
Abstract
Introduction
Fixed-dose combinations (FDCs) of angiotensin II receptor blockers, calcium channel blockers, and statins are conventional therapeutic interventions prescribed for cardiovascular diseases. This study aimed at drawing a comparison between the pharmacokinetics and safety of an FDC and the corresponding individual formulations in healthy subjects.
Methods
A randomized, open-label, single-dose, three-sequence, three-period, partially repeated crossover study was conducted with a cohort of healthy volunteers. A 14-day washout period was maintained between each of the three periods. In this study, candesartan cilexetil, amlodipine, and atorvastatin was administered orally as FDCs of 16/10/40 mg in study 1 and 16/5/20 mg in study 2. The maximum plasma concentration (Cmax) and area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration (AUClast) of candesartan, amlodipine, and atorvastatin were estimated as the geometric mean ratios (GMRs) and 90% confidence intervals (CIs) of the FDC to individual formulations. If the within-subject coefficient of variation (CVwr) of Cmax was greater than 0.3, the bioequivalence (BE) range calculated using the reference-scaled average bioequivalence was used to assess whether the 90% CI was within the BE range.
Results
The GMRs (90% CIs) for the AUClast for candesartan and amlodipine were 0.9612 (0.9158–1.0089)/0.9965 (0.9550–1.0397) and 1.0033 (0.9800–1.0271)/1.0067 (0.9798–1.0344), and the GMRs (90% CIs) for Cmax were 0.9600 (0.8953–1.0294)/0.9851 (0.9368–1.0359) and 1.0198 (0.9950–1.0453)/1.0003 (0.9694–1.0321) in studies 1 and 2, respectively. The extended BE ranges calculated from the CVwr of the Cmax of atorvastatin were 0.7814–1.2797 and 0.7415–1.3485, respectively. The GMRs (90% CIs) for the AUClast of atorvastatin were 1.0532 (1.0082–1.1003)/1.0252 (0.9841–1.0680), and the GMRs (90% CIs) for Cmax were 1.0630 (0.9418–1.1997)/0.9888 (0.8792–1.1120) in studies 1 and 2, respectively.
Conclusion
The Cmax and AUClast values of candesartan cilexetil/amlodipine/atorvastatin 16/10/40 mg and 16/5/20 mg, respectively, were within the BE ranges. There were no clinically significant differences in safety between the two formulations.
Trial Registration
ClinicalTrials.gov identifier, study 1: NCT04478097; study 2: NCT04627207.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
There is a limited availability of licensed fixed-dose combinations of angiotensin receptor blockers, calcium channel blockers, and statins globally for patients with cardiovascular disease. |
The phase 1 clinical trial was conducted to compare the pharmacokinetics and safety of a fixed-dose combination of candesartan cilexetil, amlodipine, and atorvastatin and the co-administration of the corresponding individual formulations. |
What was learned from the study? |
The systemic exposure of candesartan cilexetil/amlodipine/atorvastatin 16/10/40 mg and 16/5/20 mg fixed-dose combination and that of the co-administered corresponding individual formulations were within the pharmacokinetic bioequivalence ranges. |
The fixed-dose combination of candesartan cilexetil/amlodipine/atorvastatin 16/10/40 mg and 16/5/20 mg demonstrates a favorable safety profile and is anticipated to be utilized in real-world patients with cardiovascular disease. |
Introduction
Cardiovascular diseases (CVD) have exhibited a decline in their mortality rates over the past two decades; nonetheless, hypertension continues to be the predominant chronic disorder in the elderly population [1, 2]. The incidence of CVD is expected to increase in tandem with the aging population, which consequently warrants management through the implementation of therapeutic interventions. The benefits and health risks associated with prolonged drug therapy for elderly patients with hypertension remain controversial. On average, elderly patients are prescribed 5.3–6.9 medications, with approximately 44.2–57.7% of the patients being administered more than 5 medications, and 9.1–23.2% consuming more than 10 medications [3,4,5,6,7,8,9]. The elderly population is more susceptible to polypharmacy owing to age-related modifications in the pharmacokinetics and pharmacodynamics of prescribed drugs, which consequently heighten the likelihood of adverse drug reactions (ADRs) [10]. Nevertheless, extant literature suggests that it is advisable to administer extended periods of active treatment for the management of hypertension, despite the possibility of ADRs [11]. Although polypharmacy poses a variety of potential risks, it should not serve as a deterrent to the continuation of long-term treatment for patients. Therefore, the effective management of patients presenting with complex diseases and the potential induction of polypharmacy pose a substantial challenge for clinicians and healthcare facilities worldwide [12, 13]. Consequently, extensive research has been conducted to investigate the correlation between medication regimen complexity and clinical outcomes, wherein regimen complexity has been empirically associated with medication adherence and clinical outcomes [14, 15]. The Guidelines of the European Society of Hypertension promote the use of simplified fixed-dose combination (FDC) therapies. This recommendation is especially pertinent for managing lower blood pressure targets and enhancing long-term treatment adherence [16]. Rea et al. reported in their analysis of large databases that FDCs enhance adherence compared to individual formulations, which in turn reduces the risk of CVD and healthcare costs [17]. This supports the promotion of the Guidelines of the European Society of Hypertension.
This study aimed to develop an FDC comprising candesartan cilexetil, amlodipine, and atorvastatin by evaluating specific dosages (8/5/10 mg, 8/5/20 mg, 16/5/10 mg, 16/5/20 mg, and 16/10/40 mg) to mitigate the escalating risk of polypharmacy. Antihypertensive and statin therapies are the most prevalent therapeutic interventions prescribed for CVDs and, owing to the availability of low-cost generic drugs, represent cost-effective remediation for a myriad of risks associated with CVDs [18,19,20,21]. Angiotensin-converting enzyme inhibitors (ACEI), angiotensin II receptor blockers (ARB), and calcium channel blockers (CCB) are conventionally recommended as first-line treatments for hypertensive disorders [22]. Candesartan cilexetil is the esterified prodrug of candesartan, which serves as an ARB, and exhibits a dosage range of 4–32 mg. Following oral administration, candesartan cilexetil is enzymatically hydrolyzed to candesartan during absorption in the gastrointestinal tract, resulting in undetectable candesartan cilexetil plasma concentrations. Therefore, the therapeutic effect of candesartan cilexetil is solely attributable to candesartan [23, 24]. Candesartan exhibits the most pronounced angiotensin 1 (AT1) receptor binding affinity among ARBs, which confers it the beneficial capability of lowering blood pressure at lower doses relative to alternative ARBs [25, 26]. Furthermore, this has proven to be an advantage in the development of FDCs. Amlodipine belongs to the CCB family and has been reported to be more effective compared to other CCBs [27, 28]. Statin therapy is a treatment to manage CVD risk by lowering low-density lipoprotein cholesterol to treat dyslipidemia and is used in combination with ARBs and CCBs [28]. The leading drug, atorvastatin, has been reported to have the lowest rate of fatal rhabdomyolysis among statins [29]. Atorvastatin has the advantage of being dose-selective based on patient severity, with moderate-to-high intensity regimens available depending on the dose (10–80 mg) [30]. In addition, atorvastatin is administered in the active acid form and is metabolized to o-hydroxyatorvastatin (2-OH atorvastatin), p-hydroxyatorvastatin, and their lactone metabolites, primarily in the liver via cytochrome P450 3A4. In vitro, the active metabolites are reported to have 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitory activity equipotent to that of the parent drug, with approximately 70% of the HMG-CoA reductase inhibitory activity attributed to the active metabolites [31, 32]. It has the advantage of a long half-life compared to other statins (e.g., lovastatin, fluvastatin, and simvastatin), with the half-life of atorvastatin reported to be approximately 7 h. However, the half-life of its HMG-CoA reductase inhibitory activity is approximately 13–16 h, owing to the action of its active metabolite [32,33,34].
Accordingly, a phase I clinical trial was conducted to develop FDCs of candesartan cilexetil/amlodipine/atorvastatin at doses of 16/10/40 mg and 16/5/20 mg. The primary purpose of this study was to evaluate the pharmacokinetics and safety of these FDCs and to compare them with those of individual formulations.
Methods
Ethical Approval
These studies were conducted in accordance with the Declaration of Helsinki and Korean Good Clinical Practice and the International Conference on Harmonization and the study protocol was approved by the Ministry of Food and Drug Safety (MFDS) and the Chungnam National University Hospital Institutional Review Board ([IRB] study 1: IRB No. CNUH 2020-06-031, study 2: IRB No. CNUH 2020-09-117). Both Studies 1 and 2 were registered at ClinicalTrials.gov (study 1: NCT04478097; study 2: NCT04627207). All study participants gave informed consent. The informed consent form was reviewed and approved by the MFDS and the IRB.
Subjects
Studies 1 and 2 recruited 51 and 50 healthy adult volunteers, respectively, aged between 19 and under 55 years, with a minimum weight requirement of 55 kg for men and 45 kg for women. Additionally, the participants were required to possess a body mass index (BMI) within the range of 17.5–30.5 kg/m2. The inclusion of study participants was based on the evaluation of their vital signs, physical examination, clinical laboratory tests, and 12-lead electrocardiography (ECG). Additionally, we collected information on medical history, family history, and current or scheduled medication use through interviews, which was utilized in the screening. The predominant criteria for exclusion included a deviation of blood pressure from the reference range (systolic blood pressure 90–140 mmHg, diastolic blood pressure 60–90 mmHg), measured after 5 min of rest in a seated position, a creatine phosphokinase (CPK) level exceeding three times the upper limit of the reference range, and aspartate transaminase and alanine transferase levels surpassing two times the upper limit of the reference range. Individuals with prior incidences of angioedema as ADRs to ACEIs or ARBs were also excluded. All volunteers provided written informed consent prior to the commencement of the trial.
Study Design
This study was conducted at the Chungnam National University Hospital Clinical Trial Center (Daejeon, Republic of Korea) in compliance with the principles of the Declaration of Helsinki. The experimental protocols of studies 1 and 2 were identical, except for the variation in the ratios of the drugs administered.
The present investigation constituted a phase 1 clinical trial that employed a randomized, open-label, single-dose, three-sequence, three-period, partially repeated crossover methodology. The study participants were randomized into three sequences in a 1:1:1 ratio. The order of drug administration varied between the distinct sequences; nevertheless, all individuals received two doses of the individual formulations and a single dose of the FDC. The study design is depicted in Fig. 1. The participants fasted for a minimum of 10 h prior to each administration. A 14-day washout period was maintained between each period, the duration of which exceeded the previously reported half-life of the candesartan/amlodipine/atorvastatin by five times [35,36,37].

Study design. Notes: Individual formulations, individual formulations of candesartan/amlodipine + atorvastatin; fixed-dose combination, fixed-dose combination of candesartan/amlodipine/atorvastatin
Blood was drawn at 0 (pre-dose), 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 48, and 72 h post administration and collected in dipotassium ethylenediaminetetraacetic acid anticoagulation tubes. The blood samples were centrifuged at 1950–1980×g for 10 min at 4 °C and the plasma was stored in Eppendorf tubes at temperatures below − 60 °C for subsequent analysis.
Investigational Products
Study 1
A 16/10 mg Cantabell Tab (16 mg candesartan cilexetil and 10 mg amlodipine; Chong Kun Dang Pharm. Corp, Seoul, Republic of Korea) and a 40 mg Lipitor Tab (40 mg atorvastatin; Pfizer Inc., Seoul, Republic of Korea) were employed as reference drugs (individual formulations). A 16/5/40 mg dose of CKD-333 (16 mg candesartan cilexetil, 10 mg amlodipine, and 40 mg atorvastatin), manufactured by Chong Kun Dang Pharm. Corp (Seoul, Republic of Korea), was used as the test drug (FDC) in study 1. The investigational product, Cantabell Tab, used in these studies was confirmed in previous clinical trials to have pharmacokinetic characteristics bioequivalent to its individual formulations (ClinicalTrials.gov identifier NCT02548286), and no pharmacokinetic interactions have been observed between candesartan cilextil and amlodipine [38]. Therefore, although Cantabell Tab is not an individual formulation in the strict sense, we assumed that it has the same pharmacokinetics properties as the individual formulation.
Study 2
A 16/5 mg Cantabell Tab (16 mg candesartan cilexetil and 5 mg amlodipine) and a 20 mg Lipitor Tab (20 mg atorvastatin) were used as reference drugs and a 16/5/20 mg dose of CKD-333 (16 mg candesartan cilexetil, 5 mg amlodipine, and 20 mg atorvastatin) was used as the test drug (FDC) in study 2.
Determination of Plasma Concentrations
The bioassays for determining the plasma concentrations of the test and reference drugs were performed at Bioinfra Co., Ltd. (Yongin, Republic of Korea), which is a designated clinical trial sample analysis institution with excellent clinical laboratory practice certification.
Candesartan
Blood was drawn for the analysis of candesartan at 0 (pre-dose), 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, and 48 h post dose. The plasma concentrations of candesartan were measured via ultra-high-performance liquid chromatography (UPLC) using a Waters ACQUITY UPLC™ System (Waters, Milford, MA, USA) coupled with a tandem mass spectrometer (Waters Xevo™ TQ MS; Waters, Milford, MA, USA). Samples were prepared by protein precipitation preceding separation and injected into the LC–MS/MS system comprising a 1.7-μm Waters ACQUITY UPLC® BEH C18 column (2.1 mm ID × 50 mm L; Waters, Milford, MA, USA). A multiple reaction monitoring (MRM) mode was used for the quantification of candesartan at m/z 441.00 → 263.18 and candesartan-d5 at m/z 446.10 → 268.20. The linear calibration curve ranged from 5 to 1000 ng/mL. The concentrations of candesartan in the quality control samples were 15, 400, and 750 ng/mL. The determination coefficient (r2) for the calibration curves was ≥ 0.99. The bioassay for candesartan was validated on the basis of the within-batch and between-batch precision and accuracy of the quality control samples. The relative standard deviation was less than 15%.
Amlodipine
Blood was drawn for the analysis of amlodipine at 0 (pre-dose), 1, 2, 3, 4, 5, 6, 7, 8, 10, 12, 24, 48, and 72 h post dose. The plasma concentrations of amlodipine were measured using a Waters ACQUITY UPLC™ System coupled with a Waters Xevo™ TQ-XS mass spectrometer. Samples were prepared by protein precipitation prior to separation and injected into the 1.7-μm Waters ACQUITY UPLC® BEH C18 (2.1 mm ID × 50 mm L) column of the LC–MS/MS system. An MRM mode was used for the quantification of amlodipine at m/z 409.20 → 238.15 and amlodipine-d4 at m/z 413.20 → 238.15. The linear calibration curves ranged from 0.05 to 50 ng/mL. The concentrations of amlodipine in the quality control samples were 0.15, 15, and 37.5 ng/mL. The bioassay for amlodipine was validated on the basis of the within-batch and between-batch precision and accuracy of the quality control samples. The relative standard deviation was less than 15%.
Atorvastatin and 2-OH Atorvastatin
Blood was drawn for the analyses of atorvastatin and 2-OH atorvastatin levels at 0 (pre-dose), 0.17, 0.33, 0.5, 0.75, 1, 1.5, 2, 3, 4, 5, 6, 8, 12, 24, and 48 h post dose. The plasma concentrations of amlodipine were measured using a Waters ACQUITY UPLC™ System coupled with a Waters Xevo™ TQ-S mass spectrometer. Samples were prepared by protein precipitation prior to separation and injected into the 1.7-μm Waters ACQUITY UPLC® BEH C18, 1.7 μm (2.1 mm ID × 50 mm L) column of the LC–MS/MS system. An MRM mode was used for the quantification of atorvastatin at m/z 559.26 → 440.26, atorvastatin-d5 at m/z 564.28 → 445.28, 2-OH atorvastatin at m/z 575.26 → 440.26, and 2-OH atorvastatin-d5 at m/z 580.28 → 445.28. The linear calibration curves ranged from 0.1 to 200 ng/mL for atorvastatin and 0.05 to 100 ng/mL for 2-OH atorvastatin. The concentrations of atorvastatin and 2-OH atorvastatin in quality control samples were 0.15, 15, and 37.5 ng/mL, and 0.15, 50, and 75 ng/mL, respectively. The determination coefficient (r2) for the calibration curves was ≥ 0.99. The bioassays for atorvastatin and 2-OH atorvastatin were validated on the basis of the within-batch and between-batch precision and accuracy of the quality control samples. The relative standard deviations were less than 15%.
Pharmacokinetic Analysis
The pharmacokinetic (PK) analysis set included only those participants who had completed all PK sampling procedures and were not liable for any protocol violations. The PK analyses were performed in compliance with actual blood sampling times. The area under the plasma concentration–time curve from time zero to the time of the last quantifiable concentration (AUClast) and the maximum plasma concentrations (Cmax) of candesartan, amlodipine, and atorvastatin were deemed the primary PK endpoints. Conversely, the area under the plasma concentration–time curve from times zero to infinity (AUCinf), time of achievement of maximum plasma concentration (Tmax), the terminal elimination half-lives (t1/2) of candesartan, amlodipine, and atorvastatin, and the AUClast, Cmax, Tmax, and t1/2 for 2-OH atorvastatin were considered the secondary PK endpoints. PK analyses were performed via a non-compartmental method using the WinNonlin® software (version 8.3; Pharsight, Mountain View, CA, USA). AUC was calculated using the linear trapezoidal method. Cmax and Tmax were determined directly from the observed concentration–time data.
Statistical Analysis
Statistical analyses were performed using the SAS® software (version 9.4, SAS Institute Inc., Cary, NC, USA). In the context of the individual formulations group, the PK data recorded in the two distinct periods were regarded as separate datasets, even if they belonged to the same person. All PK parameters were summarized using descriptive statistics. The primary PK endpoints were estimated as the geometric mean ratios (GMRs) and the 90% confidence intervals (CIs) of the FDC to individual formulations. The average bioequivalence (ABE) approach was employed to assess the primary endpoints for candesartan and amlodipine in order to determine whether the 90% CI was constrained within the BE range of 0.8–1.25. Previous reports have portrayed atorvastatin as a potential highly variable drug (HVD), and a mixed-scaled average BE strategy was used in compliance with the local regulatory criteria stipulated by the MFDS (Republic of Korea) [39, 40]. On the basis of the ABE, the confirmation of the 90% CI within the range of 0.8–1.25 was examined to determine the AUClast, independent of the within-subject coefficient of variation of the reference drug (CVwr). To measure Cmax, ABE was employed if the CVwr was less than or equal to 0.3 as calculated from the Cmax of the reference (individual formulations) drugs at two distinct periods. In contrast, if the CVwr was greater than 0.3, the BE range calculated by the reference-scaled average bioequivalence (RSABE) was used to assess whether the 90% CI was limited within the BE range.
Safety
The safety and tolerability of the test and reference drugs were assessed depending on the incidence of adverse events (AEs), vital signs, physical examination, clinical laboratory tests (complete blood cell count, blood chemistry tests, namely liver and kidney function tests, and urinalysis), and 12-lead ECG of the participants. AEs were coded using the system organ class and preferred term in accordance with MedDRA version 20.1. AEs were segregated into the treatment groups based on the number of individuals, incidence, and number of events. Furthermore, the severity and relationship between AEs were evaluated and summarized.
Results
Subject Demographics
Study 1
A cohort of 51 participants was recruited and randomized; however, two individuals withdrew prior to the initial drug administration, resulting in a total of 49 participants receiving the drugs. Subsequently, another six participants discontinued, leaving only 43 to complete the trial. The average age, height, weight, and BMI of study participants who were administered with the drugs were 25.2 ± 3.7 years, 174.7 ± 5.8 cm, 76.3 ± 10.1 kg, and 24.9 ± 2.6 kg/m2, respectively (mean ± standard deviation). The demographics between the individual sequence groups did not exhibit any statistically significant differences (Table 1).
Study 2
A cohort of 50 participants was recruited and randomized. However, three individuals withdrew prior to the initial drug administration, resulting in a total of 47 participants receiving the drugs. Subsequently, one participant discontinued, leaving only 46 to complete the trial. The average age, height, weight, and BMI of study participants who were administered with the drugs were 24.9 ± 4.9 years, 175.1 ± 5.9 cm, 72.4 ± 8.0 kg, and 23.6 ± 2.5 kg/m2, respectively (mean ± standard deviation). The demographics between the sequence groups did not exhibit any statistically significant differences (Table 1).
PKs
Study 1
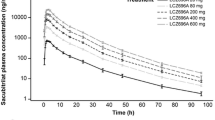
Study 1 evaluated the pharmacokinetics of candesartan cilexetil (16 mg), amlodipine (10 mg), and atorvastatin (40 mg) in 43 participants. The mean plasma concentration–time profiles of candesartan, amlodipine, and atorvastatin are illustrated in Fig. 2. Table 2 documents the descriptive statistics for the PK parameters. The GMRs (90% CIs) of the FDC to the individual formulations of candesartan, amlodipine, and atorvastatin for AUClast were 0.9612 (0.9158–1.0089), 1.0033 (0.9800–1.0271), and 1.0532 (1.0082–1.1003), respectively. The GMRs (90% CIs) of the FDC to the individual formulations of candesartan, amlodipine, and atorvastatin for Cmax were 0.9600 (0.8953–1.0294), 1.0198 (0.9950–1.0453), and 1.0630 (0.9418–1.1997), respectively. The CVwr of the Cmax for atorvastatin was computed to be 33.32%. On the basis of the RSABE, the extended BE ranged between 0.7814 and 1.2797. The 90% CIs for Cmax and AUClast of candesartan, amlodipine, and atorvastatin were constrained within the BE range (0.8–1.25) depending on the ABE (Table 3).

Mean plasma concentration–time profiles of a candesartan, b amlodipine, c atorvastatin, and d 2-OH atorvastatin after a single-dose administration of the fixed-dose combination (test drug) or co-administration of the corresponding individual formulations (reference drugs). Notes: R1, individual formulations of candesartan 16 mg/amlodipine 10 mg + atorvastatin 40 mg; T1, fixed-dose combination of candesartan 16 mg/amlodipine 10 mg/atorvastatin 40 mg; R2: candesartan 16 mg/amlodipine 5 mg + atorvastatin 20 mg; T2, fixed-dose combination of candesartan 16 mg/amlodipine 5 mg/atorvastatin 20 mg
Study 2
Study 1 evaluated the pharmacokinetics of candesartan cilexetil (16 mg), amlodipine (5 mg), and atorvastatin (20 mg) in 46 participants. The mean plasma concentration–time profiles of candesartan, amlodipine, and atorvastatin are portrayed in Fig. 2. Table 2 delineates the descriptive statistics for the PK parameters. The GMRs (90% CIs) of the FDC to the individual formulations of candesartan, amlodipine, and atorvastatin for AUClast were 0.9965 (0.9550–1.0397), 1.0067 (0.9798–1.0344), and 1.0252 (0.9841–1.0680), respectively. The GMRs (90% CIs) of the FDC to the individual formulations of candesartan, amlodipine, and atorvastatin for Cmax were 0.9851 (0.9368–1.0359), 1.0003 (0.9694–1.0321), and 0.9888 (0.8792–1.1120), respectively. The CVwr of the Cmax for atorvastatin was calculated to be 40.92%. On the basis of the RSABE, the extended BE ranged between 0.7415 and 1.3485. The 90% CIs for Cmax and AUClast of candesartan, amlodipine, and atorvastatin were constrained within the BE range (0.8–1.25) depending on the ABE (Table 3).
Safety
The safety analysis set comprised a total of 49 participants in study 1 (individual formulations group, n = 48; FDC group, n = 45) and 47 participants in study 2 (individual formulations group, n = 47; FDC group, n = 46), all of whom were treated with at least single dose of the drugs. A total of six and three AEs were reported in nine (18.4%, 9/49) and three participants (6.4%, 3/47) in studies 1 and 2, respectively. In FDC group, five AEs were observed in four subjects in study 1 and single AE was observed in one subject in study 2. In individual formulations group, eight AEs were documented in 11 subjects in study 1 and two AEs were documented in two subjects in study 2. All AEs were deemed to possess a causal relationship with the surveyed study drugs. In study 1, an elevation in the blood CPK levels was detected to be the predominant AE out of six other AEs reported in five individuals (10.2%, 5/49). Conversely, in study 2, all AEs were identical, exhibiting only one occurrence and no discernible trend (Table 4). No serious AE were reported in either of the studies, and all AEs were alleviated without sequelae.
Discussion
FDCs have the disadvantage of offering a limited array of doses. Therefore, the FDCs (candesartan cilexetil, amlodipine, and atorvastatin) were developed in five different doses (8/5/10 mg, 8/5/20 mg, 16/5/10 mg, 16/5/20 mg, and 16/10/40 mg) to minimize this constraint on dosage availability. This article documents two studies that endeavored to compare the pharmacokinetics and safety of an FDC of candesartan cilexetil/amlodipine/atorvastatin (at doses of 16/10/40 mg and 16/5/20 mg) with those of individual formulations.
Application of the extended BE range estimated using RSABE in a replicated crossover design can reduce the number of subjects required while minimizing the statistical error due to variability compared to the traditional design (2 × 2 crossover design) using ABE. Many regulatory agencies recommend the use of the extended BE range of RSABE with a partial or full replication crossover design to evaluate the bioequivalence (BE) of HVDs [45,46,47]. Therefore, we conducted two-treatment, three-sequence, three-period, partial replication crossover design studies to estimate the extended BE range with RSABE, considering atorvastatin as an HVD. The BE studies were preceded by a drug–drug interaction (DDI) study (Clinicaltrials.gov identifier: NCT03017950) in which PK parameters were evaluated after repeated doses of candesartan cilexetil 16 mg, amlodipine 10 mg, and atorvastatin 40 mg in 80 subjects. In a DDI study, the CVwr for candesartan, amlodipine, atorvastatin, and 2-OH atorvastatin were 14.52%, 7.55%, 16.32%, and 18.13% for the area under the plasma concentration–time curve for dose interval at steady state (AUCτ) and 12.41%, 6.34%, 48.82%, and 40.13% for the maximum plasma concentration at steady state (Cmax,ss), respectively. Based on the CVwr of 48.82% for the Cmax of atorvastatin, which was the largest PK parameter variation, the within-subject standard deviation of the reference drug (Swr) was calculated to be 0.4624, and the BE range using the RSABE method was 0.7037–1.4210 [41,42,43,44,45].
Therefore, assuming a CVwr of 48.82%, a T/R ratio of 0.9, and a BE range of 0.7037–1.4210, the number of participants needed to achieve α = 0.05 and power = 80% was 35, with a total target number of 51 after assuming the dropout rate. This study was conducted in 2020 and considered a high dropout rate (approximately 30%) owing to the national quarantine policies for COVID-19 infection. However, contrary to our assumptions, the dropout rates were not high, with 43 and 46 participants completing the studies, respectively. As a result, the 90% CIs for Cmax and AUClast for candesartan, amlodipine, and atorvastatin in both studies were within the BE range (0.8–1.25) based on ABE. Based on the highest CVwr of 40.92% calculated from the results of these studies, the number of participants needed for a 2 × 2 crossover design rather than a replication crossover design was calculated to be 55 subjects (significance level = 0.05, power = 80%, BE range = 0.8–1.25), and in conclusion, the number of data needed was 110, so a total of 129 participants were obtained for study 1 and 138 participants were obtained for study 2. Therefore, it was possible to evaluate BE using ABE. In another BE study of atorvastatin, 90% CIs were within the extended BE range calculated using RSABE, despite the small number of subjects, and studies with large numbers of subjects were within the BE range using ABE, despite the high CVwr [39, 40]. This tendency is consistent with our results.
Atorvastatin has extensive first-pass metabolism, meaning that much of it is metabolized in the intestine and liver before entering systemic circulation, resulting in a low bioavailability of 14%. Therefore, the US Food and Drug Administration recommends that the active metabolite of atorvastatin be evaluated as an adjunct in BE studies [46]. In studies 1 and 2, the AUC ratios (atorvastatin/2-OH atorvastatin) were calculated to be 1.08 and 1.12, and 1.31 and 1.30 for the FDC and individual formulations, respectively. There were no statistically significant differences in the AUC ratios between the FDC and the individual formulations (p = 0.7177, p = 0.8118). This was consistent with the lack of statistically significant differences between the FDCs and individual formulas with different ingredients reported in previous studies (p = 0.8357) [39, 47, 48]. The AUC ratios of atorvastatin/2-OH atorvastatin used in the comparison are summarized in Table 5 and Fig. 3.

Summary of the AUC ratio atorvastatin/2-OH atorvastatin. Bars represent standard deviations. Notes: AUC area under the plasma concentration–time curve
Gundlach et al. reported that there was no statistically significant interaction between candesartan, amlodipine, and atorvastatin and that only candesartan reduced AUCτ and Cmax,ss by approximately 20% [49]. However, when comparing our results with the PK profile of candesartan 16 mg alone, our results differed, with an increase of approximately 20% in both AUCinf and Cmax [50].
In studies 1 and 2, the incidences of AEs were 18.4% and 6.4%, respectively, indicating a tendency towards higher AE rates in study 1. In studies 1 and 2, the doses of candesartan cilexetil were the same; however, the doses of amlodipine and atorvastatin were 10/5 and 40/20 mg, respectively. There was a trend towards more AEs with the higher-dose formulation. Specifically, the AEs that differed in incidence were all clinical laboratory abnormalities, with a trend towards more liver function tests (8 cases vs. 1 case) and a CPK increase (6 cases vs. 0 cases) (Table 4). These are typical AEs of statins and appear to be dose-related. However, because these studies were conducted on a small number of healthy subjects, there is limited evidence to conclude that the incidence of AEs is dose-dependent. The meta-analyses reported to date suggest that atorvastatin has a dose-related effect only on liver dysfunction and no dose-related effects on other safety signals [29, 51].
Conclusions
The Cmax and AUClast values of candesartan cilexetil/amlodipine/atorvastatin 16/10/40 mg and 16/5/20 mg were within the BE range determined using ABE. There were no clinically significant differences in safety between the two formulations.
Data Availability
The datasets for this study will not be shared because it is possessed by Chong Kun Dang Pharm. Corp, Seoul, Republic of Korea).
References
Federal Interagency Forum on Aging-Related Statistics. Older Americans 2016: key indicators of well-being. 2016. https://agingstats.gov/docs/LatestReport/Older-Americans-2016-Key-Indicators-of-WellBeing.pdf. Accessed 2 Apr 2024.
Collerton J, Davies K, Jagger C, et al. Health and disease in 85 year olds: baseline findings from the Newcastle 85+ cohort study. BMJ. 2012;345:e4462–e4462. https://doi.org/10.1136/bmj.e4462.
Wauters M, Elseviers M, Vaes B, et al. Polypharmacy in a Belgian cohort of community-dwelling oldest old (80+). Acta Clin Belg. 2016;71(3):158–66. https://doi.org/10.1080/17843286.2016.1148298.
Herr M, Robine JM, Pinot J, Arvieu JJ, Ankri J. Polypharmacy and frailty: prevalence, relationship, and impact on mortality in a French sample of 2350 old people. Pharmacoepidemiol Drug Saf. 2015;24(6):637–46. https://doi.org/10.1002/pds.3772.
Hovstadius B, Petersson G, Hellström L, Ericson L. Trends in inappropriate drug therapy prescription in the elderly in Sweden from 2006 to 2013: assessment using national indicators. Drugs Aging. 2014;31(5):379–86. https://doi.org/10.1007/s40266-014-0165-5.
Guerriero F, Orlando V, Tari DU, et al. How healthy is community-dwelling elderly population? Results from Southern Italy. Transl Med UniSA. 2015;13:59–64.
Halvorsen KH, Selbaek G, Ruths S. Trends in potentially inappropriate medication prescribing to nursing home patients: comparison of three cross-sectional studies. Pharmacoepidemiol Drug Saf. 2017;26(2):192–200. https://doi.org/10.1002/pds.4142.
Tsoi CS, Chow JY, Choi KS, et al. Medical characteristics of the oldest old: retrospective chart review of patients aged 85+ in an academic primary care centre. BMC Res Notes. 2014;7:340. https://doi.org/10.1186/1756-0500-7-340.
Junius-Walker U, Theile G, Hummers-Pradier E. Prevalence and predictors of polypharmacy among older primary care patients in Germany. Fam Pract. 2007;24(1):14–9. https://doi.org/10.1093/fampra/cml067.
Corsonello A, Pedone C, Incalzi RA. Age-related pharmacokinetic and pharmacodynamic changes and related risk of adverse drug reactions. Curr Med Chem. 2010;17(6):571–84. https://doi.org/10.2174/092986710790416326.
SPRINT Research Group, Wright JT Jr, Williamson JD, et al. A randomized trial of intensive versus standard blood-pressure control. N Engl J Med. 2015;373(22):2103–16. https://doi.org/10.1056/NEJMoa1511939.
Barnett K, Mercer SW, Norbury M, Watt G, Wyke S, Guthrie B. Epidemiology of multimorbidity and implications for health care, research, and medical education: a cross-sectional study. Lancet. 2012;380(9836):37–43. https://doi.org/10.1016/S0140-6736(12)60240-2.
Guthrie B, Payne K, Alderson P, McMurdo ME, Mercer SW. Adapting clinical guidelines to take account of multimorbidity. BMJ. 2012;345:e6341. https://doi.org/10.1136/bmj.e6341.
Wimmer BC, Cross AJ, Jokanovic N, et al. Clinical outcomes associated with medication regimen complexity in older people: a systematic review. J Am Geriatr Soc. 2017;65(4):747–53. https://doi.org/10.1111/jgs.14682.
Chapman RH, Benner JS, Petrilla AA, et al. Predictors of adherence with antihypertensive and lipid-lowering therapy. Arch Intern Med. 2005;165(10):1147–52. https://doi.org/10.1001/archinte.165.10.1147.
Mancia G, Kreutz R, Brunström M, et al. 2023 ESH Guidelines for the management of arterial hypertension The Task Force for the management of arterial hypertension of the European Society of Hypertension: endorsed by the International Society of Hypertension (ISH) and the European Renal Association (ERA). J Hypertens. 2023;41(12):1874–2071. https://doi.org/10.1097/HJH.0000000000003480.
Rea F, Morabito G, Savaré L, Pathak A, Corrao G, Mancia G. Adherence and related cardiovascular outcomes to single pill vs. separate pill administration of antihypertensive triple-combination therapy. J Hypertens. 2023;41(9):1466–73. https://doi.org/10.1097/HJH.0000000000003497.
Bress AP, Bellows BK, King JB, et al. Cost-effectiveness of intensive versus standard blood-pressure control. N Engl J Med. 2017;377(8):745–55. https://doi.org/10.1056/NEJMsa1616035.
Richman IB, Owens DK, Goldhaber-Fiebert J. Cost-effectiveness of intensive blood pressure management—is there an additional price to pay?—reply. JAMA Cardiol. 2017;2(5):581–2. https://doi.org/10.1001/jamacardio.2016.5837.
Heller DJ, Coxson PG, Penko J, et al. Evaluating the impact and cost-effectiveness of statin use guidelines for primary prevention of coronary heart disease and stroke. Circulation. 2017;136(12):1087–98. https://doi.org/10.1161/CIRCULATIONAHA.117.027067.
Mihaylova B, Briggs A, Hlatky M, et al. Heart Protection Study Collaborative Group Statin cost-effectiveness in the United States for people at different vascular risk levels. Circ Cardiovasc Qual Outcomes. 2009;2(2):65–72. https://doi.org/10.1161/CIRCOUTCOMES.108.808469.
McCormack T, Krause T, O’Flynn N. Management of hypertension in adults in primary care: NICE guideline: NICE guideline. Br J Gen Pract. 2012;62(596):163–4. https://doi.org/10.3399/bjgp12X630232.
Delacrétaz E, Nussberger J, Biollaz J, Waeber B, Brunner HR. Characterization of the angiotensin II receptor antagonist TCV-116 in healthy volunteers. Hypertension. 1995;25(1):14–21. https://doi.org/10.1161/01.hyp.25.1.14.
Burnier M, Buclin T, Biollaz J, Nussberger J, Waeber B, Brunner HR. Pharmacokinetic-pharmacodynamic relationships of three angiotensin II receptor antagonists in normal volunteers. Kidney Int Suppl. 1996;55:S24–9.
Unger T. Pharmacology of AT1-receptor blockers. Blood Press Suppl. 2001;3(3):5–10. https://doi.org/10.1080/08037050152518302.
Lacourcière Y, Asmar R, Investi CLS. A comparison of the efficacy and duration of action of candesartan cilexetil and losartan as assessed by clinic and ambulatory blood pressure after a missed dose, in truly hypertensive patients: a placebo-controlled, forced titration study. Am J Hypertens. 1999;12(12):1181–7. https://doi.org/10.1016/S0895-7061(99)00142-9.
Zhang L, Yang J, Li L, et al. Comparison of amlodipine versus other calcium channel blockers on blood pressure variability in hypertensive patients in China: a retrospective propensity score-matched analysis. J Comp Eff Res. 2018;7(7):651–60. https://doi.org/10.2217/cer-2017-0063.
Kim SE, Jo SH, Han SH, et al. Comparison of calcium-channel blockers for long-term clinical outcomes in patients with vasospastic angina. Korean J Intern Med. 2021;36(1):124–34. https://doi.org/10.3904/kjim.2019.308.
Arca M. Atorvastatin: a safety and tolerability profile. Drugs. 2007;67(suppl 1):63–9. https://doi.org/10.2165/00003495-200767001-00007.
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines. Circulation. 2019;139(25):e1082–143. https://doi.org/10.1161/CIR.0000000000000625.
Food and Drug Administration. Lipitor® (atorvastatin calcium) label. Washington, DC: FDA; 1996.
Kantola T, Kivistö KT, Neuvonen PJ. Effect of itraconazole on the pharmacokinetics of atorvastatin. Clin Pharmacol Ther. 1998;64(1):58–65. https://doi.org/10.1016/S0009-9236(98)90023-6.
Lilja JJ, Kivistö KT, Neuvonen PJ. Grapefruit juice increases serum concentrations of atorvastatin and has no effect on pravastatin. Clin Pharmacol Ther. 1999;66(2):118–27. https://doi.org/10.1053/cp.1999.v66.100453001.
McIver LA, Siddique MS. Atorvastatin. [Updated 2022 Nov 13]. Treasure Island: StatPearls; 2023.
Gleiter CH, Mörike KE. Clinical pharmacokinetics of candesartan. Clin Pharmacokinet. 2002;41(1):7–17. https://doi.org/10.2165/00003088-200241010-00002.
Meredith PA, Elliott HL. Clinical pharmacokinetics of amlodipine. Clin Pharmacokinet. 1992;22(1):22–31. https://doi.org/10.2165/00003088-199222010-00003.
Lennernäs H. Clinical pharmacokinetics of atorvastatin. Clin Pharmacokinet. 2003;42(13):1141–60. https://doi.org/10.2165/00003088-200342130-00005.
Kim JR, Kim S, Huh W, Ko JW. No pharmacokinetic interactions between candesartan and amlodipine following multiple oral administrations in healthy subjects. Drug Des Dev Ther. 2018;12:2475–83. https://doi.org/10.2147/DDDT.S172568.
Kim S, Ko JW, Kim JR. Pharmacokinetic and safety profiles of a fixed-dose combination of amlodipine, valsartan, and atorvastatin: a 3-period replicate crossover study. Clin Pharmacol Drug Dev. 2020;9(3):386–94. https://doi.org/10.1002/cpdd.727.
Hwang JG, Yu KS, Lee S. Comparison of the pharmacokinetics of highly variable drugs in healthy subjects using a partial replicated crossover study: a fixed-dose combination of fimasartan 120 mg and atorvastatin 40 mg versus separate tablets. Drug Des Dev Ther. 2020;14:1953–61. https://doi.org/10.2147/DDDT.S233732.
Committee for Medicinal Products for Human Use. Guideline on the investigation of bioequivalence. London: European Medicines Agency; 2010.
Food and Drug Administration. Guidance for industry: statistical approaches to establishing bioequivalence. Washington, DC: FDA; 2001.
Ministry of Food and Drug Safety. Standards on the equivalency study of drug. Cheongju: MFDS; 2023.
Haidar SH, Davit B, Chen ML, et al. Bioequivalence approaches for highly variable drugs and drug products. Pharm Res. 2008;25(1):237–41. https://doi.org/10.1007/s11095-007-9434-x.
Midha KK, Rawson MJ, Hubbard JW. The bioequivalence of highly variable drugs and drug products. Int J Clin Pharmacol Ther. 2005;43(10):485–98. https://doi.org/10.5414/cpp43485.
Food and Drug Administration. Draft guidance on atorvastatin calcium. Washington, DC: FDA; 2010.
Ghim JL, Phuong NTT, Kim MJ, et al. Pharmacokinetics of fixed-dose combination of atorvastatin and metformin compared with individual tablets. Drug Des Dev Ther. 2019;13:1623–32. https://doi.org/10.2147/DDDT.S193254.
Choi YK, Park SE, Kim EY, et al. Pharmacokinetics of atorvastatin and sustained-release metformin fixed-dose combination tablets: two randomized, open-label, 2-way crossover studies in healthy male subjects under fed conditions. Transl Clin Pharmacol. 2017;25(4):190–5. https://doi.org/10.12793/tcp.2017.25.4.190.
Gundlach K, Wolf K, Salem I, Randerath O, Seiler D. Safety of candesartan, amlodipine, and atorvastatin in combination: interaction study in healthy subjects. Clin Pharmacol Drug Dev. 2021;10(2):190–7. https://doi.org/10.1002/cpdd.787.
Jeon JY, Im YJ, Kim Y, et al. Pharmacokinetic properties and bioequivalence of candesartan cilexetil in Korean healthy volunteers. Drug Dev Ind Pharm. 2013;39(9):1296–9. https://doi.org/10.3109/03639045.2012.725732.
Cai T, Abel L, Langford O, et al. Associations between statins and adverse events in primary prevention of cardiovascular disease: systematic review with pairwise, network, and dose-response meta-analyses. BMJ. 2021;374:n1537. https://doi.org/10.1136/bmj.n1537.
Acknowledgements
We express our gratitude to all participants of the studies.
Editorial Assistance.
This manuscript received English language editing support from Editage (www.editage.com). The editorial assistance fee was funded by Chong Kun Dang Pharm. Corp., Seoul, Republic of Korea.
Funding
This study was sponsored by Chong Kun Dang Pharm. Corp.; Seoul, Republic of Korea. The journal’s publication fee was funded by Chong Kun Dang Pharm. Corp., Seoul, Republic of Korea.
Author information
Authors and Affiliations
Contributions
Conceptualization, Jung Sunwoo; methodology, Jung Sunwoo and Jin-Gyu Jung; formal analysis, Jae Hoon Kim and Ji Hye Song; investigation, Jae Hoon Kim, Ji Hye Song, MinYoung Kim, Jang Hee Hong, Jin-Gyu JUNG, and Jung Sunwoo; resources, MinYoung Kim; data curation, Jae Hoon Kim, Ji Hye Song, and Jang Hee Hong; writing—original draft preparation, Jae Hoon Kim; writing—review and editing, Jung Sunwoo and Jin-Gyu Jung; visualization, Jae Hoon Kim; supervision, Jung Sunwoo and Jin-Gyu Jung; project administration, Jang Hee Hong; funding acquisition, Jang Hee Hong. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of Interest
MinYoung Kim is an employee of Chong Kun Dang Pharm. Corp., Yongin, Republic of Korea. The other authors (Jae Hoon Kim, Ji Hye Song, Jang Hee Hong, Jung Sunwoo and Jin-Gyu Jung) declare no conflict of interest in this study.
Ethical Approval
These studies were conducted in accordance with the Declaration of Helsinki and Korean Good Clinical Practice and the International Conference on Harmonization and the study protocol was approved by the MFDS and the Chungnam National University Hospital IRB (study 1: IRB No. CNUH 2020-06-031, study 2: IRB No. CNUH 2020-09-117). Both studies 1 and 2 were registered at ClinicalTrials.gov (study 1: NCT04478097; study 2: NCT04627207). All study participants gave informed consent. The informed consent form was reviewed and approved by the MFDS and the IRB.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Kim, J.H., Song, J.H., Kim, M. et al. Pharmacokinetic Comparison of a Fixed-Dose Combination of Candesartan Cilexetil/Amlodipine/Atorvastatin Versus Co-administration of Individual Formulations in Healthy Participants. Adv Ther 41, 2808–2825 (2024). https://doi.org/10.1007/s12325-024-02869-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-024-02869-y