traína, S.J.: Sorption of Zn2+ and Cd2+ on hydroxyapatite surfaces. Environ. Sci. Technol. 28, 1472–1480 (1994)
Suzuki, T., Hatsushika, T., Hayakawa, Y.: Synthetic hydroxyapatites employed as inorganic cation-exchangers. J. Chem. Soc. 77, 1059–1062 (1981). https://doi.org/10.1039/F19817701059
Barrett, E.P.: Trends in the development of granular adsorbents for sugar refining. Adv. Carbohydr. Chem. 6, 205–230 (1951). https://doi.org/10.1016/S0096-5332(08)60068-5
Bosch, K., Freebourn, A., Parker, K., Scherman, M., Zodrow, K.R., Hutchins, D.: Biochar for enhanced surface water quality. In: Proceedings of the 2021 Waste-management Education Research Conference (WERC), pp. 1–9 (2021). https://doi.org/10.1109/WERC52047.2021.9477543
Chen, Y., Zhang, X., Chen, W., Yang, H., Chen, H.: The structure evolution of biochar from biomass pyrolysis and its correlation with gas pollutant adsorption performance. Biores. Technol. 246, 101–109 (2017). https://doi.org/10.1016/j.biortech.2017.08.138
Rouquerol, J., Rouquerol, F., Llewellyn, P., Maurin, G., Sing, K.S.W.: Adsorption by Powders and Porous Solids: Principles, Methodology and Applications, 2nd edn. Academic Press, Cambridge (2013)
Vallet-Regí, M., González-Calbet, J.M.: Calcium phosphates as substitution of bone tissues. Prog. Solid State Chem. 32, 1–31 (2004)
Niu, L., Shen, C., Yan, L., Zhang, J., Lin, Y., Gong, Y., Li, C., Sun, C.Q., Xu, S.: Waste bones derived nitrogen-doped carbon with high micropore ratio towards supercapacitor applications. J. Colloid Interface Sci. (2019). https://doi.org/10.1016/j.jcis.2019.03.097
Fowler, B.O., Moreno, E.C., Brown, W.E.: Infra-red spectra of hydroxyapatite, octacalcium phosphate and pyrolysed octacalcium phosphate. Arch. Oral Biol. 11, 477–492 (1966). https://doi.org/10.1016/0003-9969(66)90154-3
Haberko, K., Bućko, M.M., Brzezińska-Miecznik, J., Haberko, M., Mozgawa, W., Panz, T., Pyda, A., Zarębski, J.: Natural hydroxyapatite: its behaviour during heat treatment. J. Eur. Ceram. Soc. 26, 537–542 (2006). https://doi.org/10.1016/j.jeurceramsoc.2005.07.033
Szymański, G.S., Karpiński, Z., Biniak, S., Świa̧tkowski, A.: The effect of the gradual thermal decomposition of surface oxygen species on the chemical and catalytic properties of oxidized activated carbon. Carbon N Y. 40, 2627–2639 (2002). https://doi.org/10.1016/S0008-6223(02)00188-4
Conway, N.M.J., Ferrari, A.C., Flewitt, A.J., Robertson, J., Milne, W.I., Tagliaferro, A., Beyer, W.: Defect and disorder reduction by annealing in hydrogenated tetrahedral amorphous carbon. Diam. Relat. Mater. 9, 765–770 (2000). https://doi.org/10.1016/S0925-9635(99)00271-X
Bounouh, Y., Thèye, M.L., Dehbi-Alaoui, A., Matthews, A., Stoquert, J.P.: Influence of annealing on the hydrogen bonding and the microstructure of diamondlike and polymerlike hydrogenated amorphous carbon films. Phys. Rev. B 51, 9597 (1995). https://doi.org/10.1103/PhysRevB.51.9597
Ivanova, T.I., Frank-Kamenetskaya, O.V., Koltsov, A.B., Ugolkov, V.L.: Crystal structure of calcium-deficient carbonated hydroxyapatite thermal decomposition. J. Solid State Chem. 160, 340–349 (2001). https://doi.org/10.1006/JSSC.2000.9238
Pasteris, J.D., Wopenka, B., Freeman, J.J., Rogers, K., Valsami-Jones, E., van der Houwen, J.A.M., Silva, M.J.: Lack of OH in nanocrystalline apatite as a function of degree of atomic order: implications for bone and biomaterials. Biomaterials 25, 229–238 (2004). https://doi.org/10.1016/S0142-9612(03)00487-3
Davies, E., Müller, K.H., Wong, W.C., Pickard, C.J., Reid, D.G., Skepper, J.N., Duer, M.J.: Citrate bridges between mineral platelets in bone. Proc. Natl. Acad. Sci. 111, E1354–E1363 (2014). https://doi.org/10.1073/PNAS.1315080111
Wise, E.R., Maltsev, S., Davies, M.E., Duer, M.J., Jaeger, C., Loveridge, N., Murray, R.C., Reid, D.G.: The organic−mineral interface in bone is predominantly polysaccharide. Chem. Mater. 19, 5055–5057 (2007). https://doi.org/10.1021/CM702054C
Ibsen, C.J.S., Chernyshov, D., Birkedal, H.: Apatite formation from amorphous calcium phosphate and mixed amorphous calcium phosphate/amorphous calcium carbonate. Chem. Eur. J. 22, 12347–12357 (2016). https://doi.org/10.1002/CHEM.201601280
Wang, Y., von Euw, S., Fernandes, F.M., Cassaignon, S., Selmane, M., Laurent, G., Pehau-Arnaudet, G., Coelho, C., Bonhomme-Coury, L., Giraud-Guille, M.-M., Babonneau, F., Azaïs, T., Nassif, N.: Water-mediated structuring of bone apatite. Nat. Mater. 2013(12), 1144–1153 (2013). https://doi.org/10.1038/nmat3787
Elliott, J.C.: Calcium phosphate biominerals. Rev. Miner. Geochem. 48, 427–453 (2002). https://doi.org/10.2138/RMG.2002.48.11
Wilson, E.E., Awonusi, A., Morris, M.D., Kohn, D.H., Tecklenburg, M.M.J., Beck, L.W.: Three structural roles for water in bone observed by solid-state NMR. Biophys. J . 90, 3722–3731 (2006). https://doi.org/10.1529/BIOPHYSJ.105.070243
Fleet, M.E., Liu, X.: Coupled substitution of type A and B carbonate in sodium-bearing apatite. Biomaterials 28, 916–926 (2007). https://doi.org/10.1016/J.BIOMATERIALS.2006.11.003
Fleet, M.E.: Infrared spectra of carbonate apatites: evidence for a connection between bone mineral and body fluids. Am. Miner. 102, 149–157 (2017). https://doi.org/10.2138/AM-2017-5704
Madupalli, H., Pavan, B., Tecklenburg, M.M.J.: Carbonate substitution in the mineral component of bone: discriminating the structural changes, simultaneously imposed by carbonate in A and B sites of apatite. J. Solid State Chem. 255, 27–35 (2017). https://doi.org/10.1016/j.jssc.2017.07.025
Rey, C., Collins, B., Goehl, T., Dickson, I.R., Glimcher, M.J.: The carbonate environment in bone mineral: a resolution-enhanced fourier transform infrared spectroscopy study. Calcif. Tissue Int. 1989(45), 157–164 (1989). https://doi.org/10.1007/BF02556059
Bouzy, P., O’grady, S., Madupalli, H., Tecklenburg, M., Rogers, K., Palombo, F., Morgan, M.P., Stone, N.: A time-course Raman spectroscopic analysis of spontaneous in vitro microcalcifications in a breast cancer cell line. Lab. Investig. 101, 1267–1280 (2021). https://doi.org/10.1038/s41374-021-00619-0
Deymier, A.C., Nair, A.K., Depalle, B., Qin, Z., Arcot, K., Drouet, C., Yoder, C.H., Buehler, M.J., Thomopoulos, S., Genin, G.M., Pasteris, J.D.: Protein-free formation of bone-like apatite: new insights into the key role of carbonation. Biomaterials 127, 75–88 (2017). https://doi.org/10.1016/J.BIOMATERIALS.2017.02.029
Boskey, A.L.: Mineralization of bones and teeth. Elements 3, 385–391 (2007). https://doi.org/10.2113/GSELEMENTS.3.6.385
Fleet, M.E.: Infrared spectra of carbonate apatites: ν2-Region bands. Biomaterials 30, 1473–1481 (2009). https://doi.org/10.1016/J.BIOMATERIALS.2008.12.007
Young, R.A., Holcomb, D.W.: Calcified tissue international role of acid phosphate in hydroxyapatite lattice expansion*. Calcif. Tissue Int. 36, 60–63 (1984)
Von Euw, S., Wang, Y., Laurent, G., Drouet, C., Babonneau, F., Nassif, N., Azais, T.: Bone mineral: new insights into its chemical composition OPEN. Sci. Rep. (2019). https://doi.org/10.1038/s41598-019-44620-6
Gadaleta, S.J., Paschalis, E.P., Betts, F., Mendelsohn, R., Boskey, A.L.: Fourier transform infrared spectroscopy of the solution-mediated conversion of amorphous calcium phosphate to hydroxyapatite: new correlations between X-ray diffraction and infrared data. Calcif. Tissue Int. 1996(58), 9–16 (1996). https://doi.org/10.1007/BF02509540
Markovic, M., Brownla, W.E.: Octacalcium phosphate carboxylates. 2. Characterization and structural considerations. Chem. Mater. 5, 1406–1416 (1993)
Rey, C., Shimizu, M., Collins, B., Glimcher, M.J.: Resolution-enhanced fourier transform infrared spectroscopy study of the environment of phosphate ions in the early deposits of a solid phase of calcium-phosphate in bone and enamel, and their evolution with age I: Investigations in thev 4 PO4 domain. Calcif. Tissue Int. 1990(46), 384–394 (1990). https://doi.org/10.1007/BF02554969
Rey, C., Miquel, J.L., Facchini, L., Legrand, A.P., Glimcher, M.J.: Hydroxyl groups in bone mineral. Bone 16, 583–586 (1995). https://doi.org/10.1016/8756-3282(95)00101-I
LeGeros, R.Z., Trautz, O.R., Klein, E., LeGeros, J.P.: Two types of carbonate substitution in the apatite structure. Experientia 25, 5–7 (1969). https://doi.org/10.1007/BF01903856
Marques, M.P.M., Mamede, A.P., Vassalo, A.R., Makhoul, C., Cunha, E., Gonçalves, D., Parker, S.F., Batista-De-Carvalho, L.A.E.: Heat-induced bone diagenesis probed by vibrational spectroscopy OPEN. Sci. Rep. (2018). https://doi.org/10.1038/s41598-018-34376-w
Taylor, E.A., Donnelly, E.: Raman and Fourier transform infrared imaging for characterization of bone material properties. Bone 139, 115490 (2020). https://doi.org/10.1016/J.BONE.2020.115490
Lin-Vien, D., Colthup, N.B., Fateley, W.G., Grasselli, J.G.: The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, p. 503. Elsevier, Amsterdam (1991)
Gibson, I.R., Bonfield, W.: Novel synthesis and characterization of an AB-type carbonate-substituted hydroxyapatite. J. Biomed. Mater. Res. 59, 697–708 (2002). https://doi.org/10.1002/JBM.10044
Paschalis, E.P., Gamsjaeger, S., Klaushofer, K.: Vibrational spectroscopic techniques to assess bone quality. Osteoporos. Int. 28, 2275 (2017). https://doi.org/10.1007/s00198-017-4019-y
Courtland, H.-W., Nasser, P., Goldstone, A.B., Spevak, L., Boskey, A.L., Jepsen, K.J.: Fourier transform infrared imaging microspectroscopy and tissue-level mechanical testing reveal intraspecies variation in mouse bone mineral and matrix composition. Calcif. Tissue Int. 2008(83), 342–353 (2008). https://doi.org/10.1007/S00223-008-9176-8
Pleshko, N., Boskey, A., Mendelsohn, R.: Novel infrared spectroscopic method for the determination of crystallinity of hydroxyapatite minerals. Biophys. J. 60, 786–793 (1991). https://doi.org/10.1016/S0006-3495(91)82113-0
Boskey, A.L.: Bone composition: relationship to bone fragility and antiosteoporotic drug effects. BoneKEy Rep. (2013). https://doi.org/10.1038/BONEKEY.2013.181
Spevak, L., Flach, C.R., Hunter, T., Mendelsohn, R., Boskey, A.: Fourier transform infrared spectroscopic imaging parameters describing acid phosphate substitution in biologic hydroxyapatite. Calcif. Tissue Int. 2013(92), 418–428 (2013). https://doi.org/10.1007/S00223-013-9695-9
Rey, C., Combes, C., Drouet, C., Sfihi, H., Barroug, A.: Physico-chemical properties of nanocrystalline apatites: implications for biominerals and biomaterials. Mater. Sci. Eng. C 27, 198–205 (2007). https://doi.org/10.1016/J.MSEC.2006.05.015
Wu, V.M., Uskoković, V.: Is there a relationship between solubility and resorbability of different calcium phosphate phases in vitro? Biochim. Biophys Acta. 1860, 2157 (2016). https://doi.org/10.1016/J.BBAGEN.2016.05.022
Sugiyama, S., Ichii, T., Fujisawa, M., Kawashiro, K., Tomida, T., Shigemoto, N., Hayashi, H.: Heavy metal immobilization in aqueous solution using calcium phosphate and calcium hydrogen phosphates. J. Colloid Interface Sci. 259, 408–410 (2003). https://doi.org/10.1016/S0021-9797(02)00211-4
Liao, C.-J., Lin, F.-H., Chen, K.-S., Sun, J.-S.: Thermal decomposition and reconstitution of hydroxyapatite in air atmosphere. Biomaterials 20, 1807–1813 (1999). https://doi.org/10.1016/S0142-9612(99)00076-9
Etok, S.E., Valsami-Jones, E., Wess, T.J., Hiller, J.C., Maxwell, C.A., Rogers, K.D., Manning, D.A.C., White, M.L., Lopez-Capel, E., Collins, M.J., Buckley, M., Penkman, K.E.H., Woodgate, S.L.: Structural and chemical changes of thermally treated bone apatite. J. Mater. Sci. 42, 9807–9816 (2007). https://doi.org/10.1007/S10853-007-1993-Z/FIGURES/11
Barralet, J., Best, S., Bonfield, W.: Carbonate substitution in precipitated hydroxyapatite: an investigation into the effects of reaction temperature and bicarbonate ion concentration. Jpn. Soc. Biomater Aust Soc Biomater. (1998). https://doi.org/10.1002/(SICI)1097-4636(199807)41:1
Kumar, R., Prakash, K.H., Cheang, P., Khor, K.A.: Temperature driven morphological changes of chemically precipitated hydroxyapatite nanoparticles. Langmuir 20, 5196–5200 (2004). https://doi.org/10.1021/LA049304F
Young, R.A., Bartlett, M.L., Mackie, P.E.: Reversible high temperature exchange of carbonate and hydroxyl ions in tooth enamel and synthetic hydroxyapatite. J. Biol. Phys. 9, 1–26 (1981)
Londoño-Restrepo, S.M., Jeronimo-Cruz, R., Millán-Malo, B.M., Rivera-Muñoz, E.M., Rodriguez-García, M.E.: Effect of the nano crystal size on the x-ray diffraction patterns of biogenic hydroxyapatite from human, bovine, and porcine bones. Sci. Rep. 2019(9), 1–12 (2019). https://doi.org/10.1038/s41598-019-42269-9
Rogers, K.D., Daniels, P.: An X-ray diffraction study of the effects of heat treatment on bone mineral microstructure. Biomaterials 23, 2577–2585 (2002). https://doi.org/10.1016/S0142-9612(01)00395-7
Tehrani, K.A., Kersiene, M., Adams, A., Venskutonis, R., De Kimpe, N.: Thermal degradation studies of glucose/glycine melanoidins. J. Agric. Food Chem. 50, 4062–4068 (2002). https://doi.org/10.1021/JF0116247
Goodman, P.A., Li, H., Gao, Y., Lu, Y.F., Stenger-Smith, J.D., Redepenning, J.: Preparation and characterization of high surface area, high porosity carbon monoliths from pyrolyzed bovine bone and their performance as supercapacitor electrodes. Carbon N Y. 55, 291–298 (2013). https://doi.org/10.1016/J.CARBON.2012.12.066
Niu, J., Shao, R., Liu, M., Liang, J., Zhang, Z., Dou, M., Huang, Y., Wang, F.: Porous carbon electrodes with battery-capacitive storage features for high performance Li-ion capacitors. Energy Storage Mater. 12, 145–152 (2018). https://doi.org/10.1016/J.ENSM.2017.12.012
Huang, S.-H., Chen, D.-H.: Rapid removal of heavy metal cations and anions from aqueous solutions by an amino-functionalized magnetic nano-adsorbent. J. Hazard. Mater. 163, 174–179 (2009). https://doi.org/10.1016/j.jhazmat.2008.06.075
Zhu, Z., Zhang, M., Wang, W., Zhou, Q., Liu, F.: Efficient and synergistic removal of tetracycline and Cu(II) using novel magnetic multi-amine resins. Sci. Rep. 2018(8), 1–9 (2018). https://doi.org/10.1038/s41598-018-23205-9
Zhang, J., Anson, F.C.: Electrocatalysts for the reduction of O2 and H2O2 based on complexes of Cu(II) with the strongly adsorbing 2,9-dimethyl-1,10-phenanthroline ligand. Electrochim. Acta 38, 2423–2429 (1993). https://doi.org/10.1016/0013-4686(93)85111-B
Hirohama, T., Kuranuki, Y., Ebina, E., Sugizaki, T., Arii, H., Chikira, M., Selvi, P.T., Palaniandavar, M.: Copper(II) complexes of 1,10-phenanthroline-derived ligands: studies on DNA binding properties and nuclease activity. J. Inorg. Biochem. 99, 1205–1219 (2005). https://doi.org/10.1016/J.JINORGBIO.2005.02.020
Chaiyo, S., Chailapakul, O., Sakai, T., Teshima, N., Siangproh, W.: Highly sensitive determination of trace copper in food by adsorptive stripping voltammetry in the presence of 1,10-phenanthroline. Talanta 108, 1–6 (2013). https://doi.org/10.1016/J.TALANTA.2013.02.031
Rodil, S.E., Milne, W.I., Robertson, J., Brown, L.M.: Maximized sp 3 bonding in carbon nitride phases. Appl. Phys. Lett. 77, 1458–1460 (2000)
Ferrari, A.C., Rodil, S.E., Robertson, J.: Interpretation of infrared and Raman spectra of amorphous carbon nitrides. Phys. Rev. B 67, 155306 (2003). https://doi.org/10.1103/PhysRevB.67.155306
Stani, C., Vaccari, L., Mitri, E., Birarda, G.: FTIR investigation of the secondary structure of type I collagen: New insight into the amide III band. Spectrochim. Acta A. 229, 118006 (2020). https://doi.org/10.1016/J.SAA.2019.118006
Bacon, J.F., van der Maas, J.H.: The identification of structural fragments of enolic monoterpenes, enolic pheromones, and related natural products, from OH stretching vibration frequencies and relative intensities. Can. J. Chem. 67, 250–256 (2011). https://doi.org/10.1139/V89-041
Ferrari, A.C., Robertson, J.: Interpretation of Raman spectra of disordered and amorphous carbon. Phys. Rev. B 61, 14095–14107 (2000). https://doi.org/10.1103/PhysRevB.61.14095
Ferrari, A.C., Basko, D.M.: Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 8, 235–246 (2013). https://doi.org/10.1038/NNANO.2013.46
Bourke, J., Manley-Harris, M., Fushimi, C., Dowaki, K., Nunoura, T., Antal, M.J.: Do all carbonized charcoals have the same chemical structure? 2. A model of the chemical structure of carbonized charcoal. Ind. Eng. Chem. Res. 46, 5954–5967 (2007). https://doi.org/10.1021/ie070415u
Mészáros, E., Jakab, E., Várhegyi, G., Bourke, J., Manley-Harris, M., Nunoura, T., Antal, M.J.: Do all carbonized charcoals have the same chemical structure? 1. Implications of thermogravimetry-mass spectrometry measurements. Ind. Eng. Chem. Res. 46, 5943–5953 (2007). https://doi.org/10.1021/ie0615842
Xiao, X., Chen, Z., Chen, B.: H/C atomic ratio as a smart linkage between pyrolytic temperatures, aromatic clusters and sorption properties of biochars derived from diverse precursory materials. Sci. Rep. 6, 22644 (2016). https://doi.org/10.1038/srep22644
Harris, P.J.F.: Structure of non-graphitising carbons. Int. Mater. Rev. 42, 206–218 (1997). https://doi.org/10.1179/imr.1997.42.5.206
Franklin, R.: Crystallite growth in graphitizing and non-graphitizing carbons. Proc. R. Soc. Lond. Ser. A. 209, 196–218 (1951). https://doi.org/10.1098/rspa.1951.0197
Harris, P.J.F., Liu, Z., Suenaga, K.: Imaging the atomic structure of activated carbon. J. Phys. 20, 362201 (2008)
Hasdeo, E.H., Nugraha, A.R.T., Dresselhaus, M.S., Saito, R.: Breit-Wigner-Fano line shapes in Raman spectra of graphene. Phys. Rev. B 90, 245140 (2014). https://doi.org/10.1103/PhysRevB.90.245140
Ferrari, A.C.: Determination of bonding in diamond-like carbon by Raman spectroscopy. Diam. Relat. Mater. 11, 1053–1061 (2002). https://doi.org/10.1016/S0925-9635(01)00730-0
Ferrari, A.C., Robertson, J.: Raman spectroscopy of amorphous, nanostructured, diamond–like carbon, and nanodiamond. Philos. Trans. R. Soc. Lond. Ser. A. 362, 2477–2512 (2004). https://doi.org/10.1098/rsta.2004.1452
Ji, Q., Hu, C., Liu, H., Qu, J.: Development of nitrogen-doped carbon for selective metal ion capture. Chem. Eng. J. 350, 608–615 (2018). https://doi.org/10.1016/J.CEJ.2018.06.018
Modi, A., Bhaduri, B., Verma, N.: Facile one-step synthesis of nitrogen-doped carbon nanofibers for the removal of potentially toxic metals from water. Ind. Eng. Chem. Res. (2015). https://doi.org/10.1021/ie505016d
Yuan, X., An, N., Zhu, Z., Sun, H., Zheng, J., Jia, M., Lu, C., Zhang, W., Liu, N.: Hierarchically porous nitrogen-doped carbon materials as efficient adsorbents for removal of heavy metal ions. Process Saf. Environ. Prot. 119, 320–329 (2018). https://doi.org/10.1016/J.PSEP.2018.08.012
Majdoub, M., Amedlous, A., Anfar, Z., Jada, A., el Alem, N.: Engineering of amine-based binding chemistry on functionalized graphene oxide/alginate hybrids for simultaneous and efficient removal of trace heavy metals: towards drinking water. J. Colloid Interface Sci. 589, 511–524 (2021). https://doi.org/10.1016/J.JCIS.2021.01.029
Acknowledgements
The authors thank Combat Capabilities Development Command Army Research Laboratory for funding this project. The authors thank Gary Wyss and the Center for Advanced Materials Processing (CAMP) for access to instrumentation. Special thanks to Kristopher Bosch and Dr. David Hutchins for materials and X-ray diffraction, and to Dr. Cristina Stephanescu and Reyla Williams for additional X-ray diffraction. The authors acknowledge Dr. Wellington Muchero at Oak Ridge National Laboratory for generously donating the feedstock used in biochar preparation, and Dynalene Laboratory Services for N2 volumetric adsorption measurements and SSA/PSD calculations.
Funding
Research was sponsored by the Combat Capabilities Development Command Army Research Laboratory and was accomplished under Cooperative Agreement Number W911NF-20–2-0163. The views and conclusions contained in this document are those of the authors and should not be interpreted as representing the official policies, either expressed or implied, of the Combat Capabilities Development Command Army Research Laboratory or the U.S. Government.
Author information
Authors and Affiliations
Contributions
All Authors contributed to manuscript preparation and have read and approved the final manuscript. JEM: Conceptualization, Methodology, Formal Analysis, Investigation, Data Curation, Writing—Original Draft, Visualization.: DP-C: Conceptualization, Methodology, Validation, Resources, Writing—Review and Editing, Supervision, Funding Acquisition, Project Administration.: RLD: Resources, Writing—Review and Editing, Funding Acquisition, Project Administration.: JK: Methodology, Validation, Resources, Writing—Review and Editing, Supervision.
Corresponding author
Ethics declarations
Competing interests
The authors have no competing interests to declare that are relevant to the content of this article.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Muretta, J.E., Prieto-Centurion, D., LaDouceur, R. et al. Unique Chemistry and Structure of Pyrolyzed Bovine Bone for Enhanced Aqueous Metals Adsorption. Waste Biomass Valor 14, 703–722 (2023). https://doi.org/10.1007/s12649-022-01895-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12649-022-01895-7