
Abstract
The identification of protein–protein interactions (PPIs) can lead to a better understanding of cellular functions and biological processes of proteins and contribute to the design of drugs to target disease-causing PPIs. In addition, targeting host–pathogen PPIs is useful for elucidating infection mechanisms. Although several experimental methods have been used to identify PPIs, these methods can yet to draw complete PPI networks. Hence, computational techniques are increasingly required for the prediction of potential PPIs, which have never been seen experimentally. Recent high-performance sequence-based methods have contributed to the construction of PPI networks and the elucidation of pathogenetic mechanisms in specific diseases. However, the usefulness of these methods depends on the quality and quantity of training data of PPIs. In this brief review, we introduce currently available PPI databases and recent sequence-based methods for predicting PPIs. Also, we discuss key issues in this field and present future perspectives of the sequence-based PPI predictions.
Similar content being viewed by others


Introduction
Proteins are biological macromolecules composed of one or more chains of amino acid residues and play many critical roles in living cells, participating in a variety of biological processes, such as catalyzing chemical reactions, synthesizing and repairing DNA, and receiving and sending chemical signals. In order to perform their biological functions, they interact with other molecules such as ions, membrane lipids, DNA, and proteins by making direct physical contact through their specific residues. Interactions between proteins, i.e., protein–protein interactions (PPIs), are essential to the formation of macromolecular structures and to almost every biological process (Braun and Gingras 2012; Caterino et al. 2017; Dos Santos Vasconcelos et al. 2018; Liu et al. 2019). Those interactions are made through non-covalent contacts, electrostatic forces, or hydrophobic effects, between specific residues on proteins (De Las and Fontanillo 2010).
The identification of essential proteins required for the survival and development of the cell is important in understanding cell life and will help us better understand diseases and develop new drugs. Also, since almost every biological process involves one or more PPIs, the identification of PPIs in an organism is useful for understanding the molecular mechanisms underlying specific biological processes and for elucidating biological functions of proteins. In addition, comprehensive PPI networks associated with normal and abnormal physiological conditions are necessary not only for understanding physiological mechanisms but also for drug discovery for specific disorders, such as neurological disorders including Alzheimer disease and Creutzfeldt-Jacob disease (Qi et al. 2006; von Mering et al. 2002; Pedamallu and Posfai 2010). Furthermore, the identification of interspecies PPIs, such as virus-host PPIs, is also useful for understating infection mechanisms and for the design of new antiviral drugs and the treatment of infected patients. For example, studies of PPI networks of SARS-CoV-2 and (H1N1) influenza, which have similar clinical symptoms, have shown that virus-human PPIs are involved in multiple heterogeneous processes, including protein trafficking, translation, transcription, and ubiquitination (Khojasteh et al. 2022). These studies can help reveal similarities and difference between the two viruses.
There are two types of experimental methods for identifying PPIs: large-scale and high-throughput experiment methods and target-specific methods. The former screens large-scale PPIs by expressing each protein and exhaustively probes interactions between proteins of interest, such as yeast two-hybrid system, tandem affinity purification mass spectrometry, protein chip technology, and phage display. The latter determines a complex structure of a specific PPI of interest, such as X-ray crystallography, nuclear magnetic resonance (NMR) spectroscopy, and cryo-electron microscopy. The latter can determine interactions at the atomic level.
Although many experimental methods have been developed to identify PPIs, our knowledge of the whole set of PPIs in a particular cell or organism, i.e., interactome, is still incomplete, as numerous factors affect the detection of potential PPIs by those experiments; key factors include transient interactions, post-translational modifications (PTMs) (Seet et al. 2006; Duan and Walther 2015), intrinsically disordered regions (Acuner Ozbabacan et al. 2011; Lua et al. 2014; Meszaros et al. 2009; Babu et al. 2012), and other physiological conditions. Moreover, experimental methods are costly, time-consuming, and labor-intensive. Hence, the development of reliable computational methods for predicting PPIs is required. These computational methods can confirm or supplement experimentally detected PPIs, and add extra information, allowing us, for example, to prioritize therapeutically relevant PPIs as a target or an off-target.
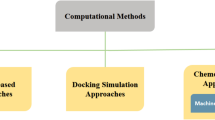
There are mainly two types of computational methods for predicting PPIs: data-driven methods and molecular docking. The former predicts PPIs based on various features of protein pairs, such as interlog (Yu et al. 2004), the protein sequences (Huang et al. 2016; Eid et al. 2016), physicochemical properties (Romero-Molina et al. 2019), evolutionary profiles (Hamp and Rost 2015), and structural information (Zhang et al. 2012), with statistical models or machine learning (ML), which discover relations among the training data of known PPIs. Various ML algorithms have been used in this field, such as random forest (RF), support vector machine (SVM), and ensemble classifiers. In recent publications, deep learning (DL), which is a subset of ML methods based on artificial neural networks, has been recognized as a powerful technique through benchmarking on blind data sets. The second type of methods, molecular docking, searches for the potential binding mode of proteins with surface complementarity and interaction energies (Pierce et al. 2014; Pierce and Weng 2007; Ohue et al. 2014). AlphaFold-Multimer is an extension of AlphaFold 2.0, specifically built for predicting protein complexes with high accuracy (Evans et al. 2022). AlphaFold 2.0 is the first computational method capable of predicting monomeric protein structures with near-experimental accuracy (Jumper et al. 2021). These methods may be considered reliable tools for the prediction of protein structures or protein complexes and will be implemented for the prediction of multimeric protein complexes (Al-Janabi 2022). AlphaPulldown is a Python package for screening PPIs and high-throughput modeling of higher-order oligomers using AlphaFold-Multimer (Yu et al. 2022). However, AlphaPulldown requires structural templates for each query protein, and we still need to know which protein pairs will interact without depending on the presence or absence of protein structures or structural templates. Furthermore, proteins are dynamic and can change their conformations, for example, under different pH conditions (Warwicker 2022). Therefore, the data-driven methods for predicting PPIs, especially sequence-based PPI predictions, which allow exhaustive PPI searches, will continue to be important. In this brief review, we will focus on this type of methods. Below, we will introduce currently available PPI databases and recent sequence-based methods for predicting PPIs. Also, we will discuss key issues in this field and present future perspectives of the sequence-based PPI predictions.
PPI databases
The development of reliable methods for predicting PPIs requires a diverse and representative dataset of known interacting protein pairs. PPIs determined experimentally have been registered as computer-readable data in a database to use in biochemical studies. There are mainly two types of PPI databases: primary databases and secondary databases.
The primary databases collect experimentally derived data which are submitted directly from researchers or collected from peer-reviewed publications. For example, DIP (Database of Interacting Proteins) stores experimentally determined PPIs, both manually curated by expert curators and automatically curated using computational approaches (Salwinski et al. 2004). IntAct (Orchard et al. 2014) and BioGRID (Oughtred et al. 2021) are open-source and comprehensive PPI databases and provide analysis tools for molecular interaction data. IntAct also provides high-quality negative PPIs and several disease-specific datasets, such as interactions associated with Alzheimer’s disease and interactions investigated in the context of cancer or coronavirus. These data are useful and unique among other PPI databases. BioGRID also includes chemical interactions between genes/proteins and bioactive small molecules, and post-translational modifications (Oughtred et al. 2019, 2021).
On the other hand, the secondary databases comprise PPI data derived from numerous primary or other secondary databases using rigorous computational approaches. For example, HitPredict is a database of experimentally determined PPIs from IntAct, BioGrid, MINT (Chatr-aryamontri et al. 2007), DIP, MatrixDB (Clerc et al. 2019), and InnateDB (Breuer et al. 2013), with confidence scores assigned (Lopez et al. 2015). Those scores are calculated based on the experimental details of each interaction and the sequence, structure, and functional annotations of the interacting proteins. PINA (Protein Interaction Network Analysis) platform is a database that integrates PPIs from IntAct, BioGrid, MINT, DIP, and HPRD and provides a variety of web tools to construct, filter, and analyze the networks of proteins of interest (Du et al. 2021). APID (Agile Protein Interaction Data Analyzer) (Alonso-Lopez et al. 2019) provides a collection of known experimentally validated PPIs for more than 400 organisms from DIP (Salwinski et al. 2004), IntAct, MINT, HPRD (Keshava Prasad et al. 2009), BioGRID, BioPlex (Huttlin et al. 2015), and also from experimentally resolved 3D structures, PDB (ww 2019) and PDBsum (Laskowski et al. 2018), indicating different quality levels, i.e., whether interactions are proven by at least one binary detection method or not. This database also provides an interactive data visualization web tool that allows the construction of subinteractomes from query lists of proteins and the exploration and analysis of the corresponding networks about PPIs of interest. HIPPE (Human Integrated Protein–Protein Interaction rEference) provides functionally annotated human PPIs integrated from 10 primary databases and manually curated PPI data with the confidence scoring of experimentally measured interactions (Alanis-Lobato et al. 2017). The parameters of this scoring scheme were jointly optimized by human experts and a computer algorithm. STRING provides known PPIs including direct (physical) and indirect (functional) associations and provides predicted PPIs from automated text-mining of the scientific literature, conserved co-expression, and genomic context predictions (Szklarczyk et al. 2021). In this database, each PPI is annotated with various scores computed based on interaction evidence from the organism of interest or systematic transfers of interaction evidence from one organism to another.
In addition, there is a database called Negatome for experimentally supported non-interacting protein pairs (non-PPIs) collected by manual curation of the literature and computational analysis of protein complexes registered in PDB, excluding interactions from IntAct (Blohm et al. 2014). This database is especially important for training PPI prediction algorithms because it is complementary to the negative data generated by other methods such as randomly selecting proteins from different cellular locations.
Furthermore, there is an international collaboration, IMEx (International Molecular Exhange Consortium), between several institutions providing PPI data in order to develop a single set of curation rules for the registration of PPI data derived from experimentally derived data, pre-prints, and peer-reviewed publications and to standardize the data formats of PPI data (Orchard et al. 2012).
The databases and the number of PPIs registered are listed in Table 1.
Preparation of PPI datasets
Preparation of a high-quality dataset is crucial for the sequence-based PPI prediction. The experimentally determined PPIs sourced from the primary or secondary databases shown in Table 1 are normally merged into a set of PPIs as positive samples, excluding interactions between similar proteins. On the other hand, the preparation of non-PPIs can be more important than that of PPIs, because the quality and quantity of non-PPIs influence the PPI predictions significantly. Most PPI prediction methods require training with both positive and negative samples. One simple method is to generate negative samples by randomly pairing proteins in the positive samples and ignoring the actual interactions, assuming that randomly paired proteins are unlikely to be positive samples (dissimilarity negative sampling). Methods with more realistic considerations have been proposed. For example, Hamp and Rost (2015) generated negative samples by randomly sampling from all the pairs in each of the four PPI datasets: one training dataset and three testing datasets C1–C3 (C1, test pairs sharing both proteins with the training dataset; C2, test pairs sharing only one protein with the training dataset; and C3, test pairs sharing neither protein with the training dataset). The need to distinguish between these classes C1–C3 was introduced by Park and Marcotte (2012). Sun et al. (2017) generated negative samples by paring proteins found in different subcellular locations, excluding proteins annotated with ambiguous or uncertain subcellular location terms and with two or more locations.
The size of the negative samples and the size balance between positive and negative samples is one issue that should be carefully considered in developing an accurate and reliable method to predict potential PPIs. One common solution is to randomly sample negative samples, keeping a ratio of positive and negative samples. Hamp and Rost (2015) sampled 10 times as many negative samples as positive samples. However, the data imbalance is an issue that needs to be discussed and solved. More details of training datasets, test datasets, and independent test datasets used in recently developed methods and their data sources are shown in Table 2.
Sequence-based prediction of PPIs
The sequence-based prediction of PPI refers to the problems of inferring, given a pair of protein sequences, the likelihood of an interaction between them, i.e., a score that represents their interacting probability. This approach can be applied to the inference of PPI networks by adding new nodes and new edges to the PPI network graph (Murakami et al. 2017; Tripathi et al. 2019). So far, many computational methods to solve this problem have been proposed as complementary to experimental methods. Even though these sequenced-based methods are less accurate than structure-based methods, they are useful in predicting PPIs involving proteins, for which structural information is unknown or which are intrinsically disordered proteins. In addition, primary structures are available for all proteins, and thus, modeling and predicting PPIs using only sequence information has long been of interest. Almost all these methods are data-driven methods and can be categorized into statistical methods, similarity-based methods, and ML-based methods; however, most of the methods used in recent years are based on similarity or ML. Currently available web servers or downloadable programs for PPI prediction are shown in Table 3, along with a brief description of their strengths and weaknesses. In addition, the reported benchmark results of PPI prediction methods developed within the last three years are shown in Table 4; however, a fair performance comparison is difficult due to the different test datasets.
Statistical methods
The statistical methods generally employed the statistical characteristics or the conserved patterns of protein sequences, assuming that functionally important proteins are conserved across organisms, such as the topological similarity between phylogenetic trees of a pair of proteins (Pazos and Valencia 2001), the co-occurrence of a fine number of short polypeptide sequences observed in known interacting protein pairs (Pitre et al. 2006), and the co-evolutionary divergence based on the assumption that protein pairs with similar substitution rates are likely to interact with each other (Hsin Liu et al. 2013). MirrorTree is a currently available server used to detect the coevolution between proteins and predicts their physical interactions (Ochoa and Pazos 2010). The underlying principle behind this method is that the co-evolution between interacting proteins can be reflected from the similarity scores from the distance matrices of the corresponding phylogenetic trees of the interacting proteins (Craig and Liao 2007).
Similarity-based methods
The similarity-based methods basically employed homologous interactions, in which two PPIs are homologous if a pair of interacting proteins is homologous to a pair of other interacting proteins. The homologous interactions basically include, but are not limited to, orthologous interactions (homologous interactions found in different organisms), i.e., interolog (Walhout et al. 2000), and paralogous interactions (homologous interactions in the same organisms). For example, BIPS (Biana Interolog Prediction Server) (Garcia-Garcia et al. 2012) is based on interolog information, assuming that the homologous proteins preserve similar functional behavior and also the same interactions (Matthews et al. 2001; Yu et al. 2004), and predicts interactions between proteins based on PPIs found in several PPI-related databases integrated using the BIONA (Biologic Interactions and Network Analysis) framework (Garcia-Garcia et al. 2010). SPRINT (Li and Ilie 2017) and PIPE4 (Dick et al. 2020) are based on the idea that a pair of query proteins (X1, X2) has an interaction if X1 and X2 are similar to either of the known interacting protein pair (P1, P2); that is, X1 is similar to P1 and X2 is similar to P2. However, these methods do not always work well in the absence of known interacting protein pairs with high-sequence similarity to the query protein pairs.
ML-based methods
The ML-based methods employ various supervised ML algorithms, such as SVM, RF, and DL. These algorithms are used for most of the existing PPI prediction methods. SVM aims to find a maximum margin hyperplane in an n-dimensional space (n is the number of features) that separates the labelled samples, i.e., maximizing the distance between samples of different classes. SVM requires computing power to train and test high-dimensional features with radial basis function (RDF) kernel that transforms linearly inseparable samples to linearly separable ones (kernel trick). RF is an ensemble learning method involving numerous decision trees (DT) for classification and outputs the class selected by most trees. RF can effectively train a large dataset of PPIs and vectors with many features and can rank the feature importance for accurate prediction. To train an RF model, the optimal value of the number of trees in the forest is usually adjusted, concerning the computational time and the accuracy. DL is an artificial neural network with multiple layers between the input and output layers. DL is considered to achieve better performance than the conventional ML-based methods in the PPI predictions. DL consists of several algorithms, such as Deep Neural Networks (DNN), Convolutional Neural Networks (CNN), Recurrent Neural Networks (RNN), and Graph Convolutional Networks (GCN). These different algorithms have been applied to the PPI predictions and require the different input forms of proteins. DNN requires a one-dimensional vector, while other algorithms input flexible forms, for example, a two-dimensional matrix such as position-specific scoring matrix (PSSM) (Hu et al. 2022). Recently developed DL-based methods are shown in Table 3.
Protein feature encoding
One important issue for the ML-based methods is how to encode protein sequences of variable lengths into fixed-length numeric feature vectors used for the development of the prediction models and the prediction of PPIs. A pair of feature vectors encoding a protein pair is generally inputted to the method either by combining them sequentially or separately. In addition, the extraction of appropriate features from protein sequences is critical for the accurate PPI prediction.
Commonly used protein feature encoding methods includes physicochemical properties of amino acids (AA) (Bock and Gough 2001; Sun et al. 2017), protein sequence profiles (evolutionary profiles) (Liu et al. 2019; Hashemifar et al. 2018; Hamp and Rost 2015), and protein sequence embedding (Alachram et al. 2021).
Various physicochemical properties of AA are used, such as hydropathy index (hydrophobic or hydrophilic properties of an AA side chain), positively or negatively charged AA, uncharged AA, and pKa value (the acid dissociation constant at logarithmic scale which is a quantitative measure of the strength of an acid in solution). These properties are available in the AAindex (https://www.genome.jp/aaindex/) database (Kawashima and Kanehisa 2000; Kawashima et al. 2008).
Protein sequence profiles are a list of preferences for each AA at each position in a given multiple sequence alignment (MSA), i.e., a PSSM, which is derived from MSA in position-specific iterative BLAST (PSI-BLAST) (Altschul et al. 1997). PSSM is informative protein feature based on evolutionary information extracted from MSA, even though an enormous search time to compute it is required by PSI-BLAST. In addition, PSSM can reproduce evolutionary conserved interactions between protein sequences through their evolutionary information. For example, DPPI (Hashemifar et al. 2018) and TransPPI (Yang et al. 2021) employ PSSM which is a \(N\times 20\) matrix M = {Mij, i = 1…N, j = 1…20}, where N is the length of a given protein sequence and each element Mij is the score of the jth AA in the ith position of the sequence.
Protein sequence embedding captures semantic information on AA residues in entire sequences. The widely used embedding methods, such as Word2Vec (Mikolov et al. 2013a) and Doc2Vec (Le and Mikolov 2014), was originally developed in the field of natural language processing in order to obtain the distributed representation of words and documents. These methods are learned from the contexts of words in each document using a shallow two-layer neural network with continuous bag-of-words model (CBOW) and the continuous skip-gram model (Skip-Gram). CBOW predicts the target words from the context of documents, while Skip-Gram predicts the target context from entire words. CBOW learns faster while Skip-Gram does more efficient with small amounts of training data and has better representations for infrequent words (Mikolov et al. 2013b). In the biological sequences, a sequence is regarded as a sentence and represented by multiple k consecutive AA (k-mers) used to train the Word2Vec or Doc2Vec models. These methods were recently applied to the prediction of human and virus protein interactions, showing that they learned the protein features well that enable the reliable prediction of huma-virus PPIs (Yang et al. 2020b; Tsukiyama et al. 2021). Furthermore, several new residue representation methods based on Word2Vec have been proposed, such as Res2vec (Yao et al. 2019) and DL2vec (Chen et al. 2021).
Current issues
False positives, overfitting, and underfitting
Prediction models generated are usually evaluated with several performance measures to compare predicted scores with the actual observed ones. To minimize false positives and false negatives, it is necessary to use performance measures that can appropriately evaluate predictive performance, even though it is difficult to distinguish between false positives and novel PPIs.
Overfitting and underfitting issues can occur when generating predictive models. The performance of a predictive model generated by a learning algorithm depends on how well it captures the underlying features of the training dataset. When the algorithm is too simple and insufficient to model the training dataset, it is called underfitting, whereas when the algorithm is too complex and the training dataset are not sufficient to constrain it, it is called overfitting (Sarkar and Saha 2019). These issues can be solved by selecting an appropriate learning algorithm, performing appropriate evaluation with independent blind test datasets, and preparing a training dataset without missing underlying features of PPIs or bias. In terms of training dataset preparation, to solve the overfitting, it is still important to appropriately reduce the redundancy of homologous sequences present in the dataset. CD-HIT (Fu et al. 2012) is generally used for clustering and comparing protein sequences with several options, such as a sequence identity threshold or an alignment coverage, to reduce redundancies in the dataset. The main advantage of this program is that it is very fast and can handle extremely large datasets. Attention should be paid to the appropriate setting of the options used in CD-HIT. Datasets containing proteins with higher sequence similarity (e.g., > 30%) may lead to overfitting and overestimate the prediction performance. To avoid overfitting and develop reliable prediction models, it is expected to use low-sequence identity cut-off (< 30%) widely used in various sequence-based methods, shown in Table 2. On the other hand, to solve the underfitting, sufficient and up-to-date PPI data that reflect the features of target PPIs should be collected from the existing databases, shown in Table 1.
ML-based methods, realistic datasets
In the ML-based prediction methods, it is important to select the optimal ML approach. Currently, various ML-based methods have been developed that differ in terms of protein representation, method complexity, various protein features, and computational cost. DL-based methods have reported higher prediction performance than other methods but are computationally expensive.
Most methods have been developed on a balanced dataset containing equal numbers of interacting protein pairs and non-interacting protein pairs or an imbalanced dataset containing non-interacting pairs several times greater than the number of interacting pairs. However, this data balance, i.e., the ratio of interacting and non-interacting protein pairs, is highly imbalanced in nature and few methods have been developed or evaluated regarding the impact of realistic datasets containing huge numbers of non-interacting pairs. The realistic datasets should be huge, so handling such datasets requires more efficient algorithms for the prediction of PPIs.
Future perspectives and conclusion
Despite advances in techniques for large-scale experimental analysis of protein interactions, our knowledge of the whole set of PPIs in a particular cell is still incomplete, considering various physicochemical factors such as transient dynamics, post-translational modification (PTM), intrinsically disordered regions, and physiological conditions. For example, a comprehensive understanding of liquid–liquid phase separation (LLPS), which has received increasing attention over the past decade, should require the construction of complete PPI networks involving phase-separated proteins and analyze the network properties of different classes of phase-separating proteins in the human interactome. LLPS is an important mechanism that drives the formation of membrane-less organelles fundamentally driven by multivalent interactions between proteins and/or nucleic acids (Li et al. 2012; Mondal et al. 2022), which can occur in proteins between multiple folded domains or are medicated by intrinsically disordered proteins (Chu et al. 2022). For example, a mutation in the Speckle-type POZ protein (SPOP), a tumor-suppressor protein, can lead to the formation of many solid tumors, including prostate, gastric, and colorectal cancers (Wang et al. 2021). A recent study revealed that substrates of SPOP can phase-separate with SPOP to form condensates in vitro and co-localize in liquid nuclear organelles in cells (Bouchard et al. 2018). Therefore, identification of PPI networks requires further improvement to uncover all possible interactions that may exist at the same cellular localization or the proteome level. In addition, it is indispensable to develop computational methods to predict potential PPIs rapidly and accurately from a large number of candidate protein pairs using only sequence information. The development of such methods will enable comprehensive PPI prediction between proteins, leading to the identification for interacting partners of proteins of interest.
In general, many ML-based PPI prediction methods discriminate whether two proteins interact or not, given output scores from those ML models, but these scores do not explain the strength of the interactions of the two proteins. In order to further improve the reliability of PPI prediction and capture PPI network properties more clearly, protein binding affinities, which are typically measured by the equilibrium dissociation constant (Kd), should be considered to assess predicted interactions. However, the determination of the protein binding affinity is generally not applicable on a large scale due to the dissociation rate dependent accuracy of experimental methods, cost and time constraints, and the need for protein complexes (Abbasi et al. 2020). Therefore, accurate computational techniques can play an important role in the protein binding affinity determination. In particular, sequence-based protein binding affinity prediction is challenging but, like sequence-based PPI prediction, is also an important research topic (Yugandhar and Gromiha 2014; Abbasi et al. 2020). In addition, further development of sequence-based interaction site prediction is also important, as detailed interaction site and residue information enables more accurate PPI prediction, more accurate prediction of protein binding affinity, and more accurate analysis of PPI network properties.
Various PPI prediction methods have been proposed and available as a web application or on an Internet hosting service for software development like GitHub (Table 2). The availability of these methods as a public resource is of great benefit to the drug discovery community as well as to further advances in the PPI prediction. ML-based methods, especially, DL-based method, are currently great success in the PPI prediction. Further improvements to these methods include development with more realistic datasets and construction of large size and independent unbiased datasets to evaluate the proposed methods, because the performance of these methods essentially depends on reliable training data with known PPIs determined experimentally. In addition, it will be necessary to develop protein encoding methods that can better capture various protein features including functional, structural, and evolutional information.
As evidenced by the recent increase in the development of sequence-based PPI prediction methods based on heterogeneous datasets (Tables 2 and 3), these methods are readily available and indispensable for solving pressing questions, such as the mechanism of infection, without relying on structural information. In addition, sequence-based PPI prediction alone cannot surpass the reliability of structure-based methods, but by leveraging recently developed protein structure prediction methods such as AlphaFold 2.0, sequence-based PPI prediction overcome the weakness, which will be expected to further improve the reliability of PPI predictions.
References
Abbasi WA, Yaseen A, Hassan FU, Andleeb S, Minhas F (2020) ISLAND: in-silico proteins binding affinity prediction using sequence information. BioData Min 13(1):20. https://doi.org/10.1186/s13040-020-00231-w
AcunerOzbabacan SE, Engin HB, Gursoy A, Keskin O (2011) Transient protein-protein interactions. Protein Eng Des Sel 24(9):635–648. https://doi.org/10.1093/protein/gzr025
Alachram H, Chereda H, Beissbarth T, Wingender E, Stegmaier P (2021) Text mining-based word representations for biomedical data analysis and protein-protein interaction networks in machine learning tasks. PLoS ONE 16(10):e0258623. https://doi.org/10.1371/journal.pone.0258623
Alanis-Lobato G, Andrade-Navarro MA, Schaefer MH (2017) HIPPIE v.20: enhancing meaningfulness and reliability of protein-protein interaction networks. Nucleic Acids Res 45(D1):D408–D414. https://doi.org/10.1093/nar/gkw985
Al-Janabi A (2022) Has DeepMind’s AlphaFold solved the protein folding problem? Biotechniques 72(3):73–76. https://doi.org/10.2144/btn-2022-0007
Alley EC, Khimulya G, Biswas S, AlQuraishi M, Church GM (2019) Unified rational protein engineering with sequence-based deep representation learning. Nat Methods 16(12):1315–1322. https://doi.org/10.1038/s41592-019-0598-1
Alonso-Lopez D, Campos-Laborie FJ, Gutierrez MA, Lambourne L, Calderwood MA, Vidal M, De Las Rivas J (2019) APID database: redefining protein-protein interaction experimental evidences and binary interactomes. Database (Oxford) 2019.https://doi.org/10.1093/database/baz005
Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25(17):3389–3402. https://doi.org/10.1093/nar/25.17.3389
Ammari MG, Gresham CR, McCarthy FM, Nanduri B (2016) HPIDB 2.0: a curated database for host-pathogen interactions. Database Oxford 2016:baw103. https://doi.org/10.1093/database/baw103
Babu MM, Kriwacki RW, Pappu RV (2012) Structural biology. Versatility from Protein Disorder. Science 337(6101):1460–1461. https://doi.org/10.1126/science.1228775
Barman RK, Saha S, Das S (2014) Prediction of interactions between viral and host proteins using supervised machine learning methods. PLoS ONE 9(11):e112034. https://doi.org/10.1371/journal.pone.0112034
Bepler T, Berger B (2019) Learning protein sequence embeddings using information from structure. proceedings of ICLR 2019 abs/1902.08661:1–17. https://doi.org/10.48550/arXiv.1902.08661
Blohm P, Frishman G, Smialowski P, Goebels F, Wachinger B, Ruepp A, Frishman D (2014) Negatome 2.0: a database of non-interacting proteins derived by literature mining, manual annotation and protein structure analysis. Nucleic Acids Res 42(Database issue):D396-400. https://doi.org/10.1093/nar/gkt1079
Bock JR, Gough DA (2001) Predicting protein–protein interactions from primary structure. Bioinformatics 17(5):455–460. https://doi.org/10.1093/bioinformatics/17.5.455
Bouchard JJ, Otero JH, Scott DC, Szulc E, Martin EW, Sabri N, Granata D, Marzahn MR, Lindorff-Larsen K, Salvatella X, Schulman BA, Mittag T (2018) Cancer mutations of the tumor suppressor SPOP disrupt the formation of active, phase-separated compartments. Mol Cell 72(1):19-36 e18. https://doi.org/10.1016/j.molcel.2018.08.027
Braun P, Gingras AC (2012) History of protein-protein interactions: from egg-white to complex networks. Proteomics 12(10):1478–1498. https://doi.org/10.1002/pmic.201100563
Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ (2013) InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res 41(Database issue):D1228-1233. https://doi.org/10.1093/nar/gks1147
Calderone A, Licata L, Cesareni G (2015) VirusMentha: a new resource for virus-host protein interactions. Nucleic Acids Res 43(Database issue):D588-592. https://doi.org/10.1093/nar/gku830
Caterino M, Ruoppolo M, Mandola A, Costanzo M, Orru S, Imperlini E (2017) Protein-protein interaction networks as a new perspective to evaluate distinct functional roles of voltage-dependent anion channel isoforms. Mol Biosyst 13(12):2466–2476. https://doi.org/10.1039/c7mb00434f
Chatr-aryamontri A, Ceol A, Palazzi LM, Nardelli G, Schneider MV, Castagnoli L, Cesareni G (2007) MINT: the Molecular INTeraction database. Nucleic Acids Res 35(Database issue):D572-574. https://doi.org/10.1093/nar/gkl950
Chatr-aryamontri A, Ceol A, Peluso D, Nardozza A, Panni S, Sacco F, Tinti M, Smolyar A, Castagnoli L, Vidal M, Cusick ME, Cesareni G (2009) VirusMINT: a viral protein interaction database. Nucleic Acids Res 37(Database issue):D669-673. https://doi.org/10.1093/nar/gkn739
Chen C, Zhang Q, Ma Q, Yu B (2019a) LightGBM-PPI: Predicting protein-protein interactions through LightGBM with multi-information fusion. Chemom Intell Lab Syst 191:54–64. https://doi.org/10.1016/j.chemolab.2019.06.003
Chen M, Ju CJ, Zhou G, Chen X, Zhang T, Chang KW, Zaniolo C, Wang W (2019b) Multifaceted protein-protein interaction prediction based on Siamese residual RCNN. Bioinformatics 35(14):i305–i314. https://doi.org/10.1093/bioinformatics/btz328
Chen J, Althagafi A, Hoehndorf R (2021) Predicting candidate genes from phenotypes, functions and anatomical site of expression. Bioinformatics 37(6):853–860. https://doi.org/10.1093/bioinformatics/btaa879
Chu X, Sun T, Li Q, Xu Y, Zhang Z, Lai L, Pei J (2022) Prediction of liquid-liquid phase separating proteins using machine learning. BMC Bioinformatics 23(1):72. https://doi.org/10.1186/s12859-022-04599-w
Clerc O, Deniaud M, Vallet SD, Naba A, Rivet A, Perez S, Thierry-Mieg N, Ricard-Blum S (2019) MatrixDB: integration of new data with a focus on glycosaminoglycan interactions. Nucleic Acids Res 47(D1):D376–D381. https://doi.org/10.1093/nar/gky1035
Craig RA, Liao L (2007) Phylogenetic tree information aids supervised learning for predicting protein-protein interaction based on distance matrices. BMC Bioinformatics 8:6. https://doi.org/10.1186/1471-2105-8-6
De Las RJ, Fontanillo C (2010) Protein-protein interactions essentials: key concepts to building and analyzing interactome networks. PLoS Comput Biol 6(6):e1000807. https://doi.org/10.1371/journal.pcbi.1000807
Dick K, Samanfar B, Barnes B, Cober ER, Mimee B, Tan LH, Molnar SJ, Biggar KK, Golshani A, Dehne F, Green JR (2020) PIPE4: fast PPI predictor for comprehensive inter- and cross-species interactomes. Sci Rep 10(1):1390. https://doi.org/10.1038/s41598-019-56895-w
Ding Y, Tang J, Guo F (2016) Predicting protein-protein interactions via multivariate mutual information of protein sequences. BMC Bioinformatics 17(1):398. https://doi.org/10.1186/s12859-016-1253-9
Dong TN, Brogden G, Gerold G, Khosla M (2021) A multitask transfer learning framework for the prediction of virus-human protein-protein interactions. BMC Bioinformatics 22(1):572. https://doi.org/10.1186/s12859-021-04484-y
Dos Santos Vasconcelos CR, de Lima CT, Rezende AM (2018) Building protein-protein interaction networks for Leishmania species through protein structural information. BMC Bioinformatics 19(1):85. https://doi.org/10.1186/s12859-018-2105-6
Du X, Sun S, Hu C, Yao Y, Yan Y, Zhang Y (2017) DeepPPI: boosting prediction of protein–protein interactions with deep neural networks. J Chem Inf Model 57(6):1499–1510. https://doi.org/10.1021/acs.jcim.7b00028
Du Y, Cai M, Xing X, Ji J, Yang E, Wu J (2021) PINA 3.0: mining cancer interactome. Nucleic Acids Res 49(D1):D1351–D1357. https://doi.org/10.1093/nar/gkaa1075
Duan G, Walther D (2015) The roles of post-translational modifications in the context of protein interaction networks. PLoS Comput Biol 11(2):e1004049. https://doi.org/10.1371/journal.pcbi.1004049
DurmusTekir S, Cakir T, Ardic E, Sayilirbas AS, Konuk G, Konuk M, Sariyer H, Ugurlu A, Karadeniz I, Ozgur A, Sevilgen FE, Ulgen KO (2013) PHISTO: pathogen-host interaction search tool. Bioinformatics 29(10):1357–1358. https://doi.org/10.1093/bioinformatics/btt137
Eid FE, ElHefnawi M, Heath LS (2016) DeNovo: virus-host sequence-based protein-protein interaction prediction. Bioinformatics 32(8):1144–1150. https://doi.org/10.1093/bioinformatics/btv737
Evans R, O’Neill M, Pritzel A, Antropova N, Senior A, Green T, Žídek A, Bates R, Blackwell S, Yim J, Ronneberger O, Bodenstein S, Zielinski M, Bridgland A, Potapenko A, Cowie A, Tunyasuvunakool K, Jain R, Clancy E, Kohli P, Jumper J, Hassabis D (2022) Protein complex prediction with AlphaFold-Multimer. DeepMind. https://doi.org/10.1101/2021.10.04.463034
Fu L, Niu B, Zhu Z, Wu S, Li W (2012) CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28(23):3150–3152. https://doi.org/10.1093/bioinformatics/bts565
Garcia-Garcia J, Guney E, Aragues R, Planas-Iglesias J, Oliva B (2010) Biana: a software framework for compiling biological interactions and analyzing networks. BMC Bioinformatics 11:56. https://doi.org/10.1186/1471-2105-11-56
Garcia-Garcia J, Schleker S, Klein-Seetharaman J, Oliva B (2012) BIPS: BIANA Interolog Prediction Server. A tool for protein-protein interaction inference. Nucleic Acids Res 40(Web Server issue):W147-151. https://doi.org/10.1093/nar/gks553
Gordon DE, Jang GM, Bouhaddou M, Xu J, Obernier K, White KM, O’Meara MJ, Rezelj VV, Guo JZ, Swaney DL, Tummino TA, Huttenhain R, Kaake RM, Richards AL, Tutuncuoglu B, Foussard H, Batra J, Haas K, Modak M, Kim M, Haas P, Polacco BJ, Braberg H, Fabius JM, Eckhardt M, Soucheray M, Bennett MJ, Cakir M, McGregor MJ, Li Q, Meyer B, Roesch F, Vallet T, Mac Kain A, Miorin L, Moreno E, Naing ZZC, Zhou Y, Peng S, Shi Y, Zhang Z, Shen W, Kirby IT, Melnyk JE, Chorba JS, Lou K, Dai SA, Barrio-Hernandez I, Memon D, Hernandez-Armenta C, Lyu J, Mathy CJP, Perica T, Pilla KB, Ganesan SJ, Saltzberg DJ, Rakesh R, Liu X, Rosenthal SB, Calviello L, Venkataramanan S, Liboy-Lugo J, Lin Y, Huang XP, Liu Y, Wankowicz SA, Bohn M, Safari M, Ugur FS, Koh C, Savar NS, Tran QD, Shengjuler D, Fletcher SJ, O’Neal MC, Cai Y, Chang JCJ, Broadhurst DJ, Klippsten S, Sharp PP, Wenzell NA, Kuzuoglu-Ozturk D, Wang HY, Trenker R, Young JM, Cavero DA, Hiatt J, Roth TL, Rathore U, Subramanian A, Noack J, Hubert M, Stroud RM, Frankel AD, Rosenberg OS, Verba KA, Agard DA, Ott M, Emerman M, Jura N, von Zastrow M, Verdin E, Ashworth A, Schwartz O, d’Enfert C, Mukherjee S, Jacobson M, Malik HS, Fujimori DG, Ideker T, Craik CS, Floor SN, Fraser JS, Gross JD, Sali A, Roth BL, Ruggero D, Taunton J, Kortemme T, Beltrao P, Vignuzzi M, Garcia-Sastre A, Shokat KM, Shoichet BK, Krogan NJ (2020) A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583(7816):459–468. https://doi.org/10.1038/s41586-020-2286-9
Guirimand T, Delmotte S, Navratil V (2015) VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res 43(Database issue):D583-587. https://doi.org/10.1093/nar/gku1121
Guo Y, Yu L, Wen Z, Li M (2008) Using support vector machine combined with auto covariance to predict protein–protein interactions from protein sequences. Nucleic Acids Res 36(9):3025–3030. https://doi.org/10.1093/nar/gkn159
Guo Y, Li M, Pu X, Li G, Guang X, Xiong W, Li J (2010) PRED_PPI: a server for predicting protein-protein interactions based on sequence data with probability assignment. BMC Res Notes 3:145. https://doi.org/10.1186/1756-0500-3-145
Hamp T, Rost B (2015) Evolutionary profiles improve protein-protein interaction prediction from sequence. Bioinformatics 31(12):1945–1950. https://doi.org/10.1093/bioinformatics/btv077
Hashemifar S, Neyshabur B, Khan AA, Xu J (2018) Predicting protein-protein interactions through sequence-based deep learning. Bioinformatics 34(17):i802–i810. https://doi.org/10.1093/bioinformatics/bty573
HitPredict version 4 (2015) Comprehensive reliability scoring of physical protein-protein interactions from more than 100 species. Database (Oxford). https://doi.org/10.1093/database/bav117
Hsin Liu C, Li KC, Yuan S (2013) Human protein-protein interaction prediction by a novel sequence-based co-evolution method: co-evolutionary divergence. Bioinformatics 29(1):92–98. https://doi.org/10.1093/bioinformatics/bts620
Hu X, Feng C, Zhou Y, Harrison A, Chen M (2021) DeepTrio: a ternary prediction system for protein-protein interaction using mask multiple parallel convolutional neural networks. Bioinformatics. https://doi.org/10.1093/bioinformatics/btab737
Hu X, Feng C, Ling T, Chen M (2022) Deep learning frameworks for protein-protein interaction prediction. Comput Struct Biotechnol J 20:3223–3233. https://doi.org/10.1016/j.csbj.2022.06.025
Huang YA, You ZH, Gao X, Wong L, Wang L (2015) Using weighted sparse representation model combined with discrete cosine transformation to predict protein-protein interactions from protein sequence. Biomed Res Int 2015:902198. https://doi.org/10.1155/2015/902198
Huang YA, You ZH, Chen X, Chan K, Luo X (2016) Sequence-based prediction of protein-protein interactions using weighted sparse representation model combined with global encoding. BMC Bioinformatics 17(1):184. https://doi.org/10.1186/s12859-016-1035-4
Huttlin EL, Ting L, Bruckner RJ, Gebreab F, Gygi MP, Szpyt J, Tam S, Zarraga G, Colby G, Baltier K, Dong R, Guarani V, Vaites LP, Ordureau A, Rad R, Erickson BK, Wuhr M, Chick J, Zhai B, Kolippakkam D, Mintseris J, Obar RA, Harris T, Artavanis-Tsakonas S, Sowa ME, De Camilli P, Paulo JA, Harper JW, Gygi SP (2015) The BioPlex network: a systematic exploration of the human interactome. Cell 162(2):425–440. https://doi.org/10.1016/j.cell.2015.06.043
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Zidek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873):583–589. https://doi.org/10.1038/s41586-021-03819-2
Kawashima S, Kanehisa M (2000) AAindex: amino acid index database. Nucleic Acids Res 28(1):374. https://doi.org/10.1093/nar/28.1.374
Kawashima S, Pokarowski P, Pokarowska M, Kolinski A, Katayama T, Kanehisa M (2008) AAindex: amino acid index database, progress report 2008. Nucleic Acids Res 36(Database issue):D202-205. https://doi.org/10.1093/nar/gkm998
Keshava Prasad TS, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, Balakrishnan L, Marimuthu A, Banerjee S, Somanathan DS, Sebastian A, Rani S, Ray S, Harrys Kishore CJ, Kanth S, Ahmed M, Kashyap MK, Mohmood R, Ramachandra YL, Krishna V, Rahiman BA, Mohan S, Ranganathan P, Ramabadran S, Chaerkady R, Pandey A (2009) Human Protein Reference Database—2009 update. Nucleic Acids Res 37(Database issue):D767-772. https://doi.org/10.1093/nar/gkn892
Khojasteh H, Khanteymoori A, Olyaee MH (2022) Comparing protein-protein interaction networks of SARS-CoV-2 and (H1N1) influenza using topological features. Sci Rep 12(1):5867. https://doi.org/10.1038/s41598-022-08574-6
Laskowski RA, Jablonska J, Pravda L, Varekova RS, Thornton JM (2018) PDBsum: structural summaries of PDB entries. Protein Sci 27(1):129–134. https://doi.org/10.1002/pro.3289
Le QV, Mikolov T (2014) Distributed representations of sentences and documents. Proc 31st Int Conf Mach Learn, PMLR 32(2):1188–1196. https://doi.org/10.48550/arXiv.1405.4053
Li Y, Ilie L (2017) SPRINT: ultrafast protein-protein interaction prediction of the entire human interactome. BMC Bioinformatics 18(1):485. https://doi.org/10.1186/s12859-017-1871-x
Li P, Banjade S, Cheng HC, Kim S, Chen B, Guo L, Llaguno M, Hollingsworth JV, King DS, Banani SF, Russo PS, Jiang QX, Nixon BT, Rosen MK (2012) Phase transitions in the assembly of multivalent signalling proteins. Nature 483(7389):336–340. https://doi.org/10.1038/nature10879
Li J, Guo M, Tian X, Wang X, Yang X, Wu P, Liu C, Xiao Z, Qu Y, Yin Y, Wang C, Zhang Y, Zhu Z, Liu Z, Peng C, Zhu T, Liang Q (2021) Virus-host interactome and proteomic survey reveal potential virulence factors influencing SARS-CoV-2 pathogenesis. Med (N Y) 2(1):99-112 e117. https://doi.org/10.1016/j.medj.2020.07.002
Li X, Han P, Wang G, Chen W, Wang S, Song T (2022) SDNN-PPI: self-attention with deep neural network effect on protein-protein interaction prediction. BMC Genomics 23(1):474. https://doi.org/10.1186/s12864-022-08687-2
Liu X, Yang Z, Sang S, Lin H, Wang J, Xu B (2019) Detection of protein complexes from multiple protein interaction networks using graph embedding. Artif Intell Med 96:107–115. https://doi.org/10.1016/j.artmed.2019.04.001
Liu-Wei W, Kafkas S, Chen J, Dimonaco NJ, Tegner J, Hoehndorf R (2021) DeepViral: prediction of novel virus-host interactions from protein sequences and infectious disease phenotypes. Bioinformatics. https://doi.org/10.1093/bioinformatics/btab147
Lua RC, Marciano DC, Katsonis P, Adikesavan AK, Wilkins AD, Lichtarge O (2014) Prediction and redesign of protein-protein interactions. Prog Biophys Mol Biol 116(2–3):194–202. https://doi.org/10.1016/j.pbiomolbio.2014.05.004
Matthews LR, Vaglio P, Reboul J, Ge H, Davis BP, Garrels J, Vincent S, Vidal M (2001) Identification of potential interaction networks using sequence-based searches for conserved protein-protein interactions or “interologs.” Genome Res 11(12):2120–2126. https://doi.org/10.1101/gr.205301
Meszaros B, Simon I, Dosztanyi Z (2009) Prediction of protein binding regions in disordered proteins. PLoS Comput Biol 5(5):e1000376. https://doi.org/10.1371/journal.pcbi.1000376
Mikolov T, Sutskever I, Chen K, Corrado G, Dean J (2013b) Distributed representations of words and phrases and their compositionality. NIPS’13: Proc 26th Int Conf Neural Inf Process Syst 2:3111–3119
Mikolov T, Chen K, Corrado G, Dean J (2013a) Efficient Estimation of Word Representations in Vector Space. Proceedings of Workshop at ICLR arXiv:1301.3781v1. https://doi.org/10.48550/arXiv.1301.3781
Mondal S, Narayan K, Botterbusch S, Powers I, Zheng J, James HP, Jin R, Baumgart T (2022) Multivalent interactions between molecular components involved in fast endophilin mediated endocytosis drive protein phase separation. Nat Commun 13(1):5017. https://doi.org/10.1038/s41467-022-32529-0
Murakami Y, Mizuguchi K (2014) Homology-based prediction of interactions between proteins using Averaged One-Dependence Estimators. BMC Bioinformatics 15:213. https://doi.org/10.1186/1471-2105-15-213
Murakami Y, Tripathi LP, Prathipati P, Mizuguchi K (2017) Network analysis and in silico prediction of protein-protein interactions with applications in drug discovery. Curr Opin Struct Biol 44:134–142. https://doi.org/10.1016/j.sbi.2017.02.005
Ochoa D, Pazos F (2010) Studying the co-evolution of protein families with the Mirrortree web server. Bioinformatics 26(10):1370–1371. https://doi.org/10.1093/bioinformatics/btq137
Ohue M, Matsuzaki Y, Uchikoga N, Ishida T, Akiyama Y (2014) MEGADOCK: an all-to-all protein-protein interaction prediction system using tertiary structure data. Protein Pept Lett 21(8):766–778. https://doi.org/10.2174/09298665113209990050
Orchard S, Kerrien S, Abbani S, Aranda B, Bhate J, Bidwell S, Bridge A, Briganti L, Brinkman FS, Cesareni G, Chatr-aryamontri A, Chautard E, Chen C, Dumousseau M, Goll J, Hancock RE, Hannick LI, Jurisica I, Khadake J, Lynn DJ, Mahadevan U, Perfetto L, Raghunath A, Ricard-Blum S, Roechert B, Salwinski L, Stumpflen V, Tyers M, Uetz P, Xenarios I, Hermjakob H (2012) Protein interaction data curation: the International Molecular Exchange (IMEx) consortium. Nat Methods 9(4):345–350. https://doi.org/10.1038/nmeth.1931
Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, Broackes-Carter F, Campbell NH, Chavali G, Chen C, Del-Toro N, Duesbury M, Dumousseau M, Galeota E, Hinz U, Iannuccelli M, Jagannathan S, Jimenez R, Khadake J, Lagreid A, Licata L, Lovering RC, Meldal B, Melidoni AN, Milagros M, Peluso D, Perfetto L, Porras P, Raghunath A, Ricard-Blum S, Roechert B, Stutz A, Tognolli M, van Roey K, Cesareni G, Hermjakob H (2014) The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res 42(Database issue):D358-363. https://doi.org/10.1093/nar/gkt1115
Oughtred R, Stark C, Breitkreutz BJ, Rust J, Boucher L, Chang C, Kolas N, O’Donnell L, Leung G, McAdam R, Zhang F, Dolma S, Willems A, Coulombe-Huntington J, Chatr-Aryamontri A, Dolinski K, Tyers M (2019) The BioGRID interaction database: 2019 update. Nucleic Acids Res 47(D1):D529–D541. https://doi.org/10.1093/nar/gky1079
Oughtred R, Rust J, Chang C, Breitkreutz BJ, Stark C, Willems A, Boucher L, Leung G, Kolas N, Zhang F, Dolma S, Coulombe-Huntington J, Chatr-Aryamontri A, Dolinski K, Tyers M (2021) The BioGRID database: a comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci 30(1):187–200. https://doi.org/10.1002/pro.3978
Pan XY, Zhang YN, Shen HB (2010) Large-scale prediction of human protein-protein interactions from amino acid sequence based on latent topic features. J Proteome Res 9(10):4992–5001. https://doi.org/10.1021/pr100618t
Park Y, Marcotte EM (2012) Flaws in evaluation schemes for pair-input computational predictions. Nat Methods 9(12):1134–1136. https://doi.org/10.1038/nmeth.2259
Pazos F, Valencia A (2001) Similarity of phylogenetic trees as indicator of protein-protein interaction. Protein Eng 14(9):609–614. https://doi.org/10.1093/protein/14.9.609
Pedamallu CS, Posfai J (2010) Open source tool for prediction of genome wide protein-protein interaction network based on ortholog information. Source Code Biol Med 5:8. https://doi.org/10.1186/1751-0473-5-8
Pierce B, Weng Z (2007) ZRANK: reranking protein docking predictions with an optimized energy function. Proteins 67(4):1078–1086. https://doi.org/10.1002/prot.21373
Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, Weng Z (2014) ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30(12):1771–1773. https://doi.org/10.1093/bioinformatics/btu097
Pitre S, Dehne F, Chan A, Cheetham J, Duong A, Emili A, Gebbia M, Greenblatt J, Jessulat M, Krogan N, Luo X, Golshani A (2006) PIPE: a protein-protein interaction prediction engine based on the re-occurring short polypeptide sequences between known interacting protein pairs. BMC Bioinformatics 7:365. https://doi.org/10.1186/1471-2105-7-365
Pitre S, Hooshyar M, Schoenrock A, Samanfar B, Jessulat M, Green JR, Dehne F, Golshani A (2012) Short co-occurring polypeptide regions can predict global protein interaction maps. Sci Rep 2:239. https://doi.org/10.1038/srep00239
Qi Y, Bar-Joseph Z, Klein-Seetharaman J (2006) Evaluation of different biological data and computational classification methods for use in protein interaction prediction. Proteins 63(3):490–500. https://doi.org/10.1002/prot.20865
Romero-Molina S, Ruiz-Blanco YB, Harms M, Munch J, Sanchez-Garcia E (2019) PPI-Detect: a support vector machine model for sequence-based prediction of protein-protein interactions. J Comput Chem 40(11):1233–1242. https://doi.org/10.1002/jcc.25780
Salwinski L, Miller CS, Smith AJ, Pettit FK, Bowie JU, Eisenberg D (2004) The Database of Interacting Proteins: 2004 update. Nucleic Acids Res 32(Database issue):D449-451. https://doi.org/10.1093/nar/gkh086
Sarkar D, Saha S (2019) Machine-learning techniques for the prediction of protein-protein interactions. J Biosci 44:(4). https://doi.org/10.1007/s12038-019-9909-z
Seet BT, Dikic I, Zhou MM, Pawson T (2006) Reading protein modifications with interaction domains. Nat Rev Mol Cell Biol 7(7):473–483. https://doi.org/10.1038/nrm1960
Sledzieski S, Singh R, Cowen L, Berger B (2021) D-SCRIPT translates genome to phenome with sequence-based, structure-aware, genome-scale predictions of protein-protein interactions. Cell Syst 12(10):969-982 e966. https://doi.org/10.1016/j.cels.2021.08.010
Song X-Y, Chen Z-H, Sun X-Y, You Z-H, Li L-P, Zhao Y (2018) An ensemble classifier with random projection for predicting protein–protein interactions using sequence and evolutionary information. Appl Sci 8(1):89. https://doi.org/10.3390/app8010089
Sun T, Zhou B, Lai L, Pei J (2017) Sequence-based prediction of protein protein interaction using a deep-learning algorithm. BMC Bioinformatics 18(1):277. https://doi.org/10.1186/s12859-017-1700-2
Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, Doncheva NT, Legeay M, Fang T, Bork P, Jensen LJ, von Mering C (2021) The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res 49(D1):D605–D612. https://doi.org/10.1093/nar/gkaa1074
Tripathi LP, Chen Y-A, Mizuguchi K, Murakami Y (2019) Network-based analysis for biological discovery. In: Ranganathan S, Gribskov M, Nakai K, Schönbach C (eds) Encyclopedia of Bioinformatics and Computational Biology. Academic Press, Oxford, pp 283–291. https://doi.org/10.1016/B978-0-12-809633-8.20674-2
Tsukiyama S, Hasan MM, Fujii S, Kurata H (2021) LSTM-PHV: prediction of human-virus protein-protein interactions by LSTM with word2vec. Brief Bioinform 22 (6). https://doi.org/10.1093/bib/bbab228
von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P (2002) Comparative assessment of large-scale data sets of protein-protein interactions. Nature 417(6887):399–403. https://doi.org/10.1038/nature750
Walhout AJ, Sordella R, Lu X, Hartley JL, Temple GF, Brasch MA, Thierry-Mieg N, Vidal M (2000) Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 287(5450):116–122. https://doi.org/10.1126/science.287.5450.116
Wang YB, You ZH, Li LP, Huang YA, Yi HC (2017) Detection of interactions between proteins by using Legendre moments descriptor to extract discriminatory information embedded in PSSM. Molecules 22(8):1366. https://doi.org/10.3390/molecules22081366
Wang B, Zhang L, Dai T, Qin Z, Lu H, Zhang L, Zhou F (2021) Liquid-liquid phase separation in human health and diseases. Signal Transduct Target Ther 6(1):290. https://doi.org/10.1038/s41392-021-00678-1
Warwicker J (2022) The physical basis for pH sensitivity in biomolecular structure and function, with application to the spike protein of SARS-CoV-2. Front Mol Biosci 9:834011. https://doi.org/10.3389/fmolb.2022.834011
wwPDBc (2019) Protein Data Bank: the single global archive for 3D macromolecular structure data. Nucleic Acids Res 47(D1):D520–D528. https://doi.org/10.1093/nar/gky949
Yang F, Fan K, Song D, Lin H (2020a) Graph-based prediction of protein-protein interactions with attributed signed graph embedding. BMC Bioinformatics 21(1):323. https://doi.org/10.1186/s12859-020-03646-8
Yang X, Yang S, Li Q, Wuchty S, Zhang Z (2020b) Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method. Comput Struct Biotechnol J 18:153–161. https://doi.org/10.1016/j.csbj.2019.12.005
Yang X, Yang S, Lian X, Wuchty S, Zhang Z (2021) Transfer learning via multi-scale convolutional neural layers for human-virus protein-protein interaction prediction. Bioinformatics. https://doi.org/10.1093/bioinformatics/btab533
Yao Y, Du X, Diao Y, Zhu H (2019) An integration of deep learning with feature embedding for protein-protein interaction prediction. PeerJ 7:e7126. https://doi.org/10.7717/peerj.7126
You ZH, Huang WZ, Zhang S, Huang YA, Yu CQ, Li LP (2019) An efficient ensemble learning approach for predicting protein-protein interactions by integrating protein primary sequence and evolutionary information. IEEE/ACM Trans Comput Biol Bioinf 16(3):809–817. https://doi.org/10.1109/TCBB.2018.2882423
Yu H, Luscombe NM, Lu HX, Zhu X, Xia Y, Han JD, Bertin N, Chung S, Vidal M, Gerstein M (2004) Annotation transfer between genomes: protein-protein interologs and protein-DNA regulogs. Genome Res 14(6):1107–1118. https://doi.org/10.1101/gr.1774904
Yu B, Chen C, Wang X, Yu Z, Ma A, Liu B (2021) Prediction of protein–protein interactions based on elastic net and deep forest. Expert Syst Appl 176:114876. https://doi.org/10.1016/j.eswa.2021.114876
Yu D, Chojnowski G, Rosenthal M, Kosinski J (2022) AlphaPulldown-a Python package for protein-protein interaction screens using AlphaFold-Multimer. Bioinformatics. https://doi.org/10.1093/bioinformatics/btac749
Yugandhar K, Gromiha MM (2014) Protein-protein binding affinity prediction from amino acid sequence. Bioinformatics 30(24):3583–3589. https://doi.org/10.1093/bioinformatics/btu580
Zhang QC, Petrey D, Deng L, Qiang L, Shi Y, Thu CA, Bisikirska B, Lefebvre C, Accili D, Hunter T, Maniatis T, Califano A, Honig B (2012) Structure-based prediction of protein-protein interactions on a genome-wide scale. Nature 490(7421):556–560. https://doi.org/10.1038/nature11503
Zhou X, Park B, Choi D, Han K (2018) A generalized approach to predicting protein-protein interactions between virus and host. BMC Genomics 19(Suppl 6):568. https://doi.org/10.1186/s12864-018-4924-2
Zhou YZ, Gao Y, Zheng YY (2011) Prediction of protein-protein interactions using local description of amino acid sequence. Advances in Computer Science and Education Applications, pp 254–262. https://doi.org/10.1007/978-3-642-22456-0_37
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Murakami, Y., Mizuguchi, K. Recent developments of sequence-based prediction of protein–protein interactions. Biophys Rev 14, 1393–1411 (2022). https://doi.org/10.1007/s12551-022-01038-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-022-01038-1