
Abstract
Nintedanib is a tyrosine kinase inhibitor approved for the treatment of idiopathic pulmonary fibrosis (IPF) and other progressive fibrosing interstitial lung diseases. Placebo-controlled trials showed that the adverse event profile of nintedanib was characterised mainly by gastrointestinal events, particularly diarrhoea. We review the data from all published real-world studies of the safety of nintedanib in patients with IPF. These real-world data were consistent with the safety profile observed in clinical trials and described in the product label. The most common adverse events were diarrhoea, nausea and vomiting, but these infrequently led to permanent treatment discontinuation. Liver enzyme elevations were observed, supporting the recommendation for regular monitoring of liver enzymes, particularly in the first few months of treatment. Bleeding and cardiovascular adverse events were rarely reported. As in clinical trials, in real-world studies, reductions of the nintedanib dose, treatment interruptions and use of anti-diarrhoeal medications were frequently employed to manage adverse events. Few data are available on the use of nintedanib in patients who are elderly or have advanced disease, but there are some data to suggest a greater rate of treatment discontinuation in these patients. Effective management of adverse events associated with nintedanib is important to minimise their impact.
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Data from real-world studies in patients with IPF support the safety profile of nintedanib observed in clinical trials. |
The most common adverse events associated with nintedanib in real-world studies were diarrhoea, nausea and vomiting, which infrequently led to treatment discontinuation. |
Liver enzymes should be monitored in patients treated with nintedanib. |
Nintedanib dose adjustments, treatment interruptions and anti-diarrhoeal medications are frequently employed to manage adverse events. |
Effective management of the adverse events associated with nintedanib in clinical practice is important to minimise their impact and help patients remain on therapy. |
Digital Features
This article is published with digital features, including a graphical abstract to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.22015745.
Introduction
Idiopathic pulmonary fibrosis (IPF) is a progressive fibrosing interstitial lung disease (ILD) associated with high morbidity and mortality [1]. Two drugs have been licensed for the treatment of IPF: nintedanib and pirfenidone. Nintedanib is a tyrosine kinase inhibitor that inhibits processes fundamental to the progression of pulmonary fibrosis, including the proliferation and transformation of lung fibroblasts and the deposition of extracellular matrix [2, 3]. The efficacy and safety of nintedanib in patients with IPF have been investigated in several randomised placebo-controlled trials [4,5,6,7], including the two INPULSIS trials involving a total of over 1000 patients [5]. In all these trials, nintedanib reduced the rate of decline in forced vital capacity (FVC), reflecting a slowing of disease progression. The safety profile of nintedanib in randomised trials and their open-label extensions was characterised mainly by gastrointestinal adverse events, particularly diarrhoea [4,5,6,7,8,9,10,11,12].
While clinical trials provide the most robust data on the safety and tolerability of a drug, they are inevitably of limited duration and exclude patients with the most advanced disease and with certain comorbidities. Real-world evidence on the adverse events observed when a therapy is used in clinical practice can provide valuable additional information on the safety profile of a drug [13,14,15]. In this article, we review the real-world evidence on the safety and tolerability of nintedanib in patients with IPF.
Literature Search
A PubMed search was conducted on 6 September 2022 using the search terms “nintedanib” and “idiopathic pulmonary fibrosis”. This identified 780 manuscripts. The titles and abstracts of these manuscripts were reviewed to identify those that may include real-world data on the safety and tolerability of nintedanib in patients with IPF obtained as part of registries, observational studies, claims databases, pharmacovigilance programmes, or expanded access/compassionate use programmes. The 52 manuscripts identified were hand-searched for data on adverse events, dose adjustments and treatment discontinuations in patients with IPF treated with nintedanib. This identified 49 manuscripts including relevant data (two from pharmacovigilance databases, four from patient registries, 43 from other observational studies). In addition, abstracts presented at the American Thoracic Society (ATS) and European Respiratory Society (ERS) international congresses in 2019, 2020, 2021 and 2022 were hand-searched for data that added value to those included in the manuscripts. Data from the two randomised, placebo-controlled INPULSIS trials of nintedanib in patients with IPF [5] were also reviewed to enable comparison of these data to the real-world data.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by the authors.
Diarrhoea
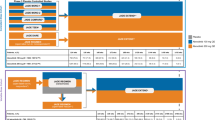
In the INPULSIS trials, diarrhoea was the most frequent adverse event associated with nintedanib, reported in 62% of patients in the nintedanib group and 18% of patients in the placebo group over 52 weeks [8]. For most patients who had diarrhoea in the INPULSIS trials, the first event occurred within 3 months of initiating treatment [8]. Pharmacovigilance data collected in the 4 years following the approval of nintedanib as a treatment for IPF also showed diarrhoea to be the most frequently reported adverse event in patients taking nintedanib (301.6 events per 1000 patient-years) [16]. Based on these pharmacovigilance data, the median time to onset of a diarrhoea event was 60 days [16]. Diarrhoea was reported much less commonly in pharmacovigilance studies than in the INPULSIS trials (Fig. 1). This likely reflects under-reporting of diarrhoea in pharmacovigilance studies, in which diarrhoea must be spontaneously reported by the patient, compared to clinical trials, in which patients are specifically asked about the adverse events that they have experienced.

Lasky JA et al.; Copyright 2020
Safety of nintedanib in the INPULSIS trials and in global pharmacovigilance data [16]. Adapted by permission from Springer Nature: Advances in Therapy; Safety of nintedanib in patients with idiopathic pulmonary fibrosis: global pharmacovigilance data;
Observational studies conducted in clinical practice have reported that diarrhoea occurred in 32–79% of patients treated with nintedanib [17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35,36,37]. In two single-centre studies, one reporting diarrhoea in 44 of 124 patients and the other reporting diarrhoea in 50 of 86 patients, the average time from starting nintedanib to the onset of diarrhoea was 2.2 months [29] and 5.4 months, respectively [38].
Exposure-safety analyses based on pooled data from clinical trials found no association between exposure to nintedanib (based on plasma concentrations) and the risk of developing diarrhoea [39]. This suggests that the risk of diarrhoea would not change based on weight. In a real-world study of 77 Japanese patients, a body mass index (BMI) < 21.6 kg/m2 was associated with an increased risk of diarrhoea (odds ratio [OR] 3.5 [95% CI 1.0, 11.5]) compared with a higher BMI, but the impact of weight was not assessed [26].
In the INPULSIS trials, the median (minimum, maximum) duration of diarrhoea events in patients treated with nintedanib was 139 (1, 473) days. Most of the diarrhoea events reported in clinical trials of nintedanib were mild or moderate in intensity [8]. Similar observations have been made in real-world studies. Among 45 patients in a Brazilian expanded access programme who experienced diarrhoea, the intensity of the worst event (in the opinion of the investigators) was mild in 49%, moderate in 29% and severe in 22% of patients [27]. No real-world data have been published on the nature of nintedanib-associated diarrhoea, such as the frequency, consistency, or urgency of bowel movements.
Specific recommendations for the management of diarrhoea were provided to the investigators in clinical trials of nintedanib, including dose adjustments, treatment interruption, and use of loperamide (Fig. 2). Similar recommendations have been provided for the management of diarrhoea in clinical practice [40,41,42]. Data from compassionate use programmes in Germany (n = 62) and Brazil (n = 57) showed that 42% and 58% of patients received loperamide, respectively [17, 27].

Copyright © 2015, Corte et al., Respir Res;16:116
Algorithm for the management of diarrhoea adverse events in the INPULSIS trials [8].
Other Gastrointestinal Adverse Events
In the INPULSIS trials, nausea was reported in 25% of patients in the nintedanib group and 7% of patients in the placebo group, while vomiting was reported in 12% of patients in the nintedanib group and 3% of patients who received placebo [8]. In real-world studies, nausea or vomiting adverse events have been reported in 2–53% of patients treated with nintedanib [17, 19,20,21,22,23, 26,27,28, 30, 31, 33,34,35,36,37, 43,44,45,46]. In a single-centre study in which five of 124 patients experienced nausea, the average time from starting nintedanib to the onset of nausea was 2.5 months [29].
In the INPULSIS trials, 9.7% of patients in the nintedanib group and 3.5% of patients in the placebo group had weight loss [8]. Based on the annual rate of decline in weight, the mean weight loss over 52 weeks was 3.3 kg in the nintedanib group and 1.5 kg in the placebo group; 37.8% and 20.1% of patients in the nintedanib and placebo groups, respectively, had a weight loss greater than 5% [47]. In real-world studies, 5–32% of patients treated with nintedanib have been reported to experience weight loss [19,20,21,22, 25, 27,28,29,30, 33, 34, 36, 37]. In three studies that reported data on discontinuations due to weight loss, this occurred in about 5% of 49, 57 and 94 patients treated with nintedanib [22, 27, 44]. Among 36 patients at a US referral centre who were treated with nintedanib for at least 12 months, mean weight loss was 6%, with 61% of patients experiencing weight loss greater than 5% [48]. Real-world studies have reported anorexia in patients treated with nintedanib (2–39% of patients) [17, 28, 29, 43]. It is unclear to what extent weight loss in patients treated with nintedanib is due to loss of appetite.
Hepatic Enzyme Elevations
In the INPULSIS trials, elevations in alanine aminotransferase (ALT) and/or aspartate aminotransferase (AST) greater than three times the upper limit of normal (ULN) were reported in 5% of patients treated with nintedanib compared with 1% of the placebo group over 52 weeks [8]. These elevations did not appear to occur at a particular time relative to the start of treatment [8]. An algorithm for the management of hepatic enzyme elevations was provided to the investigators [8]. In the majority of patients, values had returned to within the normal range by the end of treatment [8].
The product label recommends that hepatic enzymes and bilirubin be monitored prior to initiation of nintedanib, at regular intervals during the first 3 months of treatment, and then periodically or as clinically indicated [40]. Multidisciplinary panels of experts [41, 42, 49] have recommended measuring hepatic enzymes and bilirubin prior to initiating nintedanib, then every 4 weeks for the first 6 months of treatment, and then every 3 months (or as clinically indicated). In the pharmacovigilance database, elevated hepatic enzyme or bilirubin levels were reported at a rate of 31.5 events per 1000 patient-years (Fig. 1), with a median time to onset of the first elevation of 60 days [16]. In real-world studies, elevations in hepatic enzymes have been reported in 2–11% of patients treated with nintedanib [17,18,19, 21,22,23,24, 27, 28, 36, 43, 46]. Levels of these enzymes generally returned to normal following dose reduction to 100 mg twice daily (bid) or treatment interruption [17, 24, 50]. Studies from a centre in Japan (based on two cohorts of 32 and 68 patients) found an unusually high rate of hepatic enzyme elevations of greater than three times the ULN, which likely reflects the very frequent measurement of hepatic enzyme elevations at this centre [50, 51].
Exposure-safety analyses based on pooled data from clinical trials found an association between nintedanib exposure (based on plasma concentrations) and the risk of hepatic enzyme elevations, as well as a higher risk in female patients [39]. The observed association between exposure and risk suggests a potentially higher frequency of hepatic enzyme elevations in patients with higher exposure to nintedanib, such as those of low weight, and warrants close monitoring in such patients. However, no a priori dose adjustments of nintedanib are deemed necessary except in patients with mild hepatic impairment (Child–Pugh A) [39, 40].
Bleeding
As an inhibitor of the vascular endothelial growth factor receptor, nintedanib may increase the risk of bleeding [52]. Patients receiving full-dose anticoagulation should be monitored closely for bleeding, and therapy adjusted as necessary [40]. In the INPULSIS trials, bleeding events were reported in 10% of patients treated with nintedanib over 52 weeks compared to 8% of patients who received placebo [8]. The incidence of serious bleeding events was similar in the nintedanib and placebo groups (1.3% and 1.4%, respectively) [8]. However, it should be noted that patients treated with full-dose anticoagulation or with a known risk of bleeding were excluded from these trials. In the pharmacovigilance database, bleeding events were reported at a rate of 36.8 events per 1000 patient-years (Fig. 1) and most (81.0%) of the events were non-serious [16]. The most frequently reported types of bleeding event were epistaxis (25.0% of events), contusion (14.4% of events), and haematochezia (11.6% of events) [16].
Few bleeding events have been reported in real-world observational studies. Among 124 patients who received nintedanib for an average of 258 days as part of a UK patient-in-need programme, three events of haemoptysis, one of haematemesis, and one of pulmonary embolism were reported [18]. Sixteen bleeding adverse events, including haemoptysis, haematemesis and nose bleeds, were reported among 187 patients treated at three UK centres (for a median of 8 months) [19]. A multicentre study from Greece found that gastrointestinal bleeding was reported in fewer than 1% of 244 patients treated with nintedanib for a mean of 24 months [28]. In the EMPIRE registry, two of 208 patients who were receiving nintedanib in addition to anticoagulation and/or antiplatelet therapy experienced a bleeding event [53].
Cardiovascular Events
Patients with a recent history of myocardial infarction (MI), unstable angina, or thrombotic events were not enrolled in trials of nintedanib. In the INPULSIS trials, cardiac disorders were reported in 10.0% of patients treated with nintedanib and 10.6% of patients who received placebo over 52 weeks [8]. Adverse events coded as “MI” or “acute MI” based on terms in the Medical Dictionary for Regulatory Activities were reported in 1.6% and 0.5% of patients treated with nintedanib and placebo, respectively [8]. Pooled data from the TOMORROW and INPULSIS trials suggest that among 1107 patients with a history of atherosclerotic cardiovascular disease and/or at least one cardiovascular risk factor (hypertension, dyslipidaemia, BMI > 30 kg/m2, current/former smoking, diabetes) at baseline, the incidence of MI was higher (3.03 vs. 1.16 per 100 patient-years), the incidence rate of other ischaemic heart disease was lower (1.85 vs. 3.28 per 100 patient-years), and the incidence of major adverse cardiovascular events was similar (3.88 vs. 3.49 per 100 patient-years) in patients who received nintedanib vs. placebo, respectively [54]. MI and major adverse cardiovascular events were reported at lower rates in the pharmacovigilance database than in patients who received nintedanib in the INPULSIS trials [16] (Fig. 1).
Few cardiovascular events have been reported in patients treated with nintedanib in real-world studies [18,19,20,21,22,23, 28, 55, 56]. In the largest real-world study to report data on cardiovascular events, among 244 patients treated with nintedanib for an average of 24 months at seven centres in Greece, ischaemic events (MI or ischaemic stroke) were reported in 2.9% of patients [28].
Dose Adjustments and Adherence
Other than in patients with mild hepatic impairment, the recommended dose of nintedanib is 150 mg bid, with dose reductions to 100 mg bid or treatment interruptions recommended to manage adverse events [40]. In some cases, permanent discontinuation of nintedanib is required. Over 52 weeks of treatment in the INPULSIS trials, in the nintedanib and placebo groups, respectively, 28% and 4% of patients had at least one dose reduction, 24% and 10% of patients had at least one treatment interruption, and 24% and 19% of patients permanently discontinued treatment. Dose reductions due to diarrhoea occurred in 14% of patients treated with nintedanib and in no patients receiving placebo [8].
In real-world studies, nintedanib dose reductions have been reported in 13–37% of patients, and treatment interruptions in 5–42% of patients [17,18,19,20,21, 24, 27,28,29, 36, 37, 43, 55]. Dose adjustments are most frequently required because of diarrhoea. In three studies involving 62, 64 and 124 patients, with an average duration of treatment of 8–11 months, 13–31% of patients had a dose reduction and 6–16% of patients had a treatment interruption due to diarrhoea [17, 18, 20]. In the one study that looked at the duration of treatment interruption, the median duration of interruption was 17 days [17].
The rate of permanent discontinuations of nintedanib varies widely across studies, from 4% to 53% of patients, likely reflecting differences in study methodologies, patient populations and follow-up times, as well as treatment practices at particular centres [17,18,19,20,21, 23, 25, 27,28,29, 34,35,36,37, 43, 44, 55, 57,58,59,60,61,62]. In a UK study, among 69 of 154 patients who discontinued nintedanib over 12 months, 47% discontinued within the first 3 months, 17% between 3 and 6 months, and 36% between 6 and 12 months [21]. As in clinical trials, diarrhoea is the most frequent reason for permanent discontinuation of nintedanib in real-world studies [19, 20, 22, 27].
A few studies have tried to identify factors associated with discontinuation of nintedanib. In a single-centre study of 42 patients in Japan, the seven patients who discontinued nintedanib within 6 months due to adverse events had lower mean BMI (20.4 vs. 24.2 kg/m2) and FVC % predicted (51.3% vs. 73.7%) at treatment initiation than those who did not discontinue it [57]. Lower FVC % predicted and older age at treatment initiation were associated with nintedanib discontinuation within 12 months in a multicentre UK study involving 154 patients [21] (Fig. 3) while poor overall health was associated with nintedanib discontinuation within 12 months in a single-centre Japanese study [33].

Reproduced with permission of © ERS 2022. ERJ Open Res. 4:00049–2018; https://doi.org/10.1183/23120541.00049-2018 Published 19 October 2018
Proportion of patients treated with nintedanib at 1 year by baseline FVC % predicted: data from six hospitals in the UK [21].
Recent data from a retrospective analysis of a specialty pharmacy database in the USA suggest that implementation of a patient support programme reduced the risk of nintedanib discontinuation (defined as a gap of more than 60 days since the last prescription). In this study, 35% of the 3114 patients enrolled in the programme remained on treatment 1 year after their first prescription, compared with 29% of the 9388 patients who did not participate [63].
Special Populations
Elderly Patients
Based on pooled data from five clinical trials, the safety profile of nintedanib was generally similar between patients aged < 75 years (n = 1364) vs. ≥ 75 years (n = 326) at baseline, but a greater proportion of patients aged ≥ 75 years discontinued treatment because of adverse events [64]. Older age is associated with increased exposure to nintedanib (based on plasma concentrations), which may increase the risk of hepatic enzyme elevations [39, 40].
Real-world studies suggest that the adverse event profile of nintedanib is similar in patients aged ≥ 75 vs. < 75 years, although anorexia and nausea have been reported at a higher frequency among those aged ≥ 75 years in some studies conducted in Asia [31, 65,66,67] and discontinuations may be more frequent in this age group [66, 67]. Low BMI (OR 0.81 [95% CI 0.69, 0.97]) and low FVC % predicted (OR 0.97 [95% CI 0.94, 1.00]) have been associated with nintedanib discontinuation in patients aged ≥ 75 years [66]. Among 82 patients treated at a single centre in Italy, nintedanib dose reductions occurred more frequently in patients aged ≥ 80 vs. < 80 years (50% vs. 27%), independent of BMI [46].
Advanced Lung Function Impairment
Data from the INSTAGE and INPULSIS trials suggest that the adverse event profile of nintedanib is similar in patients with severely impaired gas exchange (DLco ≤ 35% predicted) as in patients with lesser impairment in gas exchange [68]. In the open-label extension of the INPULSIS trials (INPULSIS-ON), the adverse event profile of nintedanib was generally consistent between subgroups with baseline FVC ≤ 50% predicted (n = 41) vs. > 50% predicted (n = 690), but a greater proportion of patients with FVC ≤ 50% predicted discontinued treatment because of adverse events [69].
Real-world studies suggest that the adverse event profile of nintedanib is generally consistent between patients with advanced lung function impairment (i.e., FVC < 50% predicted or DLco < 30% predicted) and those with more highly preserved lung function [23, 30, 43, 45, 55, 70]. Some studies have reported no difference in the rate of nintedanib discontinuation across these groups [30, 43, 45], while in others, treatment interruptions and permanent discontinuations were more frequent in patients with advanced disease [21, 23] (Fig. 3).
Patients Switched from Pirfenidone to Nintedanib
Few data are available on the tolerability of nintedanib in patients switched from pirfenidone. However, the available data suggest that the adverse event profile of nintedanib is consistent between patients who were switched from pirfenidone to nintedanib and those who received first-line nintedanib [32, 71].
Combination Therapy with Nintedanib and Pirfenidone
Data from clinical trials suggest that the adverse event profiles of nintedanib with add-on pirfenidone [10] and of pirfenidone with add-on nintedanib [72] are as expected based on the profiles of each drug. In the INJOURNEY trial, over 12 weeks, gastrointestinal adverse events were reported in 70% of 53 patients randomised to receive nintedanib with add-on pirfenidone and 53% of 51 patients treated with nintedanib alone. Few real-world data are available on the tolerability of combination therapy with nintedanib and pirfenidone, but the available data suggest a tolerability profile consistent with observations in clinical trials [73, 74].
Patients Undergoing Lung Transplant
Given its mechanism of action, concerns have been raised that nintedanib may increase the risk of complications such as intraoperative bleeding or delayed wound healing in patients undergoing lung transplant. However, data from a number of real-world studies suggest that prior use of nintedanib does not increase intra- or post-transplant complications [75,76,77,78,79,80,81]. In a multicentre Australian study, there was a trend toward an increased rate of anastomotic dehiscence among patients with took anti-fibrotic therapy up to the day of transplant (n = 40) compared to those who did not (n = 186), but 1-year post-transplant survival was higher in the patients who continued anti-fibrotic therapy until their transplant [79].
Conclusions
The evidence available from real-world studies in patients with IPF supports the safety profile of nintedanib observed in clinical trials and described in the product label. Specific comparisons of adverse event data between real-world studies, and between real-world studies and clinical trials, should be made with caution, given the distinct patient populations, the different methods of data collection, and the high dropout rates in real-world studies. In clinical practice, adjustments of nintedanib dose and use of anti-diarrhoeal medications are frequently employed to manage adverse events. Effective management of the adverse events that may be associated with nintedanib is important to minimise their impact on patients’ lives and help patients remain on therapy.
References
Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2022;205:e18–47.
Wollin L, Wex E, Pautsch A, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. 2015;45:1434–45.
Wollin L, Distler JHW, Redente EF, et al. Potential of nintedanib in treatment of progressive fibrosing interstitial lung diseases. Eur Respir J. 2019;54:1900161.
Richeldi L, Costabel U, Selman M, et al. Efficacy of a tyrosine kinase inhibitor in idiopathic pulmonary fibrosis. N Engl J Med. 2011;365:1079–87.
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370:2071–82.
Maher TM, Stowasser S, Nishioka Y, et al. Biomarkers of extracellular matrix turnover in patients with idiopathic pulmonary fibrosis given nintedanib (INMARK study): a randomised, placebo-controlled study. Lancet Respir Med. 2019;7:771–9.
Lancaster L, Goldin J, Trampisch M, et al. Effects of nintedanib on quantitative lung fibrosis score in idiopathic pulmonary fibrosis. Open Respir Med J. 2020;14:22–31.
Corte T, Bonella F, Crestani B, et al. Safety, tolerability and appropriate use of nintedanib in idiopathic pulmonary fibrosis. Respir Res. 2015;16:116.
Kolb M, Raghu G, Wells AU, et al. Nintedanib plus sildenafil in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2018;379:1722–31.
Vancheri C, Kreuter M, Richeldi L, et al. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis. Results of the INJOURNEY trial. Am J Respir Crit Care Med. 2018;197:356–63.
Crestani B, Huggins JT, Kaye M, et al. Long-term safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis: results from the open-label extension study, INPULSIS-ON. Lancet Respir Med. 2019;7:60–8.
Lancaster L, Crestani B, Hernandez P, et al. Safety and survival data in patients with idiopathic pulmonary fibrosis treated with nintedanib: pooled data from six clinical trials. BMJ Open Respir Res. 2019;6: e000397.
Talbot JC, Nilsson BS. Pharmacovigilance in the pharmaceutical industry. Br J Clin Pharmacol. 1998;45:427–31.
Food and Drug Administration. Guidance for Industry. Good pharmacovigilance practices and pharmacoepidemiologic assessment, 2005. https://www.fda.gov/media/71546/download. Accessed 1 Apr 2022.
Blonde L, Khunti K, Harris SB, et al. Interpretation and impact of real-world clinical data for the practicing clinician. Adv Ther. 2018;35:1763–74.
Lasky JA, Criner GJ, Lazarus HM, et al. Safety of nintedanib in patients with idiopathic pulmonary fibrosis: global pharmacovigilance data. Adv Ther. 2020;37:4209–19.
Bonella F, Kreuter M, Hagmeyer L, et al. Insights from the German compassionate use program of nintedanib for the treatment of idiopathic pulmonary fibrosis. Respiration. 2016;92:98–106.
Hughes G, Toellner H, Morris H, et al. Real world experiences: pirfenidone and nintedanib are effective and well tolerated treatments for idiopathic pulmonary fibrosis. J Clin Med. 2016;5:78.
Toellner H, Hughes G, Beswick W, et al. Early clinical experiences with nintedanib in three UK tertiary interstitial lung disease centres. Clin Transl Med. 2017;6:41.
Brunnemer E, Wälscher J, Tenenbaum S, et al. Real-world experience with nintedanib in patients with idiopathic pulmonary fibrosis. Respiration. 2018;95:301–9.
Fletcher SV, Jones MG, Renzoni EA, et al. Safety and tolerability of nintedanib for the treatment of idiopathic pulmonary fibrosis in routine UK clinical practice. ERJ Open Res. 2018;4:00049–2018.
Tzouvelekis A, Karampitsakos T, Kontou M, et al. Safety and efficacy of nintedanib in idiopathic pulmonary fibrosis: a real-life observational study in Greece. Pulm Pharmacol Ther. 2018;49:61–6.
Yoon HY, Park S, Kim DS, et al. Efficacy and safety of nintedanib in advanced idiopathic pulmonary fibrosis. Respir Res. 2018;19:203.
Bargagli E, Piccioli C, Rosi E, et al. Pirfenidone and nintedanib in idiopathic pulmonary fibrosis: real-life experience in an Italian referral centre. Pulmonology. 2019;25:149–53.
Cerri S, Monari M, Guerrieri A, et al. Real-life comparison of pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis: a 24-month assessment. Respir Med. 2019;159:105803.
Kato M, Sasaki S, Nakamura T, et al. Gastrointestinal adverse effects of nintedanib and the associated risk factors in patients with idiopathic pulmonary fibrosis. Sci Rep. 2019;9:12062 (Author correction: Sci Rep. 2020;10:12080).
Pereira CAC, Baddini-Martinez JA, Baldi BG, et al. Safety and tolerability of nintedanib in patients with idiopathic pulmonary fibrosis in Brazil. J Bras Pneumol. 2019;45: e20180414.
Antoniou K, Markopoulou K, Tzouvelekis A, et al. Efficacy and safety of nintedanib in a Greek multicentre idiopathic pulmonary fibrosis registry: a retrospective, observational, cohort study. ERJ Open Res. 2020;6:00172–2019.
Cameli P, Refini RM, Bergantini L, et al. Long-term follow-up of patients with idiopathic pulmonary fibrosis treated with pirfenidone or nintedanib: a real-life comparison study. Front Mol Biosci. 2020;7: 581828.
Senoo S, Miyahara N, Taniguchi A, et al. Nintedanib can be used safely and effectively for idiopathic pulmonary fibrosis with predicted forced vital capacity ≤50%: a multi-center retrospective analysis. PLoS ONE. 2020;15: e0236935.
Takeda T, Kunimatsu Y, Tani N, et al. Improvement in subjective symptoms and tolerability in response to nintedanib treatment in elderly patients with idiopathic pulmonary fibrosis. J Clin Med. 2020;9:755.
Vianello A, Salton F, Molena B, et al. Nintedanib treatment for idiopathic pulmonary fibrosis patients who have been switched from pirfenidone therapy: a retrospective case series study. J Clin Med. 2020;9:422.
Kato M, Sasaki S, Tateyama M, et al. Clinical significance of continuable treatment with nintedanib over 12 months for idiopathic pulmonary fibrosis in a real-world setting. Drug Des Devel Ther. 2021;15:223–30.
Noor S, Nawaz S, Chaudhuri N. Real-world study analysing progression and survival of patients with idiopathic pulmonary fibrosis with preserved lung function on antifibrotic treatment. Adv Ther. 2021;38:268–77.
Wright WA, Crowley LE, Parekh D, et al. Real-world retrospective observational study exploring the effectiveness and safety of antifibrotics in idiopathic pulmonary fibrosis. BMJ Open Respir Res. 2021;8: e000782.
Harari S, Pesci A, Albera C, et al. Nintedanib in IPF: post hoc analysis of the Italian FIBRONET observational study. Respiration. 2022;101(6):577–84.
Majewski S, Białas AJ, Barczyk A, et al. A real-world multicenter retrospective observational study on Polish experience with nintedanib therapy in patients with IPF: the PolExNIB study. Am J Respir Crit Care Med. 2022;205:A2428.
Hirasawa Y, Abe M, Terada J, et al. Tolerability of nintedanib-related diarrhea in patients with idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2020;62:101917.
Schmid U, Weber B, Sarr C, et al. Exposure-safety analyses of nintedanib in patients with chronic fibrosing interstitial lung disease. BMC Pulm Med. 2021;21:244.
Boehringer Ingelheim Pharmaceuticals, Inc. OFEV® (nintedanib) prescribing information, 2022. https://docs.boehringer-ingelheim.com/Prescribing%20Information/PIs/Ofev/ofev.pdf. Accessed 10 Sep 2022.
Bendstrup E, Wuyts W, Alfaro T, et al. Nintedanib in idiopathic pulmonary fibrosis: practical management recommendations for potential adverse events. Respiration. 2019;97:173–84.
Rahaghi F, Belperio JA, Fitzgerald J, et al. Delphi consensus recommendations on management of dosing, adverse events, and comorbidities in the treatment of idiopathic pulmonary fibrosis with nintedanib. Clin Med Insights Circ Respir Pulm Med. 2021;15:11795484211006050.
Galli JA, Pandya A, Vega-Olivo M, et al. Pirfenidone and nintedanib for pulmonary fibrosis in clinical practice: tolerability and adverse drug reactions. Respirology. 2017;22:1171–8.
Barratt SL, Mulholland S, Al Jbour K, et al. South-West of England’s experience of the safety and tolerability pirfenidone and nintedanib for the treatment of idiopathic pulmonary fibrosis (IPF). Front Pharmacol. 2018;9:1480.
Nakamura M, Okamoto M, Fujimoto K, et al. A retrospective study of the tolerability of nintedanib for severe idiopathic pulmonary fibrosis in the real world. Ann Transl Med. 2019;7:262.
Harari S, Specchia C, Lipsi R, et al. Older idiopathic pulmonary fibrosis male patients are at a higher risk of nintedanib dose reduction. Respiration. 2020;99:646–8.
Jouneau S, Crestani B, Thibault R, et al. Analysis of body mass index, weight loss and progression of idiopathic pulmonary fibrosis. Respir Res. 2020;21:312.
Perelas A, Glennie J, van Kerkhove K, et al. Choice of antifibrotic medication and disease severity predict weight loss in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2019;59:101839.
Cottin V, Bonniaud P, Cadranel J, et al. Recommandations pratiques pour le diagnostic et la prise en charge de la fibrose pulmonaire idiopathique—Actualisation 2021. Version courte [French practical guidelines for the diagnosis and management of IPF - 2021 update, short version]. Rev Mal Respir. 2022: S0761-8425(22)00026-2.
Ikeda S, Sekine A, Baba T, et al. Hepatotoxicity of nintedanib in patients with idiopathic pulmonary fibrosis: a single-center experience. Respir Investig. 2017;55:51–4.
Ikeda S, Sekine A, Baba T, et al. Low body surface area predicts hepatotoxicity of nintedanib in patients with idiopathic pulmonary fibrosis. Sci Rep. 2017;7:10811.
Chen HX, Cleck JN. Adverse effects of anticancer agents that target the VEGF pathway. Nat Rev Clin Oncol. 2009;6:465–77.
Kolonics-Farkas AM, Šterclová M, Mogulkoc N, et al. Anticoagulant use and bleeding risk in central European patients with idiopathic pulmonary fibrosis (IPF) treated with antifibrotic therapy: real-world data from EMPIRE. Drug Saf. 2020;43:971–80.
Noth I, Wijsenbeek M, Kolb M, et al. Cardiovascular safety of nintedanib in subgroups by cardiovascular risk at baseline in the TOMORROW and INPULSIS trials. Eur Respir J. 2019;54:1801797.
Barczi E, Starobinski L, Kolonics-Farkas A, et al. Long-term effects and adverse events of nintedanib therapy in idiopathic pulmonary fibrosis patients with functionally advanced disease. Adv Ther. 2019;36:1221–32.
Fournier D, Jouneau S, Bouzillé G, et al. Real-world safety profiles of pirfenidone and nintedanib in idiopathic pulmonary fibrosis patients. Pulm Pharmacol Ther. 2022;76:102149.
Nishiyama O, Yamazaki R, Saeki S, et al. Early permanent discontinuation of nintedanib due to adverse events in patients with IPF in a real-life setting. Am J Respir Crit Care Med. 2019;199: A4764 (Poster presented at the American Thoracic Society (ATS) International Conference 2019).
Holtze CH, Freiheit EA, Limb SL, et al. Patient and site characteristics associated with pirfenidone and nintedanib use in the United States; an analysis of idiopathic pulmonary fibrosis patients enrolled in the Pulmonary Fibrosis Foundation Patient Registry. Respir Res. 2020;21:48.
Salisbury ML, Conoscenti CS, Culver DA, et al. Antifibrotic drug use in patients with idiopathic pulmonary fibrosis. Data from the IPF-PRO Registry. Ann Am Thorac Soc. 2020;17:1413–23.
Thillai M, Harris E, Harding K, et al. Antifibrotic persistence in idiopathic pulmonary fibrosis: a real-world experience of 447 patients. Am J Respir Crit Care Med. 2020;201:A3058 (Poster presented at the American Thoracic Society (ATS) International Conference 2020).
Belhassen M, Dalon F, Nolin M, Van Ganse E. Comparative outcomes in patients receiving pirfenidone or nintedanib for idiopathic pulmonary fibrosis. Respir Res. 2021;22:135.
Takehara K, Koga Y, Hachisu Y, et al. Differential discontinuation profiles between pirfenidone and nintedanib in patients with idiopathic pulmonary fibrosis. Cells. 2022;11:143.
Near AM, Burudpakdee C, Viswanathan S, et al. Effect of a patient support program for idiopathic pulmonary fibrosis patients on medication persistence: a retrospective database analysis. Adv Ther. 2021;38:3888–99.
Glaspole I, Bonella F, Bargagli E, et al. Efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis who are elderly or have comorbidities. Respir Res. 2021;22:125.
Ochi Y, Kato M, Sasaki S, et al. Nintedanib treatment for elderly patients with idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2019;199:A4775 (Poster presented at the American Thoracic Society (ATS) International Conference 2019).
Uchida Y, Ikeda S, Sekine A, et al. Tolerability and safety of nintedanib in elderly patients with idiopathic pulmonary fibrosis. Respir Investig. 2021;59:99–105.
Komatsu M, Yamamoto H, Ichiyama T, et al. Tolerability of nintedanib in the elderly with idiopathic pulmonary fibrosis: a single-center retrospective study. PLoS One. 2022;17: e0262795.
Richeldi L, Kolb M, Jouneau S, et al. Efficacy and safety of nintedanib in patients with advanced idiopathic pulmonary fibrosis. BMC Pulm Med. 2020;20:3.
Wuyts WA, Kolb M, Stowasser S, et al. First data on efficacy and safety of nintedanib in patients with idiopathic pulmonary fibrosis and forced vital capacity of ≤50% of predicted value. Lung. 2016;194:739–43.
Abe M, Tsushima K, Sakayori M, et al. Utility of nintedanib for severe idiopathic pulmonary fibrosis: a single-center retrospective study. Drug Des Devel Ther. 2018;12:3369–75.
Ntolios P, Archontogeorgis K, Anevlavis S, et al. Feasibility and safety of treatment switch from pirfenidone to nintedanib in patients with idiopathic pulmonary fibrosis: a real-world observational study. Eur Rev Med Pharmacol Sci. 2021;25:6326–32.
Flaherty KR, Fell CD, Huggins JT, et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur Respir J. 2018;52:1800230.
Hagmeyer L, Treml M, Priegnitz C, Randerath WJ. Successful concomitant therapy with pirfenidone and nintedanib in idiopathic pulmonary fibrosis: a case report. Respiration. 2016;91:327–32.
Hisata S, Bando M, Homma S, et al. Safety and tolerability of combination therapy with pirfenidone and nintedanib for idiopathic pulmonary fibrosis: a multicenter retrospective observational study in Japan. Respir Investig. 2021;59:819–26.
Delanote I, Wuyts WA, Yserbyt J, et al. Safety and efficacy of bridging to lung transplantation with antifibrotic drugs in idiopathic pulmonary fibrosis: a case series. BMC Pulm Med. 2016;16:156.
Leuschner G, Stocker F, Veit T, et al. Outcome of lung transplantation in idiopathic pulmonary fibrosis with previous anti-fibrotic therapy. J Heart Lung Transplant. 2017:S1053-2498(17)31886-7
Balestro E, Solidoro P, Parigi P, et al. Safety of nintedanib before lung transplant: an Italian case series. Respirol Case Rep. 2018;6: e00312.
Lambers C, Boehm P, Lee S, et al. Effect of antifibrotics on short-term outcome after bilateral lung transplantation: a multicentre analysis. Eur Respir J. 2018;51:pii:1800503.
Mackintosh JA, Munsif M, Ranzenbacher L, et al. Risk of anastomotic dehiscence in patients with pulmonary fibrosis transplanted while receiving anti-fibrotics: experience of the Australian Lung Transplant Collaborative. J Heart Lung Transplant. 2019;38:553–9.
Astor TL, Goldberg HJ, Snyder L, et al. Impact of pre-transplant anti-fibrotic therapy for IPF upon lung transplant outcomes. Am J Respir Crit Care Med. 2020;201:A2844.
Combs MP, Fitzgerald LJ, Wakeam E, Lyu DM, O’Dwyer DN. Pre-transplant antifibrotic therapy is associated with resolution of primary graft dysfunction. Ann Am Thorac Soc. 2022;19:335–8.
Acknowledgements
Funding
The authors did not receive payment for development of this article. The journal’s Rapid Service Fee and the Open Access Fee for this article has been funded by Boehringer Ingelheim.
Medical Writing, Editorial and Other Assistance
Julie Fleming and Wendy Morris of FleishmanHillard, London, UK, provided writing assistance, which was contracted and funded by Boehringer Ingelheim Pharmaceuticals, Inc. (BIPI). Boehringer Ingelheim was given the opportunity to review the article for medical and scientific accuracy as well as intellectual property considerations.
Authorship
The authors meet criteria for authorship as recommended by the International Committee of Medical Journal Editors (ICMJE).
Author Contributions
Both authors contributed to the design of the work, to the interpretation of the data and to the writing of the manuscript. Both authors were responsible for the decision to publish the manuscript. Both authors read and approved the final manuscript.
Disclosures
Anna J. Podolanczuk reports grants from the American Lung Association and National Heart, Lung, and Blood Institute (NHLBI); consulting fees from Boehringer Ingelheim (BI), IMVARIA, Roche, Regeneron; payment for a CME lecture from the National Association for Continuing Education; payments for content review from EBSCO/DynaMed. Vincent Cottin reports an unrestricted grant paid to his institution from BI; consulting fees from AstraZeneca, BI, Celgene/Bristol Myers Squibb, CSL Behring, Galapagos, PureTech, RedX, Roche, Sanofi, Shionogi; fees for lectures and support for attending meetings from BI and Roche; he has served on Data and Safety Monitoring Boards for Galapagos, Galecto, Roche and on an adjudication committee for FibroGen.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Podolanczuk, A.J., Cottin, V. A Narrative Review of Real-World Data on the Safety of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis. Adv Ther 40, 2038–2050 (2023). https://doi.org/10.1007/s12325-023-02454-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02454-9