
Abstract
Pectin, a naturally occurring biopolymer has been found to have increasing applications in the pharmaceutical and biotechnology industry. Sugars with their three-dimensional structures are important for many biological functions. We report preparation of negatively charged pectin nanoparticles encapsulating paclitaxel, a broad-spectrum anticancer drug for possible therapeutic applications. The mean diameter of the nanoparticles was ~300–350 nm with an encapsulation efficiency ~17 %. The TEM studies indicated that the particles were spherical in shape and their sizes are in unison with the DLS size spectra. The surface charge of pectin polymer was −5 mV and of nanoparticles were ~ −32 mV. The enhanced surface charge shows greater stability. The high electrophoretic mobility of ~3.5 to 1.5 μmcm/Vs confirmed the nano-size of particles. We compared the cytotoxic effect of paclitaxel (Pax) per se, pectin nanoparticles (PPN), and pectin chains on Hep G2, hepatic carcinoma cell line. Dose-dependent cytotoxicity was observed, wherein ~21.7 ± 3.2 % cytotoxicity was observed by PPN, but Pax per se showed ~55.6 ± 3.5 % cytotoxicity in a 72-h assay. The pharmacokinetics and biodistribution studies on Balb/c mice indicated that the nanoparticles had prolonged plasma retention of the drug with major accumulation in liver tissue after an i.v. tail vein injection of 20 mg/kg drug. The rank order of concentration are as follows, i.e., liver > kidney > lung > spleen for the PPN and spleen > liver > kidney ≥ lung for Pax per se. The in vitro studies clearly indicated that the efficacy of the drug was not compromised by encapsulation, making it a good candidate to deliver biopharmaceuticals. Nanoparticles produced free radicals in the free cell system and this ability caused oxidative stress, which may give rise to inflammation, cell destruction, and genotoxicity. Thus, the results obtained in this study holds great promise for pectin nanoparticles to be exploited for passive delivery of paclitaxel to tumor tissues, in particular, liver cancers.
Similar content being viewed by others
1 Introduction
Pectin has gained wide acceptance as matrix materials because of their inherent biocompatibility, polyfunctionality, biodegradable nature, flexible structural networks, non-toxicity, and low production costs (May 1990). The physiological properties, functionality, and application of pectin are dependent on the degree of methoxylation (Thibault et al. 1993; Garnier et al. 1993). Pectin is used in food industry as a thickening agent, a gelling agent and a colloidal stabilizer, and a carrier and coating material in pharmaceutical sciences (Endress 1991). Pectin is highly hydrophilic in nature, and hydrophilic matrices are generally used for the oral drug delivery and for the preparation of modified release formulation (Sinha and Kumria 2003). However, the major challenge of using pectin for the development of modified drug formulation was to overcome its solubility in aqueous medium which can contribute to the undesirable premature and local release of the active principle from the polysaccharide matrix (Sande 2005; Wu et al. 2008).
We have previously reported that coordination bond between the platinum(II) atom and the carboxylic group in the galacturonate of pectin causes spontaneous folding to form nanoconjugates (Verma and Sachin 2008), as well as formation of pectin nanoparticles prepared by inducing the gelification and cross-linking of pectin solution with calcium chloride and l-asparagine at low pH (Verma et al. 2011). At higher pH values, the polycarboxylate groups that are present can be cross-linked by the addition of calcium ions which leads to macromolecular aggregates (Kohn 1975). It has been widely reported that thiolation of a polymer such as chitosan (Kast and Bernkop-Schnürch 2001) poly(acrylic acid) (Marschütz and Bernkop-Schnürch 2002), and alginates (Bernkop-Schnürch et al. 2001), enhances its cohesive properties leading to oxidative cross-linking of thiol groups (Oosterveld et al. 2000). In continuation, our present investigation focuses on the in vitro, cytotoxicity, and in vivo pharmacokinetics and biodistribution of the negatively charged pectin nanoparticles.
2 Experimental
2.1 In vitro cytotoxicity
HepG2, hepatic carcinoma cell line was procured from American Type Culture Collection, (Rockville, MD) and maintained at 37 °C in 5 % CO2 at 95 % relative humidity. Pectin nanoparticles (PPN) were used for cytotoxicity as per our previously published protocol (Verma et al. 2005) using a standard MTT assay in a 24-, 48-, and 72-h assay. MTT assay is a nonradioactive assay done routinely to assess the viability of the cell culture. Briefly, 5 × 103cells/well were incubated in RPMI-1640 supplemented with 10 % FCS with paclitaxel (Pax) per se and paclitaxel encapsulated formazan crystals formed by the cellular reduction of MTT were dissolved in 150 μl of DMSO and plates were read on an ELISA reader using 570-nm filter. The relative cell viability (percent) related to control wells containing cells without nanoparticles was calculated by: [A]control − [A]test/[A]control × 100
Where [A]test is absorbance of the test sample and [A]control is the absorbance of the control sample.
2.2 Animal studies: pharmacokinetics and biodistribution
Studies were conducted under standardized procedures on inbred, male 6–8-week-old Balb/c (18–20 g) mice (Verma et al. 2012). Destructive sampling method was adopted for acquisition of biological samples (blood and tissues) for analysis. The animals were anesthetized with ether inhalation and paclitaxel per se and re-dispersed PPN containing drug at a dose of 20 mg/kg body weight, and PBS as control were then administered intravenously (i.v.). Blood was drawn from tail vein at predetermined intervals at 5, 10, 15, 30, 60, 90, and 120 min. Mice were sacrificed by cervical dislocation and dissected to obtain various tissues (kidney, liver, lung, and spleen), each tissue was weighed and stored vial at −20 °C for further analysis. Frozen tissue samples were thawed for 45 min and homogenized.
2.3 Determination of paclitaxel
The drug was extracted from biological samples with tert-butyl methyl ether (Xu et al. 2005). The sample was mixed with the organic reagent at a 1:1 ratio on a vortex mixture. After centrifugation, the organic layer was transferred to a clean tube and evaporated. The residue was redissolved in ethanol and analyzed by HPLC.
3 Results and discussion
Pectin nanoparticles were prepared by ionotropic gelation of degraded pectin chains with calcium ions (Verma et al. 2011). The objective of this modification was to reduce the polarity of pectin by reducing the number of free hydroxyl groups. Pectin nanoparticles thus formed and assumed an oval shape with clear edges as can be clearly observed in TEM pictures.
The sizing of pectin nanoparticles was done by dynamic light scattering setup by Brookhaven 90 Plus Particle Size Analyzer using a 35-mW He-Ne laser source at a scattering angle of 90°. The zeta potential (ξ) value was of −32 mV.
The mean diameter (mean RH) of the samples was in the range 350–500 nm with a polydispersity index of 0.3 (Fig. 1). ξ value of −32 mV at physiological pH was sufficient to keep the negatively charged particles under repelling effect thus bringing stability to the nanoparticle dispersion. Chains were cross-linked with Ca++ and folded into particulate shape in the presence of L-Asn (Fig 2), and the intrinsic viscosity [η] that indicates the hydrodynamic volume of a polymer molecule was measured and found to be reduced more than six times (Verma et al. 2011). A reduced [η] for nanoparticles as compared with that of pectin chains indicated that they can be administered at a much higher concentration in vivo. The nanoparticle solution was lyophilized to a white amorphous powder for further experiments.

TEM image of pectin nanoparticles. A representative graph of the size distribution spectra (~350 nm with PDI = 0.30) and zeta potential (−26 mv)

Flow chart of preparation of pectin nanoparticles. Ca2+ cross-links in a co-operating binding fashion between carboxylate group of adjacent chains enabling nanoparticle formation
We compared the cytotoxic effect of Pax per se, PPN, and pectin chains on Hep G2, hepatic carcinoma cell line. Dose-dependent cytotoxicity was observed, wherein ~21.7 ± 3.2 % cytotoxicity was observed by PPN (Fig. 3), but Pax per se showed ~55.6 ± 3.5 % cytotoxicity in a 72-h assay. The slow release of the drug can be attributed to the cytotoxicity observed as a function of time. Pectin chains showed negligible toxicity even at a concentration of 5 mg/ml.

Dose-dependant cytotoxicity of Pax and PPN on HepG2 (Conc., 0.02 μM). Pure pectin (5 mg/ml)
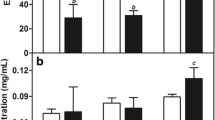
The plasma concentration–time curve of Pax and PPN at a dose of 20 mg/kg body weight of Balb/c mice has been shown in Fig. 4. A non-compartmental analysis of the pharmacokinetics graph indicates a biphasic pattern with initial rapid phase followed by a slower decay of Pax with the level falling beyond detection limit by 48 h. It was noticed that the blood profile remains much higher for PPN as compared to Pax per se.

Pharmacokinetic profile of Pax post-i.v. injection (20 mg/kg PPN) in Balb/c
Tissue biodistribution of Pax and PPN was also evaluated after an i.v. dose of 20 mg/kg PPN. Pax accumulates rapidly in various tissues with a maximum concentration found in the kidney. However, after 48 h, Pax accumulated in the lymphatic tissues, spleen, and liver. More than 90 % of Pax binds rapidly and extensively to plasma proteins (Bodmeier and Paeratakul 1991). On the contrary, the biodistribution was not altered as explicitly as with PPN at an early point of analysis, i.e., after 2 h, accumulation in kidney was markedly less. Forty-eight hours post-injection, tissue specificity changes (Fig. 5) from the difference in the rank order of concentration, i.e., liver > kidney > lung > spleen for the PPN and spleen > liver > kidney ≥ lung for Pax per se. Higher accumulation of Pax in the spleen may be attributed to splenic filtration (Liu and Krishnan 1999).

Biodistribution profile of paclitaxel and PPN after a 2 h and b 48 h of an i.v. infusion of 20 mg/kg in Balb/c mice. Mean ± SD (n = 4)
4 Conclusion
The biomedical potential of the negatively charged pectin nanoparticles has been assessed. Paclitaxel was encapsulated in the aqueous nanoparticle dispersion with high efficiency. The in vivo studies reflected that the nanoparticles significantly delayed plasma clearance with detection of Pax possible up to 48 h. It was evident from the biodistribution studies that enhanced PPN accumulation was observed in the liver but a greater accumulation of Pax per se in the kidney. The reported pectin nanoparticles form a good candidate for passive delivery of paclitaxel to liver tissue and warrant further investigation to ascertain its efficacy to regress tumor implants in mice.
References
Bernkop-Schnürch A, Kast CE, Richter MF (2001) Improvement in the mucoadhesive properties of alginate by the covalent attachment of cysteine. J Control Release 71:277–285
Bodmeier R, Paeratakul O (1991) A novel multiple-unit sustained release indomethacin hydroxylpropyl methylcellulose delivery system prepared by ionotropic gelation of sodium alginate at elevated temperatures. Carb Polym 16:399–408
Endress HU (1991) Nonfood uses of pectin. In: Walter RH, Taylor S (eds) The chemistry and technology of pectin. Academic, San Diego, pp 251–68
Garnier C, Axelos MAV, Thibault JF (1993) Phase diagrams of pectin-calcium systems: influence of pH, ionic strength, and temperature on the gelation of pectins with different degrees of methylation. Carbohydr Res 240:219–232
Kast CE, Bernkop-Schnürch A (2001) Thiolated polymers—thiomers: development and in vitro evaluation of chitosan–thioglycolic acid conjugates. Biomaterials 22:2345–52
Kohn K (1975) Ion binding on polyuronates—alginate and pectin. Pure Appl Chem 42:371–97. doi:10.1351/pac197542030371
Liu P, Krishnan TR (1999) Alginate-pectin-poly L-lysine particulate as a potential controlled release formulation. J Pharm Pharmacol 51:141–149
Marschütz MK, Bernkop-Schnürch A (2002) Thiolated polymers: self-crosslinking properties of thiolated 450 kDa poly(acrylic acid) and their influence on mucoadhesion. Eur J Pharm Sci 15:387–394
May CD (1990) Industrial pectins: sources, production and applications. Carbohydr Polym 12:79–99. doi:10.1016/0144-8617(90)90105-2
Oosterveld A, Beldman G, Voragen AGJ (2000) Oxidative cross-linking of pectic polysaccharides from sugar beet pulp. Carbohydr Res 328:199–207
Sande SA (2005) Pectin-based oral drug delivery to the colon. Expert Opin Drug Deliv 2:441–450
Sinha VR, Kumria R (2003) Microbiologically triggered drug delivery to the colon. Eur J Pharm Sci 18:3–18
Thibault JF, Renard CMGC, Axelos MAV, Roger P, Crépau MJ (1993) Studies of the length of homogalacturonic regions in pectins by acid hydrolysis. Carbohydr Res 238:287–339. doi:10.1016/0008-6215(93)87019-O
Verma AK, Sachin K (2008) Novel hydrophilic drug polymer nano-conjugates of cisplatin showing long blood retention profile—its release kinetics. Cellular uptake and bio-distribution. Curr Drug Deliv 5:120–126
Verma AK, Sachin K, Saxena A, Bohidar HB (2005) Release kinetics from bio-polymeric nanoparticles encapsulating protein synthesis inhibitor- cycloheximide, for possible therapeutic applications. Curr Pharm Biotechnol 6:121–130
Verma, A. K., Chanchal, A. and Kumar, A. (2011) Potential of negatively charged pectin nanoparticles encapsulating paclitaxel; preparation and characterization. Proceedings of the IEEE Xplore.
Verma AK, Chanchal A, Chuttani K (2012) Augmentation of anti-tumour activity of cisplatin by pectin nano-conjugates in B-16 mouse model: pharmacokinetics and in-vivo biodistribution of radio-labeled, hydrophillic nano-conjugates. Int J Nanotechnol 9:pp.872–886
Wu B, Deng D, Lu Y, Wu W (2008) Biphasic release of indomethacin from HPMC/pectin/calcium matrix tablet: II. Influencing variables, stability and pharmacokinetics in dogs. Eur j Pharm Biopharm 69(1):294–302
Xu Z, Gu W, Huang J, Sui H, Zhou Z, Yang Y, Yan Z, Li Y (2005) In vitro and in vivo evaluation of actively targetable nanoparticles for paclitaxel delivery. Int J Pharm 288(2):361–368
Acknowledgments
This work was supported by a DBT grant for Nano Science and NanoTechnology Research Initiative, GOI, (BT/PR/9895/NNT/28/55/2007).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Verma, A.K., Kumar, A. Pharmacokinetics and biodistribution of negatively charged pectin nanoparticles encapsulating paclitaxel. Cancer Nano 4, 99–102 (2013). https://doi.org/10.1007/s12645-013-0041-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12645-013-0041-8