
Abstract
Aggregation of the amyloid-β (Aβ) peptide is strongly correlated with Alzheimer’s disease (AD). Recent research has improved our understanding of the kinetics of amyloid fibril assembly and revealed new details regarding different stages in plaque formation. Presently, interest is turning toward studying this process in a holistic context, focusing on cellular components which interact with the Aβ peptide at various junctures during aggregation, from monomer to cross-β amyloid fibrils. However, even in isolation, a multitude of factors including protein purity, pH, salt content, and agitation affect Aβ fibril formation and deposition, often producing complicated and conflicting results. The failure of numerous inhibitors in clinical trials for AD suggests that a detailed examination of the complex interactions that occur during plaque formation, including binding of carbohydrates, lipids, nucleic acids, and metal ions, is important for understanding the diversity of manifestations of the disease. Unraveling how a variety of key macromolecular modulators interact with the Aβ peptide and change its aggregation properties may provide opportunities for developing therapies. Since no protein acts in isolation, the interplay of these diverse molecules may differentiate disease onset, progression, and severity, and thus are worth careful consideration.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction: what’s in a plaque?
Amyloid plaques, first identified over 100 years ago (Alzheimer 1911), have become an indicative sign of protein misfolding diseases, of which 50 are now identified (Sipe et al. 2016). As the population of the developed world ages, amyloid pathologies are becoming an increasingly grave problem. In 2016, a reported 5.4 million Americans were living with Alzheimer’s disease (AD), perhaps the most well-known amyloid disease, with this number predicted to rise to 13.8 million by 2050 (Assoc. 2016). Thus, understanding the molecular basis of amyloid diseases is of critical importance and has recently been named one of the grand challenges of protein folding, misfolding, and degradation (Goloubinoff 2014).
Alzheimer’s disease is postulated to be caused by the formation of senile plaques from the Aβ protein, a soluble, unstructured peptide cleaved from the membrane-embedded amyloid precursor protein (APP) by β and γ secretase enzymes to a length of 38–43 amino acid residues (Knowles et al. 2014). The most well-studied forms of Aβ are the abundant 40-residue form and the highly aggregation-prone 42-residue form. The ratio of Aβ42/40 in the cerebral spinal fluid (CSF) is used as a clinical biomarker to differentiate diagnosis of AD from other forms of dementia (Wiltfang et al. 2007). The Aβ peptide is comprised of a charged N-terminal region (residues 1–22) and hydrophobic C-terminal segment (residues 23–40/42; Fig. 1). The highly hydrophobic central region, residues 16–21 (KLVFFA), is the most aggregation-prone portion of the sequence, and is alone sufficient to cause formation of insoluble fibrils (Gorevic et al. 1987; Preston et al. 2012). Aggregation of both Aβ40 and Aβ42 occur in a nucleation-dependent manner (Meisl et al. 2014), in which several copies of the unstructured peptide contact one another, presumably through hydrophobic (Kim and Hecht 2006) and/or electrostatic (Buell et al. 2013) interactions, forming oligomers and eventually an oligomeric nucleus, which is highly dependent on protein concentration and cellular conditions. Oligomers of Aβ42 in particular (which may be transient or long-lived) have been implicated as cytotoxic disease-causative agents in AD (Haass and Selkoe 2007). Following nucleus formation, aggregation proceeds rapidly through higher-order oligomers to insoluble fibrils, which contain a characteristic cross-β structure (Bonar et al. 1969; Geddes et al. 1968). These fibrils then associate, creating dense mats called plaques, which are highly stable thermodynamic sinks comprised of Aβ40, Aβ42, and other cellular components. Amyloid deposits in the AD brain include intracellular neurofibrillary tangles, principally of the protein tau, and extracellular plaques comprised of the Aβ peptide (Selkoe 2002). Both in vitro (Paravastu et al. 2008) and in vivo (Lu et al. 2013) characterization of Aβ amyloid fibrils have revealed that they are heterogeneous in nature (Eichner and Radford 2011; Tycko 2015), with different fibril morphologies potentially responsible for differences in disease progression between individuals. Plaques are also stockpiles of a wide variety of macromolecular components (Fig. 2), which interact with amyloid fibrils in a variety of ways—both known and unknown—throughout the aggregation cascade (Alexandrescu 2005), and these non-proteinaceous components of amyloid may have important physiological ramifications.

The Aβ42 peptide and its interaction with various plaque components. The sequence of the Aβ peptide, with charged residues (positive black triangle, negative white triangle). Proposed binding sites of various species discussed in this review are colored by component, using the same coloring for component name below the sequence

Aβ amyloid plaque contents. Major categories of amyloid plaque components are listed, with particular species shown below. Proteinaceous species discussed in this review are listed, but others are also found within Aβ plaques, see (Liao et al. 2004). The TEM image is comprised of aggregated Aβ42 fibrils collected on a JEOL JEM-1400 microscope, with the scale bar indicated on the image
AD can result from mutations in the Aβ peptide, APP, or related enzymes. This manifestation, termed familial Alzheimer’s disease, is rare, and accounts for <3% of cases. More commonly, AD can arise sporadically late in life, which accounts for ~97% of cases (Masters et al. 2015). Both modes of onset result in a similar disease phenotype: progressive impairment of cognition (Mayeux et al. 2011). While plaque burden is not directly correlated with disease severity (Selkoe and Hardy 2016), the Aβ peptide is regarded as a causative agent in AD (Hardy and Higgins 1992). Particularly in sporadic AD, where the initiation factors of the disease are largely unknown, cellular components are strongly suspect as potential contributors to Aβ-mediated aggregation. Recently, a number of drugs targeting the amyloid cascade have been suggested (Aisen et al. 2006; Bergamaschini et al. 2004), drawn from a variety of engineered and natural binding partners (Fig. 3). However, one of the difficulties facing AD therapeutics includes the fact that Aβ may interact with a wide variety of macromolecules which can alter its aggregation properties or toxicity in vivo and may vary between individuals.

Proposed molecules targeting Aβ aggregation: a heparin-based N-acetyl-glucosamine monosaccharide (Kisilevsky et al. 2003); b Enoxaparin, a low-molecular-weight heparin (Bergamaschini et al. 2004); c RNA aptamer β55 (Ylera et al. 2002), with bases colored as shown; d RNA aptamer E2 (Rahimi et al. 2009); e the statin Atorvastatin (Lipitor); f doxcosahexaenoic acid (DHA); g clioquinol; h PBT2; i tripeptide H-W-H (Caballero et al. 2016)
This review provides a brief overview of the major types of non-proteinaceous macromolecules which co-localize with Aβ fibrils in amyloid plaques, and details their binding, aggregation, and cross-reactivity to explore how and why these components are found in in senile plaques. Since a major focus of current AD research involves targeting the aggregation pathway, we also discuss therapeutics inspired by these molecules and their effects on Aβ aggregation. It is worth noting that many proteins also co-localize in amyloid plaques, and these have been quantified by proteomic analysis (Liao et al. 2004; Perreau et al. 2010), but will not be discussed in detail here, aside from the proteins ApoE and serum amyloid P, which are associated with lipid and carbohydrate aggregation factors, respectively (Fig. 2). By focusing on plaques, we assess the variable and complex forces exerted on aggregation of the Aβ peptide in a cellular context, toward therapeutic intervention in AD and other amyloid diseases, and provide some recommendations for future directions.
Part 1: Carbohydrates
Proteoglycans and glycosaminoglycans
The term ‘amyloid’, first employed by Rudolf Virchow (Virchow and Chance 1860), means ‘starch-like’, based on an analysis of the first plaques for molecules that were anticipated to be the principal components: starch and cellulose (Sipe and Cohen 2000). It was determined later that the carbohydrate material in plaques consisted of sulfated proteoglycans (Bitter and Muir 1966), an integral part of basement membranes (BM), extracellular surfaces which separate cells and tissue throughout the body. Proteoglycans in the BM form a dense mesh-like network which provides structural support and cellular communication (Varki and Sharon 2009). Experiments utilizing gold-conjugated lectins and fluorescence microscopy have identified that saccharides are found in the periphery of human brain tissue AD plaques (Roher et al. 1993; Szumanska et al. 1987). In particular, the proteoglycan perlecan, which contains 1–3 linear heparan sulfate (HS) glycosaminoglycan (GAG) chains linked to the core protein (Esko et al. 2009), has been shown to bind directly to fibrillar Aβ40 and Aβ42. Other proteoglycans have also been detected in AD amyloid plaques, including the extracellular matrix proteoglycans collagen XVIII and agrin, and the cell surface proteoglycans syndecan 1–3 and glypican 1 (van Horssen et al. 2003). A detailed analysis of the perlecan-binding interface indicated that the GAG HS chains, particularly the negatively charged sulfate moieties, were critical to the interaction (Kisilevsky and Snow 1988; Snow et al. 1987). Several other GAGs which contain sulfate groups have also been detected in AD plaques, including dermatan sulfate (Snow et al. 1992) and chondroitin sulfate (Dewitt et al. 1993; Oohira et al. 2000). Much of the work on the interaction between GAGs and amyloid proteins has subsequently been performed with heparin, a highly sulfated analog of HS which can be produced synthetically (Diaz-Nido et al. 2002; Meneghetti et al. 2015). Heparin binds to Aβ40 similarly to HS and was shown by Castillo and co-workers to contain a high degree of the core sulfate-binding motif present in HS (Castillo et al. 1999).
A wide variety of amyloid proteins bind GAGs, including tau (Goedert et al. 1996), Aβ40/42 (McLaurin et al. 1999a, b), amylin (islet amyloid polypeptide, IAPP) (Jha et al. 2011; Meng and Raleigh 2011), β2-microglobulin (Borysik et al. 2007; So et al. 2017), transthyretin (Bourgault et al. 2011), serum amyloid A (SAA) (Ancsin and Kisilevsky 1999), α-synuclein (Madine et al. 2009), and prion (Vieira et al. 2014; Warner et al. 2002). Due to this apparent binding ubiquity, it has been suggested that the interaction between heparin and amyloid is electrostatically-driven, which is supported by the fact that removal of all sulfate groups from heparin impairs its binding to Aβ40 (Castillo et al. 1999). An investigation of interaction sites on all known heparin-binding proteins (Cardin and Weintraub 1989; Sobel et al. 1992) yielded several generalized heparin-binding motifs: XBBXBX, XBBBXXBX, and XBBBXXBBBXXBBX, where B is a basic residue and X is any other residue. The fragments of sequence-separating basic residues suggest a possible role for protein structure in heparin binding, allowing multiple basic residues to be brought into proximity by protein folding. In support of this hypothesis, heparin has been shown to bind with differing affinity to a variety of Aβ40 fibril morphologies comprised of an identical sequence (Madine et al. 2012; Stewart et al. 2016), indicating that GAG binding, despite its apparent ubiquity, can also exhibit specificity. Additionally, individual residues on a given amyloid chain have been shown to alter heparin binding in SAA (Ancsin and Kisilevsky 1999) and Aβ1–28 (McLaurin and Fraser 2000), indicating that binding is not generic across different basic residues. An investigation of the role of sulfate groups on binding to a specific morphology of Aβ40 fibrils indicates that the geometry of the GAG molecule is also important for binding to amyloid fibrils (Lindahl et al. 1999) (Stewart et al., unpublished). Thus, the heparin–amyloid interaction is governed both by general electrostatic complementarity and more specific topological requirements for both the protein and GAG chain.
Considering the Aβ peptide specifically, GAGs have been shown to reduce cellular toxicity in Aβ25–35 and Aβ42 (Bravo et al. 2008; Woods et al. 1995), to stabilize fibrils against degradation in Aβ42 (Valle-Delgado et al. 2010), and to accelerate fibril formation in Aβ40 and Aβ42 (Castillo et al. 1999). GAGs have also been proposed to perform a templating role in amyloid aggregation, providing a scaffold for subunits to self-associate (Motamedi-Shad et al. 2009a; Solomon et al. 2011), and to attenuate cellular toxicity by favoring a benign, alternate aggregation pathway (Bravo et al. 2008; Motamedi-Shad et al. 2009b). GAG molecules are also intimately tied to AD plaque formation and amyloid burden. Recent work by Liu and co-workers removed a critical component of HS biosynthesis, the gene Ext1, creating a line of HS-deficient mice (Liu et al. 2016). In these animals, soluble Aβ clearance was increased and amyloid plaque deposition was reduced (Liu et al. 2016). Ext1 inactivation also reduced neuroinflammation as measured by a reduction in TNF-α and IL-6 inflammatory cytokines, in keeping with heparin’s traditional medicinal use as an anticoagulant (Bjork and Lindahl 1982). A related study overexpressing heparinase, the enzyme which degrades heparin and heparan sulfate, also reduced plaque burden (Jendresen et al. 2015). These studies indicate that GAGs are important for Aβ deposition in amyloid plaques. However, whether this outcome exacerbates or retards disease progression remains unclear.
Serum amyloid P: a lectin-binding protein
In addition to proteoglycans, Aβ amyloid plaques also contain carbohydrate-binding proteins whose levels are altered in AD. One of the most well-characterized of these components, found almost universally in amyloid plaques, is the Ca2+-dependent protein of the innate immune system, serum amyloid P (SAP) (Pepys et al. 1994). This five subunit pentraxin interacts with GAGs during its normal cellular function and is able to neutralize their anticoagulant activity (Williams et al. 1992). Additionally, SAP binds to a variety of amyloid proteins, including Aβ fibrils isolated from AD plaques, and stabilizes them from degradation (Tennent et al. 1995). Based on refolding studies using lactate dehydrogenase, SAP has been suggested to perform a chaperone-like role in reducing aggregation generally (Coker et al. 2000). Recent findings point to Ca2+-dependent binding of the SAP pentamer to Aβ40 in both monomeric and fibril forms (Ozawa et al. 2016), although the precise molecular details of these interactions are not known. Since SAP has the ability to bind both GAGs and amyloid fibrils, it is likely an important modulator of protein aggregation and plaque formation.
Short glycosaminoglycans as amyloid therapeutics
As noted above, heparin has historically been administered as an anticoagulant (Bjork and Lindahl 1982), a property which is increasingly recognized as important for AD (Akiyama et al. 2000; Heppner et al. 2015). As GAGs are small, natural biomolecules, they are able to cross the blood–brain barrier, alter Aβ aggregation, and mitigate cytotoxicity (Bergamaschini et al. 2009). Kisilevsky and co-workers have screened an array of different short GAGs comprised of one to three disaccharide units with the hope of outcompeting full-length GAGs and other negatively charged molecules for amyloidogenic monomeric peptides (Fraser et al. 2001; Kisilevsky and Szarek 2002; Kisilevsky et al. 2003). These authors have identified several GAG mimetics which inhibit SAA amyloid aggregation in a transgenic mouse model (Kisilevsky et al. 2003); one such molecule, a derivative of N-acetyl-glucosamine, is shown in Fig. 3a, and could logically be utilized additionally in targeting Aβ aggregation. Relatedly, Enoxaparin, a low-molecular;weight heparin, acting by a similar mechanism to the short GAGs, was shown to reduce plaque accumulation in an AD mouse model, while also reducing cytotoxicity and inflammation (Bergamaschini et al. 2004) (Fig. 3b). In a randomized pilot study, Enoxaparin was shown to increase the concentration of Aβ42 in cerebrospinal fluid. A recent study, however, has called into question the benefit of increased soluble Aβ in the treatment of AD (Cui et al. 2017), and future work will be needed to resolve the role of GAGs in altering AD symptoms.
Part 2: Nucleic acids
DNA was initially recognized as a molecule which affects protein aggregation by its ability to promote prion unfolding and conversion into an infective form (Nandi et al. 2002). More recently, nucleic acids have been shown to promote tau aggregation through template-assisted growth (Dinkel et al. 2015) and to bind aggregated Aβ40 (Camero et al. 2013). Nucleic acids also co-localize in amyloid plaques (Ginsberg et al. 1997), and, in particular, neuronal mRNA transcripts have been detected at high levels in these structures (Ginsberg et al. 1999). The binding affinity of RNA molecules to Aβ40 is in the low micromolar range (Rahimi et al. 2009), similar to the affinity for GAGs (Stewart et al. 2016), suggesting that the two molecules may compete for Aβ binding in vivo.
Recently, a systematic study of polyphosphate, the molecular precursor of the nucleic acid backbone, was shown to act as a universal accelerator of amyloid aggregation (Cremers et al. 2016). Using both intracellular and extracellular amyloid proteins, including Aβ42, in both in vitro and in vivo contexts, polyphosphate was shown to be able to accelerate amyloid fibril formation and alter toxicity, stability, and fibril morphology. This work and previous studies (Calamai et al. 2006) postulate that the repeating negatively charged segments of which nucleic acids are comprised act as a β-sheet-stabilizing scaffold for fibril formation, similarly to the role suggested for glycosaminoglycans. The nucleic acid/polyphosphate binding interface for the Aβ peptide, therefore, is most likely located in the same region as the putative GAG binding site, involving positively charged N-terminal residues (Fig. 1). However, whether nucleic acids are able to bind amyloid fibrils universally, or whether binding is more specific to the amyloid and/or nucleic acid structure, as shown for GAGs, remains unanswered.
Nucleic acids may play a larger role in aggregation than simply stabilizing Aβ fibrils in plaques, and have also been observed to affect the structural state of many cellular proteins under stress conditions. Audas and colleagues recently demonstrated that Aβ fibril formation can be a reversed in vivo, via recruitment of long noncoding RNAs (ncRNA), which fine-tune protein expression (Audas and Lee 2016). The authors identified over 180 different types of proteins, including Aβ, which localize in novel cellular compartments they label as ‘A-bodies’ in response to stress (Audas et al. 2016). These proteins contained a similar arginine-histidine sequence targeted by the ncRNAs, which is also found in the N-terminal region of the Aβ peptide (Fig. 1). These surprising findings suggest that ncRNA signals may be lost or compromised in aging, resulting in a prolonged duration of the aggregated stage. Thus, DNA and RNA appear to alter Aβ aggregation processes, as well as being found in plaques. Understanding this interaction more completely, both independently and in combination with possible competing factors such as GAGs, will be key to utilizing both sets of molecules to modulate AD.
RNA aptamers as amyloid therapeutics
RNA aptamers are short (<100 bp) segments of selection-enriched nucleic acid sequences which are able to bind tightly and specifically to amyloid proteins (Ellington and Szostak 1990; Robertson and Joyce 1990; Tuerk and Gold 1990), and thus can be used to target particular fibril epitopes or stages of disease progression. RNA aptamers are small relative to antibodies and lack the cross-reactivity that antibodies possess (Jayasena 1999). To date, RNA aptamers have been developed which limit prion infectivity (Proske et al. 2002; Rhie et al. 2003), change aggregation co-assembly mechanisms (Sarell et al. 2014), and target specific amyloidogenic proteins (Bunka et al. 2007). Aptamers have also been utilized to select for Aβ-binding partners which disrupt amyloid aggregation. For example, Ylera and colleagues developed RNA aptamers which bind Aβ40 fibrils with nanomolar affinity, which could potentially be utilized as therapeutic or diagnostic tools (Ylera et al. 2002) (Fig. 3c). Relatedly, RNA aptamers developed against Aβ40 fibrils were able to recognize these structures even when thioflavin T, a common amyloid fibril identifier, could not (Rahimi et al. 2009) (Fig. 3d). Aptamers thus provide a hopeful approach to identifying or targeting amyloid proteins. To date, however, despite the potentials of RNA aptamers, these molecules have not yet been shown to provide clinical benefit.
Part 3: Lipids
A recent assessment of lipid content of AD plaques in human brain tissue revealed that lipids co-localize with cross-β fibrils in amyloid plaques and differ in their organization and composition in the plaque core versus periphery (Kiskis et al. 2015). Lipid structures may therefore potentially trap early-stage amyloidogenic proteins, increasing their local concentration and promoting aggregation. Membranes may also induce pre-fibril forms of amyloid to form pore structures, leading to dysregulation of metal ions and other small molecules, and resulting in a host of downstream consequences for cell homeostasis.
Lipid rafts and gangliosides
Although a number of lipid surfaces have been shown to affect amyloid aggregation, lipid rafts have emerged as a key binding interface for Aβ40 and Aβ42 (Kim et al. 2006; Wong et al. 2009). Lipid rafts are heterogeneous collections of dynamic gangliosides, sphingolipids, and cholesterol molecules which laterally associate and are detergent-resistant (Simons and Ikonen 1997). These membranes are involved in cellular import/export and signal transduction, including neurotransmission (Colin et al. 2016). The ganglioside and cholesterol composition of lipid rafts has been shown to affect the oligomerization of Aβ42 (Kim et al. 2006), while ganglioside-enriched brain lipid rafts have been shown to accelerate Aβ40 fibril assembly, alter fibril morphology, and increase neurotoxicity (Matsuzaki et al. 2010; Okada et al. 2008). During binding, the soluble Aβ peptide is converted into a helical fold (Fletcher and Keire 1997; Shao et al. 1999) which, upon reaching a critical concentration, is then able to convert to a β-sheet conformation (Matsuzaki 2007). Similar aggregation pathways have been observed in IAPP (Wakabayashi and Matsuzaki 2009) and α-synuclein (Di Scala et al. 2016; Rao et al. 2010), suggesting a generic scaffold-like interface for multiple amyloid proteins. Using the dye diethylaminocoumarin, Ikeda and Matsuzaki showed that binding of Aβ40 to gangliosides involves both hydrophobic and hydrogen-bonding interactions (Ikeda and Matsuzaki 2008), by contrast with the electrostatic interactions which dominate RNA binding and are also involved in GAG binding. The authors of this study map the interaction using an Aβ29-40 fragment, which localizes the ganglioside-binding interface specifically to the C-terminal hydrophobic region of the full-length protein (Fig. 1).
Unlike other effectors of amyloid aggregation, membranes may not only induce cross-β aggregates, but may also facilitate novel amyloid structures, including pores (Arispe et al. 1993). Indeed, pore-like structures comprised of protofibrils have been observed in postmortem AD patients (Inoue 2008). Pore formation is particularly dangerous as it causes increased cellular toxicity, increased passive transport of small molecules, and ultimately cell death (Butterfield and Lashuel 2010). Aβ42 is slightly more hydrophobic than Aβ40, due to its extended C-terminus, and, since hydrophobicity is an important property for membrane interactions, differences between the two peptide forms have been assessed. Sera-Batiste and co-workers systematically monitored the aggregation properties of Aβ40 and Aβ42 in the presence of membranes of various composition over time using gel filtration. The authors observed that Aβ42 reconstituted in dodecylphosphocholine micelles produced homogenous oligomers which were able to form β-barrel pore structures, while Aβ40 reconstituted under the same conditions formed fibrils which lacked pore-like properties (Serra-Batiste et al. 2016). Computational modeling of Aβ42-lipid pores proposed that these structures could be composed of several hexameric units, which associate into a stable 36-stranded β-barrel with a diameter large enough to accommodate metal ions (Shafrir et al. 2010). These results suggest differences in the hydrophobicity of Aβ peptide sequences lead to differences in their behavior with membranes, which may reflect the more toxic nature of Aβ42 compared with Aβ40. Membranes, in particular gangliosides, may play a critical role in Aβ fibril assembly and toxicity. Their co-localization in Aβ plaques suggests that the composition and properties of lipids cannot be ignored as a contributing factor to AD.
Lipids may also be intimately involved with reactive oxygen species (ROS) generation, particularly as a source of oxygen radicals. ROS damage has been linked to membrane binding by both Aβ42 oligomers and fibrils in cell culture (Cenini et al. 2010), and may also occur by dysregulation of metal ions, potentially as a result of lipid-mediated Aβ42 pore formation (Perry et al. 2002). Additional implications of ROS will be discussed in “Part 4”.
Cholesterol and apolipoprotein E
Another key component of lipid rafts is cholesterol, a molecule which has garnered significant attention for its role in heart disease. High cholesterol diets have also been implicated in causing AD-like behavioral and pathological symptoms in laboratory animals, including increased Aβ42 production (Ullrich et al. 2010). Both cholesterol and apolipoprotein E (discussed below) have been observed in the core of AD plaques (but not diffuse plaques) of transgenic mice, suggesting a direct interaction with Aβ fibrils (Burns et al. 2003). While cholesterol is not required for Aβ oligomerization (Kim et al. 2006), it has been shown to accelerate binding of the Aβ5-16 fragment to gangliosides, by stabilizing an optimal ganglioside dimer conformation (Fantini et al. 2013). Additionally, cholesterol has been shown to bind directly to fragments of the Aβ peptide through C-terminal residues V24 and K28 based on in vitro and in silico measurements (Di Scala et al. 2013), highlighting the importance of both charge and hydrophobicity. A link between cholesterol and copper ions as AD risk factors has been proposed based on patient studies, although their combined role in affecting disease progression has not been fully determined (Morris et al. 2006).
Apolipoproteins are involved in cholesterol transport through the nervous system by binding to cell surface receptors including proteoglycans. Perhaps the most well-studied apolipoprotein in the context of AD is the E class (ApoE), which has been shown to affect Aβ production, deposition, and clearance in sporadic Alzheimer’s disease and is also found in senile plaques. In APP transgenic mice, knockout of ApoE prevented amyloid deposition; instead, the animals formed only diffuse plaques (Holtzman et al. 2000). Alleles of ApoE, containing different residues at positions 112 and 158 (Ɛ2: C112/C158, Ɛ3: C112/R158, and Ɛ4 R112/R158) regulate the binding preferences for high- (Ɛ2) versus low- (Ɛ4) density lipoproteins (Puglielli et al. 2003), which affects membrane composition. Recently, it was shown that ApoE alleles directly stimulate Aβ production, with Ɛ4 > Ɛ3 > Ɛ2 (Huang et al. 2017). The allelic variation of the isoforms therefore is closely linked to AD, with 40% of individuals with AD expressing the Ɛ4 isoform (Farrer et al. 1997). Direct binding between ApoE and the Aβ peptide has been suggested (Carter 2005; Strittmatter et al. 1993); however, Verghese and colleagues have utilized in vitro and in vivo measurements in cerebrospinal and interstitial fluid analyzed by gel filtration to show minimal binding between ApoE and soluble Aβ40/42 (Verghese et al. 2013). Interestingly, ApoE processing has been linked recently to iron metabolism, indicating a role for this component in the maintenance of brain metal homeostasis, with potential implications for AD, as described in “Part 4” (Belaidi and Bush 2016). Thus, ApoE and cholesterol are closely linked, affecting lipid membrane composition and ultimately Aβ aggregation and toxicity. Research continues into the nuances of this pathway and its implications in cognitive decline.
Lipids as therapeutics
Statins, which reduce the risk of cardiovascular disease by altering cholesterol levels, have been shown to lower the risk of developing AD (Jick et al. 2000) (the most highly-prescribed statin is shown in Fig. 3e). To date, studies assessing the role of statins on AD have been hampered by generalizations between various statins which vary in blood–brain barrier penetration and thus potentially their effectiveness, as well as differences in dosage and duration between experiments (Shepardson et al. 2011). A longitudinal study measuring rates of decline in cognition in adults with normal cognition and mild cognitive impairment who used statins (with no particular type of statin specified) found reduced cognitive decline over time in adults initially with normal cognition, but no effect on patients exhibiting mild cognitive decline (Steenland et al. 2013), relative to statin non-users. Thus, statins may prove to be a protective factor for AD. However, much more data are required to determine the duration statins must be administered to show a protective effect and whether this effect is universal. The natural product omega-3-fatty acids which contain doxcosahexaenoic acid (DHA) (Fig. 3f) affect lipid raft composition, size, and stability, resulting in changes in membrane permeability and receptor binding (Colin et al. 2016). A recent review highlights that DHA, while not effective in studies comprised of the general population, is a particularly potent therapeutic for carriers of the ApoE ε4 isoform (Yassine et al. 2017). This finding represents one of the first potential treatments for carriers of the most dangerous ApoE allele. DHA can be administered with relatively few side effects, making this an attractive, potentially long-term, strategy for older individuals who do not yet show symptoms of AD.
Part 4: Metal ions
One prolific area of research on AD is the binding of metal ions to Aβ, inspired by the finding that various metals are found concentrated in senile plaques, relative to other tissues (Faller 2009; Maynard et al. 2005; Tougu et al. 2011). Levels of zinc, iron, and copper ions in the brain, although normally tightly regulated, fluctuate substantially upon neuronal activation, resulting in pools of ions that may not be cleared as readily in aged individuals (Faller 2009). These ions may also play a role in ROS generation, which may occur through metal ion reduction (Huang et al. 1999). Direct binding of Aβ40 to Cu2+ and Zn2+ has been observed, implicating the peptide as an aberrant metal chelator or indirectly in causing lipid-based pore formation which alters the brain metal ion balance. Other metal ions have also been investigated in connection with AD including Ca2+, Mg2+, Mn2+, and Al3+. However, limited studies of these ions to date have pointed to roles as upstream or indirect effectors of amyloid aggregation (Hare et al. 2016; Khachaturian 1987; Li et al. 2013). The latter set of ions will not be addressed further here. Instead, we focus on three known effectors of AD which are found elevated in amyloid plaques: Cu2+, Zn2+, and Fe3+.
Copper ions
Perhaps the most extensively studied and well-characterized metal ion bound by Aβ is copper. This interaction depends on a number of factors, including the pH of the amyloid environment, concentration of metal ions relative to Aβ, and the oxidation state of the metal. Aβ40-copper binding in both the 2+ and 1+ oxidation states has been pinpointed principally to the three histidine residues at positions 6, 13, and 14 (Fig. 1). Copper ion binding occurs more readily at mildly acidic pH resulting in characteristic insoluble plaques, while at physiological pH, soluble Aβ40 and Aβ42 aggregates have been observed (Atwood et al. 1998; Mold et al. 2013). In a series of elegant studies using electron paramagnetic resonance, the binding site of Cu2+ was mapped principally to H6 and either H13 or H14, with the interaction region alternating on successive fibril strands of Aβ40 (Gunderson et al. 2012). Additional characterization showed that Cu2+ does not alter the fibril architecture or aggregation pathway (Karr et al. 2005; Karr et al. 2004), and could bind oligomeric (Karr and Szalai 2008; Sarell et al. 2010) or monomeric (Pedersen et al. 2015) Aβ40. One consequence of this binding arrangement is its ability to induce fibril–fibril association (cross-linking), as observed in aggregation experiments with substoichiometric Cu2+ at low pH (Karr and Szalai 2008; Sarell et al. 2010). In our own work, we have observed a Cu2+-specific effect on the aggregation rate of Aβ40 under low (pH 6.4), but not neutral (pH 7.4), conditions (Fig. 4a, b), in agreement with the importance of histidine protonation in this interaction. Interestingly, the GAG heparin has also been shown to bind Cu2+ ions, causing a change in heparin chain conformation, which may have additional implications for cooperativity or competition with the Aβ peptide (Rudd et al. 2008). The binding site for Cu1+ ions has also been characterized in Aβ40 and forms a linear binding arrangement involving H13 and H14 with similar stoichiometry to Cu2+ ions (Shearer and Szalai 2008).

The effects of substoichiometric quantities of various metal ions on the rate of aggregation of monomeric Aβ40. 25 μM monomeric Aβ40 in the presence of a CuCl2 at pH 7.5, b CuCl2 at pH 6.5, c ZnCl2 at pH 7.5, and d FeCl3 at pH 7.5. All contain 10 μM thioflavin T in 25 mM NaH2PO4 at 37 °C, and were analyzed quiescently on a Fluorostar Omega plate reader (BMG Labtech) with an excitation wavelength of 440 nm and an emission wavelength of 480 nm. Multiple replicates are shown in the same color. e TEM images of fibrils formed after 24 h. Border color indicates sample shown; from left to right: 25 μM Aβ40 (alone) at pH 7.5, 25 μM Aβ40 with 15 μM CuCl2 at pH 7.5, 25 μM Aβ40 with 25 μM CuCl2 at pH 6.5, 25 μM Aβ40 with 15 μM ZnCl2 at pH 7.5, 25 μM Aβ40 with 15 μM FeCl3 at pH 7.5
The interaction of copper ions with Aβ40 and Aβ42 has also been studied in regard to ROS generation, particularly with oligomeric and fibrillar Aβ species. However, whether Cu2+ binding to Aβ species increases or decreases ROS is debated. Mayes and co-workers have suggested that Aβ42 fibrils can degrade peroxide in a Cu2+-binding dependent manner, with the highest ROS generation at a 1:1 ratio of Aβ:Cu2+ (i.e. saturated binding) (Mayes et al. 2014). In contrast, Pedersen and colleagues demonstrated that ROS generated from oxygen and ascorbate was reduced in the presence of fibril forms of Aβ40 and α-synuclein compared with Cu2+ alone (Pedersen et al. 2016). This finding suggests that ROS production is initiated by free metal ions rather than aggregation of the Aβ peptide, and that ROS in AD plaques results from the prevalence of free, rather than bound, metal ions. Regardless of the initiating species, ROS generation is strongly correlated with AD, and oxidative damage is a major factor in disease progression (Huang et al. 2016; Perry et al. 2002).
Zinc ions
Zn2+ ions are also elevated in AD amyloid plaques, and have been shown both to accelerate (Bush et al. 1994) or retard (Abelein et al. 2015; Sarell et al. 2010) Aβ40 aggregation at physiological pH in vitro, depending on the conditions used. Under similar conditions to those used by Abelein, we observed an increase in the lag time of Aβ40 aggregation with increasing concentrations of Zn2+ ions (Fig. 4c). Similarly to Cu2+/1+, the Zn2+ binding site involves residues H13 and H14, and also the N-terminus of the protein, although binding does not appear to be mediated by histidine protonation as was observed for Cu2+/1+ (Yang et al. 2000). A detailed characterization of Zn2+ binding site by Rezaei-Ghaleh and co-workers by nuclear magnetic resonance (NMR) showed that other regions of the Aβ40 peptide, particularly residues D23–G29 (Fig. 1), may change conformation in response to Zn2+ ions, indicating that the binding interaction has global implications for Aβ structure (Rezaei-Ghaleh et al. 2011). Additionally, Zn2+ has been shown to promote nucleic acid association with Aβ42, with particular importance for histidine residues 6 and 13 (Khmeleva et al. 2016). Zn2+ has also been shown to play a protective role in ROS generation, by competing for Aβ40/42 fibril binding with Cu2+ (low μM/high pM for Cu2+ vs. low- to mid-μM for Zn2+ dissociation constants) (Faller and Hureau 2009). In the presence of both ions, ROS generation was shown to be reduced relative to Cu2+ alone (Mayes et al. 2014), suggesting, in agreement with other results (Cuajungco et al. 2000), that Zn2+ binding limits ROS generation.
Iron ions
Brain Fe3+ levels have been shown to be elevated in autopsy studies of AD patients (Loef and Walach 2012) and are correlated with oxidative damage (Casadesus et al. 2004), which Fe3+/2+, like Cu2+/1+, may promote (Wang and Wang 2016). The addition of a 10-fold molar excess of Fe3+ has been reported to alter Aβ42 fibril morphology, resulting in shorter, curved fibrils with elevated toxicity (Liu et al. 2011). In a study of the binding of 20-fold excess of various metal ions to the Aβ40 peptide, Clements and co-workers demonstrated that Zn2+ and Cu2+ binding were stronger than Fe3+ and Al3+, which were unable to displace Zn2+ (Clements et al. 1996). Substoichiometric amounts of Fe3+ did not alter the rate of Aβ40 aggregation in our kinetics survey, arguing against a significant role under the conditions tested (Fig. 4d). As mentioned previously, iron levels are directly correlated with the ApoE isoform. These findings indicate that individuals with the ApoE ε4 allele contain elevated levels of the iron storage protein ferritin in the cerebrospinal fluid (Ayton et al. 2015), which cause elevated brain-iron levels in AD. Therefore, while Fe2+/3+ ions play a role in amyloid pathology, they may do so indirectly in their role as a redox-active and pathway-associated metal ion, rather than as a direct binding partner of the Aβ peptide.
Metal ion chelators as amyloid therapeutics
A number of metal ion chelators have been investigated as possible therapeutics, with a focus on altering the soluble cellular pool of metal ions. Iodochlorhydroxyquin (clioquinol) (Fig. 3g), a chelator of copper and zinc ions, was able to reduce plaque burden and memory loss in animal models (Cherny et al. 2001) and in early-stage human clinical trials (Regland et al. 2001). In pilot phase 2 clinical trials, treatment with clioquinol was significant in reducing memory loss in patients with severe dementia, and was shown to reduce plasma Aβ42 levels while increasing plasma Zn2+ levels (Ritchie et al. 2003). A related chelator, PBT2 (Fig. 3h), was developed to be more tolerant in higher doses than clioquinol, and has undergone phase II clinical trials. In an initial 12-week study, a 250-mg dose was more effective at preventing cognitive decline than a 50-mg dose (Faux et al. 2010). However, in a longer 52-week trial, PBT2 did not reduce plaque burden or improve cognitive function to a statistically significant extent. Recently, Caballero and co-workers have designed peptide fragments containing one to two histidine residues which showed higher affinity for Cu2+ ions than the Aβ40 peptide, and also showed reduced amyloid toxicity and reduced copper-generated ROS (Caballero et al. 2016) (Fig. 3i). While these fragments are now only at a preliminary test phase, they may prove to be useful therapeutic scaffolds for future metal ion chelators. There has also been an increasing focus in patient studies on the role of dietary metal ions in AD. An overview of published clinical trials and cross-section studies (Loef and Walach 2012) concluded that most trials to date have produced inconclusive results, primarily due to the study duration or inability to control for dietary or lifestyle variables. As mentioned previously, a plausible link between copper ions and high cholesterol has emerged, but specific details of the interaction must be elucidated further (Morris et al. 2006). Taken together, these results indicate that altered metal ion chelation and/or consumption, while important for AD pathology, is not alone sufficiently potent to significantly inhibit AD, and must be considered alongside other factors.
Conclusions: commonalities, competition, and cross-coordination
Plaques are complicated assortments of aggregated protein and other co-effectors of the aggregation process (Fig. 2). The balance of such molecules in the cellular environment, under both healthy and disease conditions, may alter the Aβ aggregation rate and ability to interact with additional extracellular factors. Figure 1 shows the proposed binding sites on Aβ40/42 for a number of the molecules detailed in this review. Although a large number of binding partners may compete for the histidine residues in the N-terminal region of Aβ40/42, there are other binding sites distributed throughout the sequence, suggesting that the Aβ peptide may interact with multiple binding partners, exhibiting various charges or lack thereof, simultaneously or in succession. Additionally, due to differences between the aggregation propensities and intermediate states sampled in Aβ40 versus Aβ42 (Bitan et al. 2003; Meisl et al. 2014), preferences toward binding partners may differ between Aβ forms. This complicated interplay may be responsible for the variation observed in fibril morphology (Annamalai et al. 2016; Tycko 2015) and rate of disease progression, which can fluctuate in sporadic AD from months to decades (Komarova and Thalhauser 2011; Thalhauser and Komarova 2012).
To date, no therapeutic has been identified which is able to fully mitigate AD. Perhaps this is because many drugs to date (Fig. 3) have targeted a single extracellular factor, without considering the competition between these molecules, or the fact that such competition may vary greatly between individuals. Future therapeutic strategies must consider the complexity of amyloid aggregation, particularly how the delicate balance of interactions in the brain can not only affect Aβ but how these interactions can also affect one another. One key point of this analysis is how genetic factors, such as ApoE allele, or environmental factors, such as metal ion concentration or cholesterol consumption, alter the production or interaction of the Aβ peptide with other effectors of amyloid aggregation. Figure 5 presents a simplified view of some of these cross-interactions within the complexity of the cellular environment. Clearly, Aβ aggregation is not a simple, linear process. Instead, there is a multitude of factors which mitigate amyloid structure, toxicity, and clearance. Only when these cross-coordination events are considered can the intricacies of the amyloid aggregation cascade be understood and properly targeted.
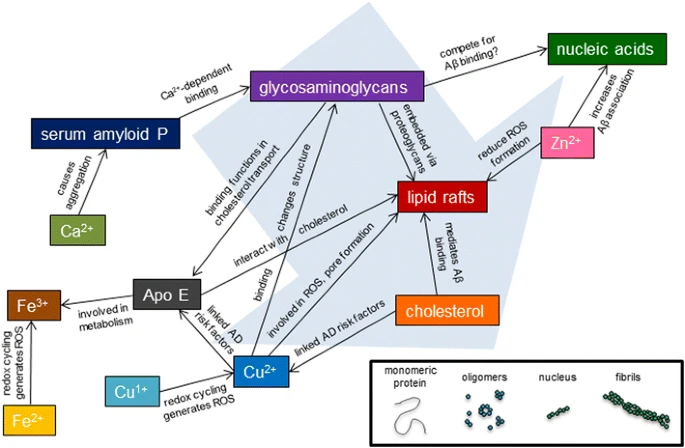
Cross-interactions of plaque components. Plaque components colored as in Fig. 1 (where applicable). Lines connecting species describe interactions. Although all these species are found in amyloid plaques (fibrils), their interactions with earlier stage Aβ is also possible. A schematic of the Aβ peptide aggregation pathway is shown at the bottom right
References
Abelein A, Graslund A, Danielsson J (2015) Zinc as chaperone-mimicking agent for retardation of amyloid beta peptide fibril formation. Proc Natl Acad Sci U S A 112:5407–5412
Aisen PS, Saumier D, Briand R, Laurin J, Gervais F, Tremblay P, Garceau D (2006) A phase II study targeting amyloid-beta with 3APS in mild to moderate Alzheimer's disease. Neurology 67:1757–1763
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL et al (2000) Inflammation and Alzheimer's disease. Neurobiol Aging 21:383–421
Alexandrescu AT (2005) Amyloid accomplices and enforcers. Protein Sci 14:1–12
Alzheimer A (1911) Concerning unsual medical cases in old age. Z Gesamte Neurol Psychiatr 4:356–385
Ancsin JB, Kisilevsky R (1999) The heparin heparan sulfate binding site on apo serum amyloid A- implications for the therapeutic intervention of amyloidosis. J Biol Chem 274:7172–7181
Annamalai K, Guhrs KH, Koehler R, Schmidt M, Michel H, Loos C, Gaffney PM, Sigurdson CJ, Hegenbart U, Schonland S et al (2016) Polymorphism of amyloid fibrils in vivo. Angew Chem Int Ed 55:4822–4825
Arispe N, Pollard HB, Rojas E (1993) Giant multilevel cation channels formed by Alzheimer's disease amyloid beta protein in bilayer membranes. Proc Natl Acad Sci U S A 90:10573–10577
Assoc., A.s (2016) 2016 Alzheimer's disease facts and figures. Alzheimers Dement 12:459–509
Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, Romano DM, Hartshorn MA, Tanzi RE, Bush AI (1998) Dramatic aggregation of Alzheimer A-beta by cu(II) is induced by conditions representing physiological acidosis. J Biol Chem 273:12817–12826
Audas TE, Audas DE, Jacob MD, Ho JJD, Khacho M, Wang ML, Perera JK, Gardiner C, Bennett CA, Head T et al (2016) Adaptation to stressors by systemic protein amyloidogenesis. Dev Cell 39:155–168
Audas TE, Lee S (2016) Stressing out over long noncoding RNA. Biochim Biophys Acta 1859:184–191
Ayton S, Faux NG, Bush AI, Initia ADN (2015) Ferritin levels in the cerebrospinal fluid predict Alzheimer's disease outcomes and are regulated by APOE. Nat Commun 6:e6760
Belaidi AA, Bush AI (2016) Iron neurochemistry in Alzheimer's disease and Parkinson's disease: targets for therapeutics. J Neurochem 139:179–197
Bergamaschini L, Rossi E, Storini C, Pizzimenti S, Distaso M, Perego C, De Luigi A, Vergani C, De Simoni MG (2004) Peripheral treatment with enoxaparin, a low molecular weight heparin, reduces plaques and beta-amyloid accumulation in a mouse model of Alzheimer's disease. J Neurosci 24:4181–4186
Bergamaschini L, Rossi E, Vergani C, De Simoni MG (2009) Alzheimer's disease: another target for heparin therapy. Sci World J 9:891–908
Bitan G, Kirkitadze MD, Lomakin A, Vollers SS, Benedek GB, Teplow DB (2003) Amyloid beta protein assembly: A-beta40 and A-beta42 oligomerize through distinct pathways. Proc Natl Acad Sci U S A 100:330–335
Bitter T, Muir H (1966) Mucopolysaccharides of whole human spleens in generalized amyloidosis. J Clin Invest 45:963–975
Bjork I, Lindahl U (1982) Mechanism of the anticoagulant action of heparin. Mol Cell Biochem 48:161–182
Bonar L, Cohen AS, Skinner MM (1969) Characterization of amyloid fibril as a cross-beta protein. Proc Soc Exp Biol Med 131:1373–1375
Borysik AJ, Morten IJ, Radford SE, Hewitt EW (2007) Specific glycosaminoglycans promote unseeded amyloid formation from beta(2)-microglobulin under physiological conditions. Kidney Int 72:174–181
Bourgault S, Solomon JP, Reixach N, Kelly JW (2011) Sulfated glycosaminoglycans accelerate transthyretin amyloidogenesis by quaternary structural conversion. Biochemistry 50:1001–1015
Bravo R, Arimon M, Valle-Delgado JJ, Garcia R, Durany N, Castel S, Cruz M, Ventura S, Fernandez-Busquets X (2008) Sulfated polysaccharides promote the assembly of A-beta(1-42) peptide into stable fibrils of reduced cytotoxicity. J Biol Chem 283:32471–32483
Buell AK, Hung P, Salvatella X, Welland ME, Dobson CM, Knowles TP (2013) Electrostatic effects in filamentous protein aggregation. Biophys J 104:1116–1126
Bunka DHJ, Mantle BJ, Morten IJ, Tennent GA, Radford SE, Stockley PG (2007) Production and characterization of RNA aptamers specific for amyloid fibril epitopes. J Biol Chem 282:34500–34509
Burns MP, Noble WJ, Olm V, Gaynor K, Casey E, LaFrancois J, Wang L, Duff K (2003) Co-localization of cholesterol, apolipoprotein E and fibrillar A-beta in amyloid plaques. Brain Res Mol Brain Res 110:119–125
Bush AI, Pettingell WH, Multhaup G, d Paradis M, Vonsattel JP, Gusella JF, Beyreuther K, Masters CL, Tanzi RE (1994) Rapid induction of Alzheimer A-beta amyloid formation by zinc. Science 265:1464–1467
Butterfield SM, Lashuel HA (2010) Amyloidogenic protein membrane interactions: mechanistic insight from model systems. Angew Chem Int Ed 49:5628–5654
Caballero AB, Terol-Ordaz L, Espargaro A, Vazquez G, Nicolas E, Sabate R, Gamez P (2016) Histidine-rich oligopeptides to lessen copper-mediated amyloid-beta toxicity. Chemistry 22:7268–7280
Calamai M, Kumita JR, Mifsud J, Parrini C, Ramazzotti M, Ramponi G, Taddei N, Chiti F, Dobson CM (2006) Nature and significance of the interactions between amyloid fibrils and biological polyelectrolytes. Biochemistry 45:12806–12815
Camero S, Ayuso JM, Barrantes A, Benitez MJ, Jimenez JS (2013) Specific binding of DNA to aggregated forms of Alzheimer's disease amyloid peptides. Int J Biol Macromol 55:201–206
Cardin AD, Weintraub HJR (1989) Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 9:21–32
Carter DB (2005) The interaction of amyloid-beta with ApoE. Subcell Biochem 38:255–272
Casadesus G, Smith MA, Zhu XW, Aliev G, Cash AD, Honda K, Petersen RB, Perry G (2004) Alzheimer's disease: evidence for a central pathogenic role of iron-mediated reactive oxygen species. J Alzheimers Dis 6:165–169
Castillo GM, Lukito W, Wight TN, Snow AD (1999) The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta-amyloid protein fibril formation. J Neurochem 72:1681–1687
Cenini G, Cecchi C, Pensalfini A, Bonini SA, Ferrari-Toninelli G, Liguri G, Memo M, Uberti D (2010) Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids 38:1101–1106
Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim YS et al (2001) Treatment with a copper-zinc chelator markedly and rapidly inhibits beta-amyloid accumulation in Alzheimer's disease transgenic mice. Neuron 30:665–676
Clements A, Allsop D, Walsh DM, Williams CH (1996) Aggregation and metal binding properties of mutant forms of the amyloid A-beta peptide of Alzheimer's disease. J Neurochem 66:740–747
Coker AR, Purvis A, Baker D, Pepys MB, Wood SP (2000) Molecular chaperone properties of serum amyloid P component. FEBS Lett 473:199–202
Colin J, Gregory-Pauron L, Lanhers MC, Claudepierre T, Corbier C, Yen FT, Malaplate-Armand C, Oster T (2016) Membrane raft domains and remodeling in aging brain. Biochimie 130:178–187
Cremers CM, Knoefler D, Gates S, Martin N, Dahl JU, Lempart J, Xie LH, Chapman MR, Galvan V, Southworth DR et al (2016) Polyphosphate: a conserved modifier of amyloidogenic processes. Mol Cell 63:768–780
Cuajungco MP, Goldstein LE, Nunomura A, Smith MA, Lim JT, Atwood CS, Huang XD, Farrag YW, Perry G, Bush AI (2000) Evidence that the beta-amyloid plaques of Alzheimer's disease represent the redox-silencing and entombment of A-beta by zinc. J Biol Chem 275:19439–19442
Cui H, King AE, Jacobson GA, Small DH (2017) Peripheral treatment with enoxaparin exacerbates amyloid plaque pathology in Tg2576 mice. J Neurosci Res 95:992–999
Dewitt DA, Silver J, Canning DR, Perry G (1993) Chondroitin sulfate poteoglycans are associated with the lesions of Alzheimer's disease. Exp Neurol 121:149–152
Di Scala C, Yahi N, Boutemeur S, Flores A, Rodriguez L, Chahinian H, Fantini J (2016) Common molecular mechanism of amyloid pore formation by Alzheimer's beta-amyloid peptide and alpha-synuclein. Sci Rep 6:e28781
Di Scala C, Yahi N, Lelievre C, Garmy N, Chahinian H, Fantini J (2013) Biochemical identification of a linear cholesterol binding domain within Alzheimer's beta-amyloid peptide. ACS Chem Neurosci 4:509–517
Diaz-Nido J, Wandosell F, Avila J (2002) Glycosaminoglycans and beta-amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 23:1323–1332
Dinkel PD, Holden MR, Matin N, Margittai M (2015) RNA binds to tau fibrils and sustains template-assisted growth. Biochemistry 54:4731–4740
Eichner T, Radford SE (2011) A diversity of assembly mechanisms of a generic amyloid fold. Mol Cell 43:8–18
Ellington AD, Szostak JW (1990) In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–822
Esko JD, Kimata K, Lindahl U (2009) Proteoglycans and sulfated glycosaminoglycans. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (eds) Essentials of glycobiology. Cold Spring Harbor, New York
Faller P (2009) Copper and zinc binding to amyloid-beta: coordination, dynamics, aggregation, reactivity and metal ion transfer. Chembiochem 10:2837–2845
Faller P, Hureau C (2009) Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-beta peptide. Dalton Trans :1080–1094
Fantini J, Yahi N, Garmy N (2013) Cholesterol accelerates the binding of Alzheimer's beta-amyloid peptide to ganglioside GM1 through a universal hydrogen bond-dependent sterol tuning of glycolipid conformation. Front Physiol 4:1–10
Farrer LA, Cupples LA, Haines JL, Hyman B, Kukull WA, Mayeux R, Myers RH, Pericak-Vance MA, Risch N, van Duijn CM (1997) Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer's disease. J Am Med Assoc 278:1349–1356
Faux NG, Ritchie CW, Gunn A, Rembach A, Tsatsanis A, Bedo J, Harrison J, Lannfelt L, Blennow K, Zetterberg H et al (2010) PBT2 rapidly improves cognition in Alzheimer's disease: additional phase II analyses. J Alzheimers Dis 20:509–516
Fletcher TG, Keire DA (1997) The interaction of beta-amyloid protein fragment (12-28) with lipid environments. Protein Sci 6:666–675
Fraser PE, Darabie AA, McLaurin J (2001) Amyloid-beta interactions with chondroitin sulfate-derived monosaccharides and disaccharides- implications for drug development. J Biol Chem 276:6412–6419
Geddes AJ, Parker KD, Atkins EDT, Beighton E (1968) Cross-beta conformation in proteins. J Mol Biol 32:343–358
Ginsberg SD, Crino PB, Hemby SE, Weingarten JA, Lee VMY, Eberwine JH, Trojanowski JQ (1999) Predominance of neuronal mRNAs in individual Alzheimer's disease senile plaques. Ann Neurol 45:174–181
Ginsberg SD, Crino PB, Lee VMY, Eberwine JH, Trojanowski JQ (1997) Sequestration of RNA in Alzheimer's disease neurofibrillary tangles and senile plaques. Ann Neurol 41:200–209
Goedert M, Jakes R, Spillantini MG, Hasegawa M, Smith MJ, Crowther RA (1996) Assembly of microtubule-associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383:550–553
Goloubinoff P (2014) Recent and future grand challenges in protein folding, misfolding, and degradation. Front Mol Biosci 1:1–3
Gorevic PD, Castano EM, Sarma R, Frangione B (1987) Ten to fourteen residue peptides of Alzheimer's disease protein are sufficient for amyloid fibril formation and its characteristic X-ray diffraction pattern. Biochem Biophys Res Commun 147:854–862
Gunderson WA, Hernandez-Guzman J, Karr JW, Sun L, Szalai VA, Warncke K (2012) Local structure and global patterning of Cu2+ binding in fibrillar amyloid-beta [Abeta(1-40)] protein. J Am Chem Soc 134:18330–18337
Haass C, Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid-beta peptide. Nat Rev Mol Cell Biol 8:101–112
Hardy JA, Higgins GA (1992) Alzheimer's disease: the amyloid cascade hypothesis. Science 256:184–185
Hare DJ, Faux NG, Roberts BR, Volitakis I, Martins RN, Bush AI (2016) Lead and manganese levels in serum and erythrocytes in Alzheimer's disease and mild cognitive impairment: results from the Australian imaging, biomarkers and lifestyle flagship study of Ageing. Metallomics 8:628–632
Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer's disease. Nat Rev Neurosci 16:358–372
Holtzman DM, Bales KR, Tenkova T, Fagan AM, Parsadanian M, Sartorius LJ, Mackey B, Olney J, McKeel D, Wozniak D et al (2000) Apolipoprotein E isoform-dependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 97:2892–2897
Huang XD, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, Cuajungco MP, Gray DN, Lim J, Moir RD et al (1999) The A-beta peptide of Alzheimer's disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry 38:7609–7616
Huang WJ, Zhang X, Chen WW (2016) Role of oxidative stress in Alzheimer's disease. Biomed Rep 4:519–522
Huang YWA, Zhou B, Wernig M, Sudhof TC (2017) ApoE2, ApoE3, and ApoE4 differentially stimulate APP transcription and A-beta secretion. Cell 168:427–441
Ikeda K, Matsuzaki K (2008) Driving force of binding of amyloid-beta protein to lipid bilayers. Biochem Biophys Res Commun 370:525–529
Inoue S (2008) In situ A-beta pores in AD brain are cylindrical assembly of A-beta protofilaments. Amyloid 15:223–233
Jayasena SD (1999) Aptamers: an emerging class of molecules that rival antibodies in diagnostics. Clin Chem 45:1628–1650
Jendresen CB, Cui H, Zhang X, Vlodavsky I, Nilsson LNG, Li JP (2015) Overexpression of heparanase lowers the amyloid burden in amyloid-beta precursor protein transgenic mice. J Biol Chem 290:5053–5064
Jha S, Patil SM, Gibson J, Nelson CE, Alder NN, Alexandrescu AT (2011) Mechanism of amylin fibrillization enhancement by heparin. J Biol Chem 286:22894–22904
Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA (2000) Statins and the risk of dementia. Lancet 356:1627–1631
Karr JW, Akintoye H, Kaupp LJ, Szalai VA (2005) N-terminal deletions modify the Cu2+ binding site in amyloid-beta. Biochemistry 44:5478–5487
Karr JW, Kaupp LJ, Szalai VA (2004) Amyloid-beta binds Cu2+ in a mononuclear metal ion binding site. J Am Chem Soc 126:13534–13538
Karr JW, Szalai VA (2008) Cu(II) binding to monomeric, oligomeric, and fibrillar forms of the Alzheimer's disease amyloid-beta peptide. Biochemistry 47:5006–5016
Khachaturian ZS (1987) Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging 8:345–346
Khmeleva SA, Radko SP, Kozin SA, Kiseleva YY, Mezentsev YV, Mitkevich VA, Kurbatov LK, Ivanov AS, Makarov AA (2016) Zinc-mediated binding of nucleic acids to amyloid-beta aggregates: role of histidine residues. J Alzheimers Dis 54:809–819
Kim W, Hecht MH (2006) Generic hydrophobic residues are sufficient to promote aggregation of the Alzheimer's A-beta42 peptide. Proc Natl Acad Sci U S A 103:15824–15829
Kim SI, Yi JS, Ko YG (2006) Amyloid-beta oligomerization is induced by brain lipid rafts. J Cell Biochem 99:878–889
Kisilevsky R, Snow A (1988) The potential significance of sulfated glycosaminoglycans as a common constituent of all amyloids. Med Hypotheses 26:231–236
Kisilevsky R, Szarek WA (2002) Novel glycosaminoglycan precursors as anti-amyloid agents, part II. J Mol Neurosci 19:45–50
Kisilevsky R, Szarek WA, Ancsin J, Bhat S, Li ZJ, Marone S (2003) Novel glycosaminoglycan precursors as anti-amyloid agents, part III. J Mol Neurosci 20:291–297
Kiskis J, Fink H, Nyberg L, Thyr J, Li JY, Enejder A (2015) Plaque-associated lipids in Alzheimer's diseased brain tissue visualized by nonlinear microscopy. Sci Rep 5:e13489
Knowles TPJ, Vendruscolo M, Dobson CM (2014) The amyloid state and its association with protein misfolding diseases. Nat Rev Mol Cell Biol 15:496–496
Komarova NL, Thalhauser CJ (2011) High degree of heterogeneity in Alzheimer's disease progression patterns. PLoS Comput Biol 7:e1002251
Li W, Yu J, Liu Y, Huang X, Abumaria N, Zhu Y, Huang X, Xiong W, Ren C, Liu XG et al (2013) Elevation of brain magnesium prevents and reverses cognitive deficits and synaptic loss in Alzheimer's disease mouse model. J Neurosci 33:8423–8441
Liao L, Cheng D, Wang J, Duong DM, Losik TG, Gearing M, Rees HD, Lah JJ, Levey AI, Peng J (2004) Proteomic characterization of postmortem amyloid plaques isolated by laser capture microdissection. J Biol Chem 279:37061–37068
Lindahl B, Westling C, Gimenez-Gallego G, Lindahl U, Salmivirta M (1999) Common binding sites for beta-amyloid fibrils and fibroblast growth factor-2 in heparan sulfate from human cerebral cortex. J Biol Chem 274:30631–30635
Liu B, Moloney A, Meehan S, Morris K, Thomas SE, Serpell LC, Hider R, Marciniak SJ, Lomas DA, Crowther DC (2011) Iron promotes the toxicity of amyloid-beta peptide by impeding its ordered aggregation. J Biol Chem 286:4248–4256
Liu CC, Zhao N, Yamaguchi Y, Cirrito JR, Kanekiyo T, Holtzman DM, Bu GJ (2016) Neuronal heparan sulfates promote amyloid pathology by modulating brain amyloid-beta clearance and aggregation in Alzheimer's disease. Sci Transl Med 8:e332ra344
Loef M, Walach H (2012) Copper and iron in Alzheimer's disease: a systematic review and its dietary implications. Br J Nutr 107:7–19
Lu JX, Qiang W, Yau WM, Schwieters CD, Meredith SC, Tycko R (2013) Molecular structure of beta-amyloid fibrils in Alzheimer’s disease brain tissue. Cell 154:1257–1268
Madine J, Clayton JC, Yates EA, Middleton DA (2009) Exploiting a C13-labelled heparin analogue for in situ solid-state NMR investigations of peptide-glycan interactions within amyloid fibrils. Org Biomol Chem 7:2414–2420
Madine J, Pandya MJ, Hicks MR, Rodger A, Yates EA, Radford SE, Middleton DA (2012) Site-specific identification of an a fibril-heparin interaction site by using solid-state NMR spectroscopy. Angew Chem Int Ed 51:13140–13143
Masters CL, Bateman R, Blennow K, Rowe CC, Sperling RA, Cummings JL (2015) Alzheimer's disease. Nat Rev Dis Primers 1:15056–15074
Matsuzaki K (2007) Physicochernical interactions of amyloid peptide with lipid bilayers. Biochim Biophys Acta-Biomembr 1768:1935–1942
Matsuzaki K, Kato K, Yanagisawa K (2010) A-beta polymerization through interaction with membrane gangliosides. Biochim Biophys Acta-Mol Cell Biol 1801:868–877
Mayes J, Tinker-Mill C, Kolosov O, Zhang H, Tabner BJ, Allsop D (2014) Beta-amyloid fibrils in Alzheimer's disease are not inert when bound to copper ions but can degrade hydrogen peroxide and generate reactive oxygen species. J Biol Chem 289:12052–12062
Mayeux R, Reitz C, Brickman AM, Haan MN, Manly JJ, Glymour MM, Weiss CC, Yaffe K, Middleton L, Hendrie HC et al (2011) Operationalizing diagnostic criteria for Alzheimer's disease and other age-related cognitive impairment, part 1. Alzheimers Dement 7:15–34
Maynard CJ, Bush AI, Masters CL, Cappai R, Li QX (2005) Metals and amyloid-beta in Alzheimer's disease. Int J Exp Pathol 86:147–159
McLaurin J, Franklin T, Kuhns WJ, Fraser PE (1999a) A sulfated proteoglycan aggregation factor mediates amyloid-beta peptide fibril formation and neurotoxicity. Amyloid 6:233–243
McLaurin J, Franklin T, Zhang XQ, Deng JP, Fraser PE (1999b) Interactions of Alzheimer amyloid-beta peptides with glycosaminoglycans - effects on fibril nucleation and growth. Eur J Biochem 266:1101–1110
McLaurin J, Fraser PE (2000) Effect of amino acid substitutions on Alzheimer's amyloid-beta peptide-glycosaminoglycan interactions. Eur J Biochem 267:6353–6361
Meisl G, Yang XT, Hellstrand E, Frohm B, Kirkegaard JB, Cohen SIA, Dobson CM, Linse S, Knowles TPJ (2014) Differences in nucleation behavior underlie the contrasting aggregation kinetics of the A-beta40 and A-beta42 peptides. Proc Natl Acad Sci U S A 111:9384–9389
Meneghetti MC, Hughes AJ, Rudd TR, Nader HB, Powell AK, Yates EA, Lima MA (2015) Heparan sulfate and heparin interactions with proteins. J R Soc Interface 12:e20150589
Meng FL, Raleigh DP (2011) Inhibition of glycosaminoglycan-mediated amyloid formation by islet amyloid polypeptide and proIAPP processing intermediates. J Mol Biol 406:491–502
Mold M, Ouro-Gnao L, Wieckowski BM, Exley C (2013) Copper prevents amyloid-beta(1-42) from forming amyloid fibrils under near-physiological conditions in vitro. Sci Rep 3:e1256
Morris MC, Evans DA, Tangney CC, Bienias JL, Schneider JA, Wilson RS, Scherr PA (2006) Dietary copper and high saturated and trans fat intakes associated with cognitive decline. Arch Neurol 63:1085–1088
Motamedi-Shad N, Monsellier E, Chiti F (2009a) Amyloid formation by the model protein muscle acylphosphatase is accelerated by heparin and heparan sulphate through a scaffolding-based mechanism. J Biochem 146:805–814
Motamedi-Shad N, Monsellier E, Torrassa S, Relini A, Chiti F (2009b) Kinetic analysis of amyloid formation in the presence of heparan sulfate. J Biol Chem 284:29921–29934
Nandi PK, Leclerc E, Nicole JC, Takahashi M (2002) DNA-induced partial unfolding of prion protein leads to its polymerisation to amyloid. J Mol Biol 322:153–161
Okada T, Ikeda K, Wakabayashi M, Ogawa M, Matsuzaki K (2008) Formation of toxic A-beta(1-40) fibrils on GM1 ganglioside-containing membranes mimicking lipid rafts: polymorphisms in A-beta(1-40) fibrils. J Mol Biol 382:1066–1074
Oohira A, Matsui F, Tokita Y, Yamauchi S, Aono S (2000) Molecular interactions of neural chondroitin sulfate proteoglycans in the brain development. Arch Biochem Biophys 374:24–34
Ozawa D, Nomura R, Mangione PP, Hasegawa K, Okoshi T, Porcari R, Bellotti V, Naiki H (2016) Multifaceted anti-amyloidogenic and pro-amyloidogenic effects of C-reactive protein and serum amyloid P component in vitro. Sci Rep 6:e29077
Paravastu AK, Leapman RD, Yau WM, Tycko R (2008) Molecular structural basis for polymorphism in Alzheimer's beta-amyloid fibrils. Proc Natl Acad Sci U S A 105:18349–18354
Pedersen JT, Borg CB, Michaels TC, Knowles TP, Faller P, Teilum K, Hemmingsen L (2015) Aggregation-prone amyloid-beta Cu(II) species formed on the millisecond timescale under mildly acidic conditions. Chembiochem 16:1293–1297
Pedersen JT, Chen SW, Borg CB, Ness S, Bahl JM, Heegaard NH, Dobson CM, Hemmingsen L, Cremades N, Teilum K (2016) Amyloid-beta and alpha-synuclein decrease the level of metal-catalyzed reactive oxygen species by radical scavenging and redox silencing. J Am Chem Soc 138:3966–3969
Pepys MB, Rademacher TW, Amatayakul-Chantler S, Williams P, Noble GE, Hutchinson WL, Hawkins PN, Nelson SR, Gallimore JR, Herbert J et al (1994) Human serum amyloid P component is an invariant constituent of amyloid deposits and has a uniquely homogeneous glycostructure. Proc Natl Acad Sci U S A 91:5602–5606
Perreau VM, Orchard S, Adlard PA, Bellingham SA, Cappai R, Ciccotosto GD, Cowie TF, Crouch PJ, Duce JA, Evin G et al (2010) A domain level interaction network of amyloid precursor protein and A-beta of Alzheimer's disease. Proteomics 10:2377–2395
Perry G, Nunomura A, Cash AD, Taddeo MA, Hirai K, Aliev G, Avila J, Wataya T, Shimohama S, Atwood CS et al (2002) Reactive oxygen: its sources and significance in Alzheimer's disease. Ageing Dement 62:69–75
Preston GW, Radford SE, Ashcroft AE, Wilson AJ (2012) Covalent cross-linking within supramolecular peptide structures. Anal Chem 84:6790–6797
Proske D, Gilch S, Wopfner F, Schatzl HM, Winnacker EL, Famulok M (2002) Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 3:717–725
Puglielli L, Tanzi RE, Kovacs DM (2003) Alzheimer's disease: the cholesterol connection. Nat Neurosci 6:345–351
Rahimi F, Murakami K, Summers JL, Chen CHB, Bitan G (2009) RNA aptamers generated against oligomeric A-beta40 recognize common amyloid aptatopes with low specificity but high sensitivity. PLoS ONE 4:e7694
Rao JN, Jao CC, Hegde BG, Langen R, Ulmer TS (2010) A combinatorial NMR and EPR approach for evaluating the structural ensemble of partially folded proteins. J Am Chem Soc 132:8657–8668
Regland B, Lehmann W, Abedini I, Blennow K, Jonsson M, Karlsson I, Sjogren M, Wallin A, Xilinas M, Gottfries CG (2001) Treatment of Alzheimer’s disease with clioquinol. Dement Geriatr Cogn Disord 12:408–414
Rezaei-Ghaleh N, Giller K, Becker S, Zweckstetter M (2011) Effect of zinc binding on beta-amyloid structure and dynamics: implications for A-beta aggregation. Biophys J 101:1202–1211
Rhie A, Kirby L, Sayer N, Wellesley R, Disterer P, Sylvester I, Gill A, Hope J, James W, Tahiri-Alaoui A (2003) Characterization of 2′-fluoro-RNA aptamers that bind preferentially to disease-associated conformations of prion protein and inhibit conversion. J Biol Chem 278:39697–39705
Ritchie CW, Bush AI, Mackinnon A, Macfarlane S, Mastwyk M, MacGregor L, Kiers L, Cherny R, Li QX, Tammer A et al (2003) Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting A-beta amyloid deposition and toxicity in Alzheimer's disease: a pilot phase 2 clinical trial. Arch Neurol 60:1685–1691
Robertson DL, Joyce GF (1990) Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 344:467–468
Roher AE, Palmer KC, Yurewicz EC, Ball MJ, Greenberg BD (1993) Morphological and biochemical analyses of amyloid plaque core proteins purified from Alzheimer's disease brain tissue. J Neurochem 61:1916–1926
Rudd TR, Skidmore MA, Guimond SE, Guerrini M, Cosentino C, Edge R, Brown A, Clarke DT, Torri G, Turnbull JE et al (2008) Site-specific interactions of copper(II) ions with heparin revealed with complementary (SRCD, NMR, FTIR and EPR) spectroscopic techniques. Carbohydr Res 343:2184–2193
Sarell CJ, Karamanos TK, White SJ, Bunka DHJ, Kalverda AP, Thompson GS, Barker AM, Stockley PG, Radford SE (2014) Distinguishing closely related amyloid precursors using an RNA aptamer. J Biol Chem 289:26859–26871
Sarell CJ, Wilkinson SR, Viles JH (2010) Substoichiometric levels of Cu2+ ions accelerate the kinetics of fiber formation and promote cell toxicity of amyloid-{beta} from Alzheimer disease. J Biol Chem 285:41533–41540
Selkoe DJ (2002) Alzheimer's disease is a synaptic failure. Science 298:789–791
Selkoe DJ, Hardy J (2016) The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med 8:595–608
Serra-Batiste M, Ninot-Pedrosa M, Bayoumi M, Gairi M, Maglia G, Carulla N (2016) A-beta42 assembles into specific beta-barrel pore-forming oligomers in membrane-mimicking environments. Proc Natl Acad Sci U S A 113:10866–10871
Shafrir Y, Durell S, Arispe N, Guy HR (2010) Models of membrane-bound Alzheimer's A-beta peptide assemblies. Proteins 78:3473–3487
Shao HY, Jao SC, Ma K, Zagorski MG (1999) Solution structures of micelle-bound amyloid-beta(1-40) and -beta(1-42) peptides of Alzheimer's disease. J Mol Biol 285:755–773
Shearer J, Szalai VA (2008) The amyloid-beta peptide of Alzheimer's disease binds Cu(I) in a linear bis-his coordination environment: insight into a possible neuroprotective mechanism for the amyloid-beta peptide. J Am Chem Soc 130:17826–17835
Shepardson NE, Shankar GM, Selkoe DJ (2011) Cholesterol level and statin use in Alzheimer disease I. Review of epidemiological and preclinical studies. Arch Neurol 68:1239–1244
Simons K, Ikonen E (1997) Functional rafts in cell membranes. Nature 387:569–572
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, Westermark P (2016) Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 nomenclature guidelines. Amyloid 23:209–213
Sipe JD, Cohen AS (2000) Review: history of the amyloid fibril. J Struct Biol 130:88–98
Snow AD, Mar H, Nochlin D, Kresse H, Wight TN (1992) Peripheral distribution of dermatan sulfate proteoglycans (decorin) in amyloid-containing plaques and their presence in neurofibrillary tangles of Alzheimer's disease. J Histochem Cytochem 40:105–113
Snow AD, Willmer J, Kisilevsky R (1987) Sulfated glycosaminoglycans- a common constituent of all amyloids. Lab Investig 56:120–123
So M, Hata Y, Naiki H, Goto Y (2017) Heparin-induced amyloid fibrillation of beta2-microglobulin explained by solubility and a supersaturation-dependent conformational phase diagram. Protein Sci 26:1024–1036
Sobel M, Soler DF, Kermode JC, Harris RB (1992) Localization and characterization of a heparin binding domain peptide of human von Willebrand factor. J Biol Chem 267:8857–8862
Solomon JP, Bourgault S, Powers ET, Kelly JW (2011) Heparin binds 8 kDa gelsolin cross-beta-sheet oligomers and accelerates amyloidogenesis by hastening fibril extension. Biochemistry 50:2486–2498
Steenland K, Zhao LP, Goldstein FC, Levey AI (2013) Statins and cognitive decline in older adults with normal cognition or mild cognitive impairment. J Am Geriatr Soc 61:1449–1455
Stewart KL, Hughes E, Yates EA, Akien GR, Huang TY, Lima MA, Rudd TR, Guerrini M, Hung SC, Radford SE et al (2016) Atomic details of the interactions of glycosaminoglycans with amyloid-beta fibrils. J Am Chem Soc 138:8328–8331
Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericakvance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD (1993) Binding of human apolipoprotein E to synthetic amyloid-beta peptide - isoform-specific effects and implications for late onset Alzheimer’s disease. Proc Natl Acad Sci U S A 90:8098–8102
Szumanska G, Vorbrodt AW, Mandybur TI, Wisniewski HM (1987) Lectin histochemistry of plaques and tangles in Alzheimer’s disease. Acta Neuropathol 73:1–11
Tennent GA, Lovat LB, Pepys MB (1995) Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer's disease and systemic amyloidosis. Proc Natl Acad Sci U S A 92:4299–4303
Thalhauser CJ, Komarova NL (2012) Alzheimer's disease: rapid and slow progression. J R Soc Interface 9:119–126
Tougu V, Tiiman A, Palumaa P (2011) Interactions of Zn(II) and Cu(II) ions with Alzheimer's amyloid-beta peptide. Metal ion binding, contribution to fibrillization and toxicity. Metallomics 3:250–261
Tuerk C, Gold L (1990) Systematic evolution of ligands by exponential enrichment - RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–510
Tycko R (2015) Amyloid polymorphism: structural basis and neurobiological relevance. Neuron 86:632–645
Ullrich C, Pirchl M, Humpel C (2010) Hypercholesterolemia in rats impairs the cholinergic system and leads to memory deficits. Mol Cell Neurosci 45:408–417
Valle-Delgado JJ, Alfonso-Prieto M, de Groot NS, Ventura S, Samitier J, Rovira C, Fernandez-Busquets X (2010) Modulation of A-beta(42) fibrillogenesis by glycosaminoglycan structure. FASEB J 24:4250–4261
van Horssen J, Wesseling P, van den Heuvel LP, de Waal RM, Verbeek MM (2003) Heparan sulphate proteoglycans in Alzheimer’s disease and amyloid-related disorders. Lancet Neurol 2:482–492
Varki A, Sharon N (2009) Historical background and overview. In: Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, Etzler ME (eds) Essentials of glycobiology. Cold Spring Harbor, New York
Verghese PB, Castellano JM, Garai K, Wang YN, Jiang H, Shah A, Bu GJ, Frieden C, Holtzman DM (2013) ApoE influences amyloid-beta clearance despite minimal apoE/A beta association in physiological conditions. Proc Natl Acad Sci U S A 110:E1807–E1816
Vieira TCRG, Cordeiro Y, Caughey B, Silva JL (2014) Heparin binding confers prion stability and impairs its aggregation. FASEB J 28:2667–2676
Virchow RLK, Chance F (1860) Cellular pathology as based upon physiological and pathological histology. Twenty lectures delivered in the pathological Institute of Berlin during the months of February, March, and April, 1858. Churchill, London
Wakabayashi M, Matsuzaki K (2009) Ganglioside-induced amyloid formation by human islet amyloid polypeptide in lipid rafts. FEBS Lett 583:2854–2858
Wang P, Wang ZY (2016) Metal ions influx is a double edged sword for the pathogenesis of Alzheimer's disease. Ageing Res Rev 35:265–290
Warner RG, Hundt C, Weiss S, Turnbull JE (2002) Identification of the heparan sulfate binding sites in the cellular prion protein. J Biol Chem 277:18421–18430
Williams EC, Huppert BJ, Asakura S (1992) Neutralization of the anticoagulant effects of glycosaminoglycans by serum amyloid-P component - comparison with other plasma and platelet proteins. J Lab Clin Med 120:159–167
Wiltfang J, Esselmann H, Bibl M, Hull M, Hampel H, Kessler H, Frolich L, Schroder J, Peters O, Jessen F et al (2007) Amyloid beta peptide ratio 42/40 but not A-beta 42 correlates with phospho-Tau in patients with low- and high-CSF A-beta40 load. J Neurochem 101:1053–1059
Wong PT, Schauerte JA, Wisser KC, Ding H, Lee EL, Steel DG, Gafni A (2009) Amyloid-beta membrane binding and permeabilization are distinct processes influenced separately by membrane charge and fluidity. J Mol Biol 386:81–96
Woods AG, Cribbs DH, Whittemore ER, Cotman CW (1995) Heparan sulfate and chondroitin sulfate glycosaminoglycan attenuate beta-amyloid(25-35) induced neurodegeneration in cultured hippocampal neurons. Brain Res 697:53–62
Yang DS, McLaurin J, Qin K, Westaway D, Fraser PE (2000) Examining the zinc binding site of the amyloid-beta peptide. Eur J Biochem 267:6692–6698
Yassine HN, Braskie MN, Mack WJ, Castor KJ, Fonteh AN, Schneider LS, Harrington MG, Chui HC (2017) Association of docosahexaenoic acid supplementation with Alzheimer's disease stage in apolipoprotein E epsilon4 carriers: a review. J Am Med Assoc Neurol 74:339–347
Ylera F, Lurz R, Erdmann VA, Furste JP (2002) Selection of RNA aptamers to the Alzheimer’s disease amyloid peptide. Biochem Bioph Res Commun 290:1583–1588
Acknowledgements
The authors would like to thank the BBSRC (UK; BB/K01451X/1 and BB/K015958/1) and The Wellcome Trust (089311/Z/09/Z) for funding.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interest
Katie L. Stewart declares that she has no conflicts of interest. Sheena E. Radford declares that she has no conflicts of interest.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Additional information
This article is part of a Special Issue on ‘IUPAB Edinburgh Congress’ edited by Damien Hall
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Stewart, K.L., Radford, S.E. Amyloid plaques beyond Aβ: a survey of the diverse modulators of amyloid aggregation. Biophys Rev 9, 405–419 (2017). https://doi.org/10.1007/s12551-017-0271-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12551-017-0271-9