
Abstract
Introduction
This integrated phase 3 analysis examined efficacy and cardiovascular safety for roxadustat vs erythropoiesis-stimulating agents (ESAs) in dialysis-dependent patients.
Methods
Efficacy and safety results from four phase 3, randomized, open-label studies comparing roxadustat to ESAs (PYRENEES, SIERRAS, HIMALAYAS, ROCKIES) in dialysis-dependent patients with anemia of chronic kidney disease (CKD) were evaluated by study, pooled population and in two subgroups: incident dialysis and stable dialysis. The primary efficacy endpoint per study was hemoglobin change from baseline (CFB) to weeks 28–36 using least-squares mean difference (LSMD) without rescue therapy. Pooled safety endpoints included time to major adverse cardiovascular event (MACE; myocardial infarction, stroke, and all-cause mortality [ACM]) and MACE+ (MACE plus congestive heart failure or unstable angina requiring hospitalization), ACM, and treatment-emergent adverse events (TEAEs). MACE and MACE+ were evaluated for non-inferiority at 1.8 and 1.3 margins using hazard ratios (HRs) and 95% confidence intervals (CIs). TEAEs were descriptively summarized.
Results
In total, 4714 patients were randomized (2354 roxadustat; 2360 ESA). Hemoglobin CFB to weeks 28–36 achieved non-inferiority for roxadustat vs ESA in each study. Roxadustat was non-inferior to ESA for risks for MACE and MACE+ in the entire cohort (MACE: HR 1.09, 95% CI 0.95–1.26; MACE+ : HR 0.98, 95% CI 0.86–1.11) and similar to the incident dialysis and stable dialysis subgroups; ACM results were consistent with MACE and MACE+ (HR 1.13, 95% CI 0.95–1.34). TEAEs were generally comparable between groups.
Conclusion
Roxadustat improved hemoglobin similarly to ESA while demonstrating comparable cardiovascular and overall safety profiles in a wide spectrum of dialysis-dependent patients with anemia of CKD. Roxadustat represents an oral alternative to ESAs for achieving a target hemoglobin for anemia of CKD in dialysis-dependent patients.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
In four separate trials including 4714 patients (2354 roxadustat; 2360 erythropoiesis-stimulating agent [ESA]), non-inferiority of roxadustat compared with ESAs was established for the pivotal efficacy endpoints of hemoglobin change from baseline to weeks 28–36 and proportion of patients who achieved hemoglobin response at weeks 28–36, establishing that roxadustat efficacy is comparable to standard of care |
Roxadustat was non-inferior to ESA for major adverse cardiovascular events (MACE) in the entire population, though patients in the incident dialysis subgroup may have a more favorable risk profile compared to ESA (hazard ratio 0.83, 95% confidence interval 0.57–1.19) |
Roxadustat was non-inferior to ESA for MACE+ in the entire cohort of dialysis-dependent patients and in both incident dialysis and stable dialysis subgroups with both roxadustat and ESA patients experiencing minimal hospitalizations for congestive heart failure or unstable angina |
The rate and incidence rate for all treatment-emergent adverse event outcomes were similar between roxadustat and ESA patients and were generally consistent between trials and with prior studies in dialysis-dependent patients |
These findings suggest that physicians may consider oral roxadustat as an alternative to an ESA in correcting and maintaining a target hemoglobin level for anemia of chronic kidney disease in dialysis-dependent patients |
Introduction
Anemia of CKD is associated with reduced quality of life and increased risk of hospitalization and mortality, particularly during the first months after dialysis initiation [1, 2]. These negative outcomes may partially result from anemia being an independent cardiovascular (CV) risk factor [3]. Anemia of CKD is initially managed with oral or intravenous (IV) iron supplementation and may progress to treatment with a parenterally administered erythropoiesis-stimulating agent (ESA); however, approximately 12% of dialysis-dependent (DD) patients with anemia of CKD respond inadequately to ESAs, necessitating the use of higher ESA doses to achieve the recommended hemoglobin target of 10–12 g/dl [4, 5].
Following approval of epoetin alfa and subsequently darbepoetin alfa, multiple trials were performed to determine optimal hemoglobin targets and ESA dosing [6,7,8]. Many of these trials demonstrated an increased risk of CV and/or thromboembolic events with ESAs [6,7,8,9] when aiming to achieve higher hemoglobin levels around 13 g/dl. The US Food and Drug Administration (FDA) recognized this risk profile and updated ESA product labels in 2011 with a boxed warning for a greater risk of death, serious adverse CV reaction, and stroke when targeting hemoglobin levels > 11 g/dl [10]. Further analyses suggested that higher ESA doses rather than higher hemoglobin targets may have been primarily responsible for the increased risk of CV events [11,12,13]. Thus, it is important to characterize the safety profiles of novel agents proposed for the treatment of anemia of CKD.
Roxadustat is an orally administered hypoxia-inducible factor (HIF) prolyl hydroxylase inhibitor (PHI) in development for the treatment of anemia of CKD. During normoxia, HIF prolyl hydroxylases catalyze the degradation of HIF-1α subunits [14]. When renal hypoxia develops, HIF prolyl hydroxylase activity is suppressed and HIF-1α dimerizes with HIF-1β, accumulating in the nucleus and leading to increased erythropoiesis, transferrin receptor expression, and iron absorption. The transient inhibition of HIF prolyl hydroxylases by HIF-PHIs mimics the body's natural response to hypoxia in the presence of normal oxygen levels [14]. Roxadustat has been compared to placebo and ESAs with favorable efficacy and safety findings in phase 3 studies, resulting in its approval in Chile, China, the European Union (EU), Japan, and South Korea for non-dialysis-dependent (NDD) and DD CKD [15,16,17,18,19,20,21,22,23,24,25,26]. The roxadustat development program included four studies in the DD population of which three were similarly designed (SIERRAS, HIMALAYAS, and ROCKIES) [24,25,26] and the fourth (PYRENEES) differed in comparator [23]. Although a study examining CV-related endpoints in the three studies with the same comparator has been published [27], an assessment of pooled data from all four studies provides additional information in a larger spectrum of dialysis patients. The purpose of this pooled analysis was to examine the efficacy and safety of roxadustat in the largest possible global population of DD patients with anemia of CKD.
Methods
Component Studies and Pooling Methodology
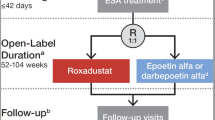
Four randomized, multicenter, open-label, active-comparator studies were included in the pooled analysis (PYRENEES, 1517-CL-0613 [NCT02278341]; HIMALAYAS, FGCL-4592–063 [NCT02052310]; SIERRAS, FGCL-4592–064 [NCT02273726]; and ROCKIES, D5740C00002 [NCT02174731]; Table 1) [23,24,25,26]. Studies were performed to evaluate efficacy and safety endpoints between roxadustat and an ESA comparator (epoetin alfa: HIMALAYAS, SIERRAS, and ROCKIES; epoetin alfa or darbepoetin alfa: PYRENEES). Studies included patients requiring hemodialysis or peritoneal dialysis of variable durations who were either ESA-naïve or pretreated with ESA up to several years. Comparable clinical data were collected for all patients, including baseline hemoglobin, iron repletion status, and dialysis type, when available. Definitions for each analysis set are provided in the Supplementary Appendix.
All study protocols were approved by relevant institutional review boards and/or ethics committees and were conducted in accordance with the tenets of the Declaration of Helsinki, the International Council for Harmonization guidelines for Good Clinical Practice, and any other applicable local health and regulatory requirements. All patients provided written informed consent before enrollment.
Participants
All eligible patients were aged ≥ 18 years, iron replete (defined as ferritin ≥ 100 ng/dl, and transferrin saturation ≥ 20%), had anemia of CKD, and were on peritoneal dialysis or hemodialysis. Key exclusion criteria included recent red blood cell transfusion, prior treatment with roxadustat or another HIF-PHI, active or chronic gastrointestinal bleeding, and anticipated elective surgery with expected blood loss. Included patients were randomized (1:1) to oral roxadustat three times weekly (TIW) or either epoetin alfa or darbepoetin alfa by the IV or subcutaneous route to maintain the same route of administration prior to randomization. Each study used different factors for stratification during randomization. Hemoglobin values prior to randomization were relatively similar between studies with most common thresholds for patients treated and untreated with ESAs at baseline as 9.0–12.0 g/dl and ≤ 10.0 g/dl, respectively. Two subgroups were considered within the entire cohort: incident dialysis (patients on incident dialysis < 4 months, largely representative of a population not receiving ESA prior to study inclusion) and stable dialysis (patients on stable dialysis ≥ 4 months, largely representative of a population receiving ESA prior to study inclusion). Additional details on participants are provided in the Supplementary Appendix.
Interventions and Rescue Therapy
In ESA-pretreated patients, initial drug dosing was based on average weekly dose of ESA prior to randomization. In ESA-untreated patients, the initial dose was based on body weight. Dose adjustments for roxadustat-treated patients were permitted every 4 weeks to maintain hemoglobin between 10–12 g/dl and were conducted in accordance with prespecified rules; treatment of patients randomized to ESA followed the local label.
Rescue therapy included receipt of a blood product transfusion and/or ESAs. ESA use was permitted in roxadustat patients (1) if the hemoglobin concentration had not adequately responded in the opinion of the treating clinician based upon the hemoglobin trend after at least two consecutive dose increases of roxadustat or the maximum dose was reached, (2) if another cause for the lack of a hemoglobin response was not clinically determined, (3) if reducing the risk of alloimmunization in transplant-eligible patients was desired, and (4) if hemoglobin on two consecutive measurements drawn at least 5 days apart was < 8.5 g/dl (only for SIERRAS and ROCKIES) or < 9.0 g/dl (only for PYRENEES). Additional details on study drug dosing and titration procedures and rescue therapy are provided in the Supplementary Appendix.
Efficacy Endpoints
Two primary efficacy endpoints were evaluated in each individual study. The primary European Union (European Medicines Agency) efficacy endpoint was mean hemoglobin change from baseline (CFB) averaged over weeks 28–36 without rescue therapy. The primary US (FDA) efficacy endpoint was mean hemoglobin CFB averaged over weeks 28–52, regardless of rescue therapy. A secondary endpoint was hemoglobin response, defined as the proportion of patients achieving mean serum hemoglobin concentrations between 10.0 and 12.0 g/dl during weeks 28–36 without having received rescue therapy within 6 weeks prior to and during weeks 28–36. Further secondary efficacy endpoints included use of rescue therapy, CFB in low-density lipoprotein (LDL) cholesterol to weeks 12–28, and monthly use of IV iron. A post hoc analysis was performed to quantify the number of patients who received no IV iron and the IV iron administrations per patient exposure year (PEY) over weeks 1–52.
Safety Endpoints
The two composite CV safety endpoints were time to first major adverse cardiovascular event (MACE)—a composite of all-cause mortality, myocardial infarction, or stroke—and time to first MACE plus (MACE+)—a composite of MACE plus unstable angina or congestive heart failure requiring hospitalization up to 7 days after the last dose [28]. Additional CV safety endpoints included all-cause mortality and time to CV-MACE and CV-MACE+, components of MACE and MACE+, respectively, that included CV-related mortality rather than all-cause mortality, and the components of MACE and MACE+ . Time to all-cause mortality and time to CV-death were evaluated. Definitions for all endpoints were based on the 2014 American College of Cardiology (ACC)/American Heart Association (AHA) Key Data Elements and Definitions for Cardiovascular Endpoint Events in Clinical Trials [29]. All CV safety endpoints were adjudicated by a central Independent Event Review Committee whose members were unaware of study group assignments and were based on the 2014 ACC/AHA Key Data Elements and Definitions for Cardiovascular Endpoint Events in Clinical Trials [29]. The adjudication process is described in the Supplementary Appendix. Malignancy-related mortality was an adjudicated exploratory endpoint.
Adverse Events
Safety was monitored by assessment of treatment-emergent adverse events (TEAEs) in the safety population (SAF; all randomized patients who receive ≥ 1 dose of study drug). TEAE was defined during the evaluation period from the first study drug administration up to 28 days after the last dose.
Statistical Analysis
According to the a priori-developed statistical analysis plan, mean hemoglobin CFBs to weeks 28–36 and weeks 28–52 were evaluated by comparing least-squares (LS) mean and LS mean difference (LSMD) and 95% confidence intervals (CIs) in each study between roxadustat and ESA. A non-inferiority margin for the difference between groups for mean hemoglobin CFB was defined as –0.75. Hemoglobin response was evaluated by comparing the difference in proportions (DOP) and 95% CI for those who achieved the target hemoglobin during weeks 28–36. The incidence rate per 100 patient-years (IR) and hazard ratio (HR) with 95% CI was determined for use of rescue therapy and packed red blood cell transfusion. The LS mean and LSMD with 95% CI between roxadustat and ESA was compared for CFB in LDL cholesterol, high-density lipoprotein (HDL) cholesterol, and LDL/HDL ratio. Clinical characteristics and demographics, as well as monthly IV iron and weekly dose of roxadustat or ESA, were presented descriptively. Pooled subgroup analyses of select efficacy endpoints were conducted for all roxadustat and ESA patients in the four studies. Pooled analyses included factors for study and the study-by-treatment interaction.
For CV safety endpoints, the analysis period was the on-treatment period in accordance with European Medicines Agency guidance [30]. This period was defined as the “on-treatment plus 7 days” time frame (OT-7), which included events that occurred after the date of the first dose and up to 7 days after the last dose of study drug [30]. The primary statistical measure to determine non-inferiority of the CV safety of roxadustat (vs ESA) was the pooled HR and its upper limit of the 95% CI in the time-to-event analysis. Considering the high CV event rate in this population, it was anticipated that the long treatment period would allow for the capture of events sufficient to characterize the risk. In addition, observation for safety continued after treatment discontinuation until the planned study end. Based on the expected number of MACE and MACE+ events, the sizes of the datasets were planned such that an upper limit of the CI for the HR against placebo and ESA (standard of care) < 1.8 would be obtained if the true HR was around 1 [31]. Given the known increased CV risk in clinical studies with standard of care (ESA) and the additional event numbers afforded by the size of the program, an improved precision for the estimate was also considered (upper limit of the CI of 1.3), which is in alignment with the CV reflection paper recommendation to consider the target population and the known CV risk profile and is informed by the FDA Guidance for Industry on Diabetes Mellitus-Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes (December 2008) [31]. Pooled hazard ratios were calculated using the inverse of variance approach over the log-transformed hazard ratios for the entire cohort and subgroup analyses.
Incidence and follow-up adjusted incidence rates (FAIRs) were calculated for safety endpoints by intervention (roxadustat or ESA), and then the pooled population HR was estimated using a meta-analytic method to combine the HRs from the individual studies using a naïve pooling process. Patient follow-up time in years (PY) was defined as the sum of each patient’s time in the analysis set from the first dose of study drug to last dose or censor date if no event of interest was observed. FAIR per 100 PY and the corresponding 95% CI were estimated for each study using Poisson regression in PROC GENMOD, modeling event count on treatment, offset by log (PY). TEAEs and malignancy-related mortality were descriptively evaluated using percentages and IRs.
The per protocol set (PPS) included all full analysis set (FAS) patients who received at least 12 weeks of treatment and did not meet any of the reasons to exclude a complete patient from the PPS. In the SIERRAS, HIMALAYAS, and ROCKIES studies, the PPS consisted of all patients in the FAS population who received at least 8 weeks of treatment, had at least one valid post-dose hemoglobin assessment, and were without any major protocol violations (Supplementary Appendix) [24,25,26]. The PPS was used for the primary endpoint analysis, selected secondary analyses, and analyses of all disposition, demography, and baseline characteristics. The safety analysis set (SAF) included all randomized subjects who received at least one dose of study drug and was used to describe demographic and baseline characteristics and all safety and tolerability related variables. Analyses were performed using SAS® Version 9.3 or higher (Cary, NC).
Results
Demographics and Baseline Characteristics
The four studies were conducted between February 2014 and September 2018 [23,24,25,26]. In total, 4714 patients were pooled for analysis in the entire cohort (roxadustat n = 2354, ESA n = 2360) of which 58.8% and 67.6% completed roxadustat and ESA therapy, respectively, with comparable results in the SAF and PPS (Table 2). Patients were more commonly male, with an approximate mean age of 56 years and mean weight of 77 kg (Table 3). Greater than 90% of patients were receiving hemodialysis at baseline with a mean dialysis vintage of > 30 months; patients in the incident dialysis subgroup had shorter dialysis exposure (Supplementary Table 1). Most patients were iron replete (approximately 79% in the incident dialysis and 90% in the stable dialysis subgroups). Mean baseline hemoglobin was < 9 g/dl in the incident dialysis subgroup and > 10 g/dl in the stable dialysis subgroup. Diabetic nephropathy and hypertensive nephropathy were the two most common etiologies of CKD.
Efficacy Endpoints
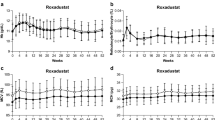
Hemoglobin CFB to weeks 28–36 achieved non-inferiority for roxadustat versus ESA in each study (Supplementary Table 2) and in the incident dialysis and stable dialysis subgroups (Table 4). In an exploratory subgroup analysis, all demographic and baseline variables were non-inferior between groups for their effects on hemoglobin CFB to weeks 28–36 in the stable dialysis subgroup (Supplementary Fig. 1) and were non-inferior for their effects on hemoglobin CFB to weeks 28–52 in the incident dialysis subgroup (Supplementary Fig. 2). The proportion of patients who achieved hemoglobin response at weeks 28–36 was similar for roxadustat and ESA in the PYRENEES, SIERRAS, and HIMALAYAS studies, but was not evaluated in the ROCKIES study (Supplementary Table 2), and was similar in the incident dialysis and stable dialysis subgroups (Table 4). The IR for rescue therapy use was similar between roxadustat and ESA groups and ranged from 7.1–9.4 (roxadustat) and 7.2–10.3 (ESA) (Supplementary Table 2). The CFB to weeks 12–28 in LDL cholesterol showed a more substantial reduction with roxadustat than ESA in all four studies (Supplementary Table 2). The CFB to weeks 12–28 in LDL/HDL ratio was improved with roxadustat compared to ESA in studies for which these data were available, whereas only one study evaluated CFB to weeks 12–28 in HDL cholesterol (Supplementary Table 2). The monthly IV iron use in roxadustat and ESA patients in incident dialysis and stable dialysis subgroups was low, with mean values < 100 mg and median values of 0, though the majority of patients (80.2–93.5%) received an IV iron infusion during the study period (Table 4). In the entire cohort, not receiving IV iron was more common for roxadustat than ESA patients over weeks 1–52 (63.5% vs 48.2%, difference 15.2%, 95% CI 12.43–18.05%). Roxadustat patients required a mean (SD) of 5.31 (16.29) IV administrations per PEY over weeks 1–52 compared with 9.64 (22.33) for ESA patients. The mean roxadustat and ESA doses decreased from the beginning to the end of the study in the incident dialysis subgroup. The mean ESA dose was increased compared with the initial dose in the stable dialysis subgroup while the mean roxadustat dose at the end of the study remained similar to the initial dose (Supplementary Fig. 3).
Safety Endpoints
Roxadustat was non-inferior to ESA for time to first event for MACE (HR 1.09, 95% CI 0.95–1.26) and MACE+ (HR 0.98, 95% CI 0.86–1.11) with consistent findings for all-cause mortality (HR 1.13, 95% CI 0.95–1.34) (Table 5) and each individual component of MACE+ in the SAF (Supplementary Table 3). Patients who received roxadustat compared with ESA in the incident dialysis subgroup had a numerically lower risk of all-cause mortality (incident dialysis: HR 0.83, 95% CI 0.57–1.19; stable dialysis: HR 1.23, 95% CI 1.02–1.49) as well as time to first event for MACE and MACE+ than those in the stable dialysis subgroup (Table 6). Roxadustat was non-inferior to ESA for time to first event for CV-MACE (HR 1.07, 95% CI 0.91–1.26) and CV-MACE+ (HR 0.94, 95% CI 0.82–1.09) in the entire cohort as well; findings for CV mortality (HR 1.14, 95% CI 0.91–1.42) were consistent with CV-MACE and CV-MACE+ (Supplementary Table 4). Malignancy-related mortality occurred infrequently in roxadustat (n = 5, 0.2%, IR 0.1/100 PY) and ESA (n = 7, 0.3%, IR 0.2/100 PY) groups.
Incidence rates of TEAEs were comparable though numerically greater in the roxadustat group compared with the ESA group (Table 7). The IR for TEAEs occurring in at least 5% of patients were generally similar between roxadustat and ESA (Supplementary Table 5). Numerically greater IRs observed for the roxadustat group compared to the ESA group included arteriovenous fistula site thrombosis (5.7 vs 3.9/100 PY), hypertension (8.3 vs 6.9/100 PY), diarrhea (7.0 vs 5.6/100 PY), headache (5.5 vs 3.8/100 PY), hypotension (5.1 vs 3.9/100 PY), nausea (5.0 vs 3.7/100 PY), and vomiting (4.3 vs 3.1/100 PY). Development of a pulmonary embolism (0.3 vs 0.3/100 PY) or deep vein thrombosis (0.8 vs 0.1/100 PY) occurred infrequently in roxadustat and ESA patients.
Discussion
In a heterogeneous cohort representative of the full spectrum of DD patients with anemia of CKD, roxadustat was non-inferior to ESA for the efficacy endpoints of hemoglobin CFB to weeks 28–36 and proportion of patients who achieved hemoglobin response at weeks 28–36 in individual studies. Roxadustat also demonstrated non-inferiority for time to first MACE and MACE+ versus ESA, and all-cause mortality was consistent with these findings.
For all four studies, non-inferiority of roxadustat compared with ESAs was found for the pivotal efficacy endpoints of hemoglobin CFB to weeks 28–36 and proportion of patients who achieved hemoglobin response at weeks 28–36, establishing that roxadustat is an effective agent compared to standard of care [32]. The considerable proportion of responders and more stable mean weekly dose to maintain target hemoglobin for patients receiving roxadustat compared to ESA in the stable dialysis subgroup warrants further investigation. Provenzano et al. reported a pooled analysis of three (SIERRAS, HIMALAYAS, and ROCKIES) of these four phase 3 studies comparing roxadustat and epoetin alfa in patients with CKD incident to dialysis (initiated on dialysis within 2 weeks to 4 months prior to randomization to roxadustat or epoetin alfa) and found roxadustat was at least as effective in hemoglobin CFB to weeks 28–52 (LSMD 0.22, 95% CI 0.05–0.40, P = 0.013) [27]. Identification of subgroups in the DD population who most benefit from roxadustat and the study of long-term efficacy remain research priorities.
Roxadustat was non-inferior to ESA for time to first MACE and MACE+ in the entire DD cohort, and exploratory all-cause mortality findings were consistent with these results and the prior pooled analysis of three studies [27]. In an exploratory analysis, results in the incident dialysis subgroup, representative of an unbiased head-to-head comparison between roxadustat and traditional ESA, showed lower HR point estimates and lower upper limit of the 95% CI for roxadustat vs traditional ESA on time to first MACE, MACE +, and all-cause mortality compared to the overall DD population. In the stable dialysis subgroup, the HR point estimates for time to first MACE, MACE+, and all-cause mortality were slightly elevated compared to the overall DD population in this exploratory analysis. Although these analyses were exploratory in nature, increased risk cannot be excluded. Risk factors for MACE and CV-MACE beyond anemia or a prior CV event require further investigation to elucidate the benefits of roxadustat in specific patients [3, 33, 34]. The overall rate and incidence of MACE was lower than previously reported in initial ESA trials, likely reflecting improved awareness of negative CV sequelae and overall improved management of anemia of CKD, including a lower target hemoglobin range [6,7,8,9].
Inclusion of a higher risk, stable dialysis population from the PYRENEES study may have affected the findings, although patients in the stable dialysis subgroup from the SIERRAS and ROCKIES studies appeared to have contributed to the possibly increased risk observed in the stable dialysis setting compared to the incident dialysis setting. Patients in the stable dialysis subgroup were pretreated with ESA on stable doses and generally had maintained hemoglobin levels within, or close to, the target range. Upon randomization and study treatment initiation, those randomized to ESA continued the same type of treatment, whereas the patients randomized to roxadustat switched to a new treatment modality with a different route of application and a different mechanism of action. Because of differences in clinical characteristics and patient demographics in the incident dialysis and stable dialysis subgroups, outcomes were evaluated separately in both populations. This conceptual difference between the study subgroups provides a plausible basis for a potentially design-related difference in CV safety outcomes between the stable dialysis and the incident dialysis subgroups that is unrelated to roxadustat. The stable dialysis subgroup represents a selected patient population with design-related one-sided fluctuations in anemia management upon switching over to treatment with roxadustat and possibly switching back again while the patients continued on ESA do not undergo these changes, potentially contributing to differences in outcomes with the active-controlled incident dialysis subgroup. The evaluation of the stable dialysis subgroup in patients on dialysis informs a differential risk for CV safety and mortality. The judicious use of roxadustat in ESA-converted patients is an important clinical consideration. In contrast, the incident dialysis subgroup represents an unselected patient population allowing an unbiased comparison between roxadustat and ESA, and the CV safety and mortality outcomes demonstrate that roxadustat is non-inferior to ESA.
The incidence and IR for all TEAE outcomes were relatively similar between roxadustat and ESA patients and were generally consistent with prior studies in DD patients and the pooled analysis of three studies [15, 17, 27, 35]. There was minimal treatment discontinuation due to a TEAE, with the numerically higher IR for discontinuation in the roxadustat group, potentially reflecting the open-label design of the four included studies [36]. While the rate and IR for common AEs were similar between roxadustat and ESA or slightly numerically higher for roxadustat than for ESA patients (e.g., nausea, vomiting, and diarrhea), these AEs have not been recognized as reasons to avoid roxadustat use [34]. Notably, though monitoring of serum potassium is recommended, hyperkalemia occurred less frequently in this substantially larger cohort than in a multicenter study by Chen and colleagues or a pooled analysis of patients incident to dialysis [15, 27, 34]. Additionally, the development of venous thromboembolism (e.g., deep vein thrombosis or arteriovenous fistula thrombosis) was infrequent, and event rates were numerically higher for roxadustat compared to ESA though the significance of this finding is unknown.
The heterogeneous cohort in this four-study pooled DD population is representative of the full spectrum of patients requiring dialysis, ranging from patients who were ESA-naïve to those pretreated with ESA for several years. Also, patients with variable duration of dialysis who were representative of the hemodialysis and peritoneal dialysis modalities were enrolled. Therefore, this dataset is representative of the overall DD population in need of anemia treatment and, at the same time, is subject to heterogeneity due to the wide range of patient characteristics. The four studies examined here evaluated comparable efficacy and safety outcomes, which improves the internal validity of this pooled analysis. Outcomes may have been affected by the frequency of dose adjustments for study medications as roxadustat-treated patients were permitted to adjust the dose every 4 weeks to maintain hemoglobin between 10–12 g/dl, whereas ESA-treated patients followed local label, and dose adjustments may not have been as frequent in all patients. Additionally, patients more frequently discontinued roxadustat versus ESA, which may reflect a potential bias of the open-label design with an investigational drug versus the standard of care. While the study cohorts were predominately white, this was a global evaluation of roxadustat that has generalizability to other populations. Specific subpopulations and variables were not evaluated beyond the stable dialysis and incident dialysis subgroups, such as iron repletion status and inflammation, but should be investigated in future analyses based on hypothesis-generating data in other cohorts [27, 37]. Though fewer patients received IV iron in the roxadustat group compared to the ESA group, this post hoc finding requires further investigation. Although post hoc, the analyses in the stable dialysis subgroup cover two-thirds of the DD pool, with the remaining one-third coming from the incident dialysis subgroup, and represent an adequate sample for assessing safety outcomes. While this pooled analysis was powered to test the non-inferiority of roxadustat to ESA, further analyses may clarify roxadustat’s place in therapy.
Conclusion
Roxadustat corrected and maintained hemoglobin similarly to ESA while demonstrating a comparable safety profile, including CV safety, in a wide spectrum of patients receiving dialysis with anemia of CKD. The results in patients switching from ESA should be interpreted with caution as patients allocated to roxadustat were switched from ESA at the start of each study, whereas patients randomized to ESA remained on the same ESA dose, and the impact of a potential risk in switching to any new treatment versus remaining on a treatment with a stabilized hemoglobin may confound the observed results; thus, any comparison of treatment effect estimates cannot be reliably established. These data are consistent with findings from previous single-study reports. Roxadustat represents an oral alternative to ESAs in correcting and maintaining a target hemoglobin level for anemia of CKD in patients receiving dialysis.
References
Locatelli F, Fishbane S, Block GA, Macdougall IC. Targeting hypoxia-inducible factors for the treatment of anemia in chronic kidney disease patients. Am J Nephrol. 2017;45(3):187–99.
Babitt JL, Lin HY. Mechanisms of anemia in CKD. J Am Soc Nephrol. 2012;23(10):1631–4.
Pereira AA, Sarnak MJ. Anemia as a risk factor for cardiovascular disease. Kidney Int Suppl. 2003;87:S32–9.
Ingrasciotta Y, Lacava V, Marciano I, Giorgianni F, Tripepi G, Graziella DA, et al. In search of potential predictors of erythropoiesis-stimulating agents (ESAs) hyporesponsiveness: a population-based study. BMC Nephrol. 2019;20(1):359.
Johnson DW, Pollock CA, Macdougall IC. Erythropoiesis-stimulating agent hyporesponsiveness. Nephrology (Carlton). 2007;12(4):321–30.
Besarab A, Bolton WK, Browne JK, Egrie JC, Nissenson AR, Okamoto DM, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339(9):584–90.
Pfeffer MA, Burdmann EA, Chen CY, Cooper ME, de Zeeuw D, Eckardt KU, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361(21):2019–32.
Singh AK, Szczech L, Tang KL, Barnhart H, Sapp S, Wolfson M, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355(20):2085–98.
Fishbane S, Schiller B, Locatelli F, Covic AC, Provenzano R, Wiecek A, et al. Peginesatide in patients with anemia undergoing hemodialysis. N Engl J Med. 2013;368(4):307–19.
Manns BJ, Tonelli M. The new FDA labeling for ESA–implications for patients and providers. Clin J Am Soc Nephrol. 2012;7(2):348–53.
Kilpatrick RD, Critchlow CW, Fishbane S, Besarab A, Stehman-Breen C, Krishnan M, et al. Greater epoetin alfa responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol. 2008;3(4):1077–83.
Zhang Y, Thamer M, Stefanik K, Kaufman J, Cotter DJ. Epoetin requirements predict mortality in hemodialysis patients. Am J Kidney Dis. 2004;44(5):866–76.
Szczech LA, Barnhart HX, Inrig JK, Reddan DN, Sapp S, Califf RM, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int. 2008;74(6):791–8.
Gupta N, Wish JB. Hypoxia-inducible factor prolyl hydroxylase inhibitors: a potential new treatment for anemia in patients with CKD. Am J Kidney Dis. 2017;69(6):815–26.
Chen N, Hao C, Liu BC, Lin H, Wang C, Xing C, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med. 2019;381(11):1011–22.
Chen N, Hao C, Peng X, Lin H, Yin A, Hao L, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381(11):1001–10.
Karaboyas A, Morgenstern H, Fleischer NL, Vanholder RC, Dhalwani NN, Schaeffner E, et al. Inflammation and erythropoiesis-stimulating agent response in hemodialysis patients: a self-matched longitudinal study of anemia management in the dialysis outcomes and practice patterns study (DOPPS). Kidney Med. 2020;2(3):286–96.
Akizawa T, Yamaguchi Y, Otsuka T, Reusch M. A phase 3, multicenter, randomized, two-arm, open-label study of intermittent oral dosing of roxadustat for the treatment of anemia in japanese erythropoiesis-stimulating agent-naive chronic kidney disease patients not on dialysis. Nephron. 2020;144(8):372–82.
Shutov E, Sulowicz W, Esposito C, Tataradze A, Andric B, Reusch M, et al. Roxadustat for the treatment of anemia in chronic kidney disease patients not on dialysis: a phase 3, randomized, double-blind, placebo-controlled study (ALPS). Nephrol Dial Transpl. 2021. https://doi.org/10.1093/ndt/gfab057.
Coyne DW, Roger SD, Shin SK, Kim SG, Cadena AA, Moustafa MA, et al. Roxadustat for CKD-related anemia in non-dialysis patients. Kidney Int Rep. 2021;6(3):624–35.
Fishbane S, El-Shahawy MA, Pecoits-Filho R, Van BP, Houser MT, Frison L, et al. Roxadustat for treating anemia in patients with CKD not on dialysis: results from a randomized phase 3 study. J Am Soc Nephrol. 2021;32(3):737–55.
Barratt J, Andric B, Tataradze A, Schomig M, Reusch M, Valluri U, et al. Roxadustat for the treatment of anaemia in chronic kidney disease patients not on dialysis: a phase 3, randomised, open-label, active-controlled study (DOLOMITES). Nephrol Dial Transpl. 2021. https://doi.org/10.1093/ndt/gfab191.
Csiky B SM, Esposito C, Barratt J, Reusch M, Valluri U, Sulowicz W. Roxadustat for the maintenance treatment of anemia in end-stage kidney disease patients on stable dialysis: a European Phase 3, Randomized, Open-Label, Active-Controlled Study (PYRENEES). Adv Therapy. 2021.
Charytan C, Manllo-Karim R, Martin ER, Steer D, Bernardo M, Dua SL, et al. A randomized trial of roxadustat in anemia of kidney failure: SIERRAS study. Kidney Int Rep. 2021. https://doi.org/10.1016/j.ekir.2021.04.007.
Provenzano R, Shutov E, Eremeeva L, Korneyeva S, Poole L, Saha G, et al. Roxadustat for anemia in patients with end-stage renal disease incident to dialysis. Nephrol Dial. 2021;25: gfab051. https://doi.org/10.1093/ndt/gfab051.
Fishbane SPC, El-Shahawy M, Escudero ET, Rastogi A, Van BP, Frison L, Houser M, Pola M, Little DJ, Guzman N, Pergola PE. Roxadustat versus epoetin alfa for treating anemia in patients with chronic kidney disease on dialysis: the ROCKIES randomized clinical trial. J Am Soc Nephrol. 2021;32:737–55.
Provenzano R, Fishbane S, Szczech L, Leong R, Saikali KG, Zhong M, et al. Pooled analysis of roxadustat for anemia in patients with kidney failure incident to dialysis. Kidney Int Rep. 2021;6(3):613–23.
Liu J, Zhang A, Hayden JC, Bhagavathula AS, Alshehhi F, Rinaldi G, et al. Roxadustat (FG-4592) treatment for anemia in dialysis-dependent (DD) and not dialysis-dependent (NDD) chronic kidney disease patients: a systematic review and meta-analysis. Pharmacol Res. 2020;155:104747.
Hicks KA, Tcheng JE, Bozkurt B, Chaitman BR, Cutlip DE, Farb A, et al. 2014 ACC/AHA key data elements and definitions for cardiovascular endpoint events in clinical trials: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Cardiovascular Endpoints Data Standards). J Am Coll Cardiol. 2015;66(4):403–69.
Blind E, Janssen H, Dunder K, de Graeff PA. The European Medicines Agency’s approval of new medicines for type 2 diabetes. Diabetes Obes Metab. 2018;20(9):2059–63.
Services USDoHaH. Guidance for industry diabetes mellitus: evaluating cardiovascular risk in new antidiabetic therapies to treat type 2 diabetes. 2021. https://www.fda.gov/media/71297/download2008. Accessed 15 Jan 2021.
Locatelli F, Barany P, Covic A, De Francisco A, Del Vecchio L, Goldsmith D, et al. Kidney Disease: Improving Global Outcomes guidelines on anaemia management in chronic kidney disease: a European Renal Best Practice position statement. Nephrol Dial Transpl. 2013;28(6):1346–59.
Pernod G, Bosson JL, Golshayan D, Barro C, Forneris G, Martina G, et al. Phenotypic and genotypic risk factors for cardiovascular events in an incident dialysis cohort. Kidney Int. 2006;69(8):1424–30.
Yap DYH, McMahon LP, Hao CM, Hu N, Okada H, Suzuki Y, et al. Recommendations by the Asian Pacific society of nephrology (APSN) on the appropriate use of HIF-PH inhibitors. Nephrology (Carlton). 2021;26(2):105–18.
Provenzano R, Besarab A, Wright S, Dua S, Zeig S, Nguyen P, et al. Roxadustat (FG-4592) versus epoetin alfa for anemia in patients receiving maintenance hemodialysis: a phase 2, randomized, 6- to 19-week, open-label, active-comparator, dose-ranging, safety and exploratory efficacy study. Am J Kidney Dis. 2016;67(6):912–24.
Manja V, Lakshminrusimha S. Epidemiology and clinical research design, part 1: study types. NeoReviews. 2014;15(12):e558–69.
Kimura S, Sato T, Ikeda S, Noda M, Nakayama T. Development of a database of health insurance claims: standardization of disease classifications and anonymous record linkage. J Epidemiol. 2010;20(5):413–9.
Acknowledgements
Roxadustat is being developed by FibroGen, AstraZeneca, and Astellas. We thank the participants of the studies.
Funding
The studies were funded by Astellas Pharma, Inc. (PYRENEES: 1517-CL-0613), FibroGen, Inc. (SIERRAS: FGCL-4592-064 and HIMALAYAS: FGCL-4592-063), and AstraZeneca (ROCKIES: D5740C00002). The publication fees were funded by Astellas Pharma, Inc.
Medical Writing, Editorial and Other Assistance
Medical writing/editorial support was provided by Patrick Tucker, PhD, Drayton Hammond, PharmD, and Elizabeth Hermans, PhD, from Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, and funded by the study sponsor.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
(1) Conception and design: MR. (2) Acquisition of data: JB, WS, MS, CE, BS. (3) Analysis and interpretation of the data: JB, WS, MS, CE, MR, JY, BC. (4) Drafting and critical revision of the article for important intellectual content: JB, WS, MS, CE, MR, JY, BC.
Disclosures
MR is an employee of Astellas Pharma Europe B.V. and JY is an employee of Astellas Pharma, Inc., Northbrook, IL, USA. JB, WS, MS, CE, and BC have nothing to disclose.
Compliance with Ethics Guidelines
All study protocols were approved by relevant institutional review boards and/or ethics committees and were conducted in accordance with the tenets of the Declaration of Helsinki, the International Council for Harmonization guidelines for Good Clinical Practice, and any other applicable local health and regulatory requirements. All patients provided written informed consent before enrollment. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Data Availability
Researchers may request access to anonymized participant level data, trial level data and protocols from Astellas sponsored clinical trials at https://www.clinicalstudydatarequest.com. For the Astellas criteria on data sharing see: https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Astellas.aspx.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Barratt, J., Sulowicz, W., Schömig, M. et al. Efficacy and Cardiovascular Safety of Roxadustat in Dialysis-Dependent Chronic Kidney Disease: Pooled Analysis of Four Phase 3 Studies. Adv Ther 38, 5345–5360 (2021). https://doi.org/10.1007/s12325-021-01903-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-021-01903-7