
Abstract
Introduction
This study was conducted to elucidate the safety of roxadustat, an oral medication, in patients with non-dialysis-dependent (NDD) or incident dialysis dialysis-dependent (ID-DD) chronic kidney disease (CKD).
Methods
Safety results from four phase 3, randomized, open-label studies comparing roxadustat to an erythropoiesis-stimulating agent (ESA) in men and women with NDD or ID-DD CKD with anemia were pooled and evaluated. Endpoints were time to major adverse cardiovascular event (MACE; myocardial infarction, stroke, and all-cause mortality) and MACE+ (MACE plus congestive heart failure or unstable angina requiring hospitalization), all-cause mortality, and treatment-emergent adverse events (TEAEs). MACE and MACE+ were evaluated for non-inferiority at 1.8- and 1.3-margins using hazard ratios (HRs) and 95% confidence intervals (CIs). TEAEs were descriptively summarized.
Results
In total, 2142 patients were evaluated (1083 roxadustat; 1059 ESA). Roxadustat was comparable to ESA for risk of MACE (HR 0.79, 95% CI 0.61–1.02), MACE+ (HR 0.78, 95% CI 0.62–0.98), and all-cause mortality (HR 0.78, 95% CI 0.57–1.05). TEAEs were comparable between roxadustat and ESA groups, including any TEAE [incidence rate per 100 (IR/100) patient-exposure years 56.1 vs. 53.5], TEAEs leading to study drug discontinuation (IR/100 patient-exposure years 6.7 vs. 5.1), and TEAEs leading to death (IR/100 patient-exposure years 6.9 vs. 7.4).
Conclusion
There was no evidence of increased risk of cardiovascular events or mortality with roxadustat compared with ESA in patients with anemia who have NDD or ID-DD CKD. Although TEAEs occurred commonly in both the roxadustat and ESA groups, patients infrequently discontinued the study drug because of an adverse event.
Clinical Trial Registration Numbers
DOLOMITES, 1517-CL-0610 [NCT02021318]; HIMALAYAS, FGCL-4592-063 [NCT02052310]; SIERRAS, FGCL-4592-064 [NCT02273726]; and ROCKIES, D5740C00002 [NCT02174731].
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The non-dialysis-dependent (NDD) and incident dialysis (ID) (i.e., initiated dialysis within the last 4 months) dialysis-dependent (DD) chronic kidney disease (CKD) population is a vulnerable population at increased risk for morbidity and mortality compared to patients without CKD, despite significant clinical advancements in recent years. |
A pooled analysis of studies comparing hemoglobin correction and then maintenance with roxadustat to hemoglobin correction and maintenance with erythropoiesis-stimulating agents in patients with NDD or ID-DD CKD is novel, and necessary to further elucidate the safety of roxadustat versus erythropoiesis-stimulating agents in a large, heterogeneous population. |
What was learned from the study? |
There was no increased cardiovascular event risk or all-cause mortality risk with roxadustat compared with erythropoiesis-stimulating agents in the NDD or ID-DD CKD populations. |
Drug-related treatment-emergent adverse events and drug-related treatment-emergent adverse events leading to discontinuation of the study drug occurred more frequently with roxadustat; however, this difference is likely influenced by investigator awareness of treatment assignment in these open-label studies. |
Introduction
The non-dialysis-dependent (NDD) and incident dialysis (ID) (i.e., initiated dialysis within the last 4 months) dialysis-dependent (DD) chronic kidney disease (CKD) population is a vulnerable population at increased risk for morbidity and mortality compared to patients without CKD, despite significant clinical advancements in recent years [1, 2]. Anemia of CKD may increase the risk of hospitalization and mortality, particularly in late-stage NDD-CKD and during the first year after dialysis initiation, because, in part, patients at these stages may need or have just initiated anemia treatment [1]. Treatment of anemia of CKD may include iron supplementation and a parenteral erythropoiesis-stimulating agent (ESA) [3].
Previous trials with ESAs in patients with NDD-CKD suggested an increased risk of cardiovascular and/or thromboembolic events when higher hemoglobin levels, such as 13 g/dL, were targeted [4,5,6,7]. Subsequent analyses identified that primary factors associated with these events may have included higher ESA doses and not achieving the desired hemoglobin target [8,9,10,11]. Those patients who do not respond adequately to an ESA are at the highest risk of mortality [12]. Therefore, in the NDD and ID-DD CKD populations that already are at an increased risk for morbidity and mortality compared to patients without CKD, further characterization of the safety profile of novel agents proposed for anemia of CKD treatment, such as hypoxia-inducible factor prolyl hydroxylase inhibitors, is needed.
Roxadustat is an orally administered hypoxia-inducible factor prolyl hydroxylase inhibitor that was developed for the treatment of CKD-related anemia. Roxadustat has had favorable or comparable efficacy in phase 3 studies versus placebo and ESAs, and has been approved in multiple countries and regions for anemia of CKD, including in Europe for the treatment of anemia of CKD in patients on dialysis or not on dialysis [13,14,15,16,17,18,19,20,21,22,23]. A pooled analysis of studies comparing hemoglobin correction and then maintenance with roxadustat to hemoglobin correction and maintenance with ESAs in patients with NDD or ID-DD CKD is novel, and necessary to further elucidate the safety of roxadustat versus ESA in a large, heterogeneous population.
Methods
Component Studies and Pooling Methodology
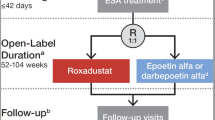
Patients from four randomized, multicenter, open-label, active-comparator studies were eligible for inclusion in the post hoc, exploratory, pooled analysis [DOLOMITES, 1517-CL-0610 (NCT02021318); HIMALAYAS, FGCL-4592-063 (NCT02052310); SIERRAS, FGCL-4592-064 (NCT02273726); and ROCKIES, D5740C00002 (NCT02174731); Table 1] [2, 21,22,23]. Comparable clinical data were collected, and safety endpoints were evaluated between roxadustat and an ESA comparator (darbepoetin alfa: DOLOMITES, epoetin alfa: HIMALAYAS, SIERRAS, and ROCKIES).
Participants
All eligible patients were aged ≥ 18 years, had anemia of CKD, and were either NDD (DOLOMITES) or were ID-DD and receiving hemodialysis or peritoneal dialysis (HIMALAYAS, SIERRAS, and ROCKIES). Patients with ID-DD CKD were defined as those who initiated dialysis ≥ 2 weeks to ≤ 4 months prior to randomization. Exclusion criteria included prior treatment with roxadustat or another hypoxia-inducible factor prolyl hydroxylase inhibitor, active or chronic gastrointestinal bleeding, recent red blood cell transfusion, and anticipated elective surgery with expected blood loss. Included patients were randomized (1:1) to oral roxadustat three times weekly or ESA, either epoetin alfa or darbepoetin alfa, by intravenous or subcutaneous route. Each study used different pre-specified factors for stratification during randomization. Patients receiving ESA at the time of randomization and randomized to the ESA treatment arm were recommended to maintain the same route of administration during the study. Additional details on participants are provided in Appendix S1.
Interventions and Rescue Therapy
In patients receiving ESA prior to randomization, initial drug dosing was based on the average weekly ESA dose prior to study inclusion. In ESA-untreated patients, the initial study drug dose was based on body weight. In general, dose adjustments for roxadustat-treated patients were permitted every 4 weeks to maintain hemoglobin between 10 and 12 g/dL, and were conducted in accordance with prespecified rules; treatment of patients randomized to ESA followed the local label. Rescue therapy included receipt of a red blood cell transfusion and/or ESAs (for roxadustat-treated patients only). Additional details on study drug dosing and titration procedures and rescue therapy are provided in Appendix S1.
Safety Endpoints
The two cardiovascular safety endpoints were time to first major adverse cardiovascular event (MACE), comprised of all-cause mortality, myocardial infarction, or stroke, and time to first MACE plus (MACE+), comprised of MACE plus unstable angina or congestive heart failure requiring hospitalization while on treatment up to 7 days after the last dose (OT-7). Time to all-cause mortality was also evaluated. These endpoints were defined based on the 2014 American College of Cardiology/American Heart Association Key Data Elements and Definitions for Cardiovascular Endpoint Events in Clinical Trials, and were adjudicated by a central Independent Event Review Committee, whose members were unaware of study group assignments [24]. The adjudication process is described in Appendix S1.
Treatment-emergent adverse events (TEAEs) were monitored and evaluated. TEAEs were defined during the evaluation period from the first study drug administration up to 28 days after the last dose (OT-28).
Statistical Analysis
Pre-specified analyses were performed in this post hoc NDD and ID-DD CKD pooled population. The safety population, which included all randomized patients who received at least one dose of study drug, was used to describe demographic and baseline characteristics and all safety- and tolerability-related variables. The analysis period was the on-treatment period (i.e., OT-7 for MACE, MACE+ , and all-cause mortality and OT-28 for TEAEs, respectively) in accordance with European Medicines Agency guidance. The pooled hazard ratio (HR) and its upper limit of the 95% confidence interval (CI) in the time-to-event analysis was used as the primary statistical measure to determine non-inferiority of the cardiovascular safety of roxadustat (vs. ESA) using previously described methods [25]. Pooled HRs were calculated for the entire cohort and subgroup analyses using the inverse of variance approach over the log-transformed HRs, and were estimated using a meta-analytic method to combine the HRs from the individual studies using a naïve pooling process. The upper limit of the 95% CI for the HR against standard of care (ESA) below 1.8 was pre-specified [26]. Additionally, an improved precision for the estimate was also considered (upper limit of the 95% CI of 1.3) because of the recognized increased cardiovascular risk in clinical studies with an ESA. The more conservative upper limit is in alignment with the known cardiovascular risk profile [26, 27].
Patient follow-up time in patient-exposure years was defined as (last dose date − first dose date) + 1/365.25. Clinical characteristics and demographics were summarized descriptively. TEAEs were descriptively evaluated using percentages and incidence rate (IR) per 100 (IR/100) patient-exposure years, defined as 100 × number of patients with events per patient-exposure year. Analyses were performed using SAS® v.9.3 or higher (SAS Institute, Cary, NC, USA).
Ethics Compliance
All study protocols for the previously published studies were approved by institutional review boards and/or ethics committees for the study sites, and were conducted in accordance with the tenets of the Declaration of Helsinki of 1964 and its later amendments, the International Council for Harmonization guidelines for Good Clinical Practice, and any other applicable local health and regulatory requirements. Individual study sites are available at https://clinicaltrials.gov/ct2/show/NCT02021318 (DOLOMITES), https://clinicaltrials.gov/ct2/show/NCT02273726 (SIERRAS), https://clinicaltrials.gov/ct2/show/NCT02052310 (HIMALAYAS), and https://clinicaltrials.gov/ct2/show/NCT02174731 (ROCKIES). As this was a pooled post hoc analysis, approval by an ethics committee was not necessary. All patients provided written informed consent before enrollment.
Results
Patients with NDD or ID-DD CKD in the safety population from the four clinical studies were pooled (roxadustat n = 1083, ESA n = 1059) [2, 21,22,23]. Patients with stable DD CKD who were randomized and included in the SIERRAS, HIMALAYAS, and ROCKIES studies were not included in this analysis (Table 1). There were no differences in the percentage of patients with ID-DD randomized to roxadustat or ESA [SIERRAS: roxadustat 36 (50.7%), ESA 35 (49.3%); HIMALAYAS: roxadustat 522 (50.2%), ESA 517 (49.8%); ROCKIES: roxadustat 202 (48.6%), ESA 214 (51.4%)]. The patient-exposure years for patients randomized to roxadustat and to ESA were 1617.5 years and 1662.0 years, respectively. Baseline demographics and clinical characteristics were generally comparable between patients randomized to roxadustat or ESA (Table 2). Most patients were White (roxadustat 75.2%, ESA 73.8%) and received care in Eastern Europe (roxadustat 53.9%, ESA 54.9%). A minority of patients received a statin (roxadustat 14.9%, ESA 15.2%) or ESA treatment (roxadustat 29.1%, ESA 30.6%) prior to randomization. Around 40% of patients had diabetes mellitus (roxadustat 43.6%, ESA 42.4%) or cardiovascular disease (roxadustat 40.8%, ESA 40.6%), while a smaller percentage of patients had cerebrovascular disease (roxadustat 8.7%, ESA 9.3%) or thromboembolic disease (roxadustat 1.2%, ESA 1.9%) at baseline. The mean baseline hemoglobin concentration was approximately 9 g/dL (roxadustat 9.04 g/dL, ESA 9.05 g/dL) in both treatment groups, and approximately one-quarter (roxadustat 27.3%, ESA 28.5%) of patients were iron-depleted at baseline. Most patients had a baseline hemoglobin < 10 g/dL regardless of prior ESA use (Supplementary Material Table S1). Less than half of patients randomized to roxadustat or ESA were receiving a concomitant agent acting on the renin-angiotensin system (roxadustat 41.8%, ESA 41.0%), an antithrombotic agent (roxadustat 48.7%, ESA 47.0%), a ß-blocking agent (roxadustat 46.4%, ESA 46.4%), a diuretic (roxadustat 34.5%, ESA 34.1%), or a lipid-modifying agent (roxadustat 32.7%, ESA 31.4%).
Most patients randomized to roxadustat (66.3%) or ESA (69.2%) completed treatment (Table 3). The most common reasons for treatment discontinuation were death (roxadustat 8.9%, ESA 8.6%), study withdrawal by the patient (roxadustat 6.8%, ESA 6.7%), and TEAEs (roxadustat 5.9%, ESA 4.5%). In total, 11 patients (0.5%) were lost to follow-up, including 6 of the 1083 patients (0.6%) who received roxadustat and 5 of the 1059 patients (0.5%) who received an ESA.
The times to first event for MACE (HR 0.79, 95% CI 0.61–1.02) and MACE+ (HR 0.78, 95% CI 0.62–0.98) demonstrated no increased risk of cardiovascular events between roxadustat and ESA. Additionally, there was no increased risk of all-cause mortality (HR 0.78, 95% CI 0.57–1.05). All cardiovascular safety endpoints suggested a numerically lower risk with patients randomized to roxadustat compared to ESA (Table 4). Comparable results for the risk of MACE, MACE+ , and all-cause mortality in subgroups stratified by baseline characteristics are presented in Supplementary Material Figs. S1–S3.
The incidence for any TEAE was similar for roxadustat and ESA (83.7% vs. 84.0%) while the incidence for drug-related TEAEs was higher for roxadustat than ESA (13.8% vs. 10.3%). Although the incidence for TEAEs leading to discontinuation of study drug was similar between roxadustat and ESA (10.0% vs. 8.0%), drug-related TEAEs leading to discontinuation of study drug occurred more frequently with roxadustat (1.6% vs. 0.3%) (Table 5). The most frequent (IR/100 patient-exposure years) TEAEs were hypertension (roxadustat 12.9, ESA 12.3); end-stage kidney disease (roxadustat 6.7, ESA 6.4), which predominately occurred in patients with NDD-CKD enrolled in the DOLOMITES study; diarrhea (roxadustat 7.1, ESA 4.9); and hyperkalemia (roxadustat 4.3, ESA 4.9) (Supplementary Material Table S2). The most frequent serious TEAEs (IR/100 patient-exposure years) were end-stage kidney disease (roxadustat 6.7, ESA 6.4), predominately in patients with NDD-CKD enrolled in the DOLOMITES study; pneumonia (roxadustat 3.5, ESA 3.1); and arteriovenous fistula thrombosis (roxadustat 3.3, ESA 1.9) (Supplementary Material Table S3), with similar IR/100 patient-exposure years between groups for grade ≥ 3 TEAEs (Supplementary Material Table S4). Drug-related serious TEAEs (IR/100 patient-exposure years, roxadustat 1.8, ESA 1.2), as well as drug-related TEAEs leading to discontinuation of the study drug (IR/100 patient-exposure years, roxadustat 1.1, ESA 0.2), occurred infrequently in both groups. While infrequent, the two most common (IR/100 patient-exposure years) TEAE categories leading to study drug discontinuation by organ class were cardiac disorders (roxadustat 1.2, ESA 1.0) and infections and infestations (roxadustat 2.0, ESA 1.3) (Supplementary Material Table S5). The IR/100 patient-exposure years for TEAEs leading to death were similar though numerically lower for roxadustat (6.9) than ESA (7.4); the three most common (IR/100 patient-exposure years) TEAE categories leading to death by organ class were cardiac disorders (roxadustat 2.0, ESA 2.2), infections and infestations (roxadustat 1.9, ESA 1.1), and general disorders and administration site conditions (roxadustat 1.7, ESA 1.4) (Supplementary Material Table S6).
Discussion
In this pooled analysis of four phase 3 clinical studies comparing roxadustat to an ESA, there was no increased cardiovascular event risk or all-cause mortality risk with roxadustat compared with ESA in the NDD or ID-DD CKD population. The combined NDD and ID-DD CKD population was chosen as this is a population representative of the hemoglobin correction population with similar needs for hemoglobin control and naïve to ESA treatment, which is a clearly defined group that requires therapy. Approximately two-thirds of patients in each group completed treatment. While 84% of patients experienced at least one TEAE of any severity, AE development was only the third most common reason for treatment discontinuation. Drug-related serious TEAEs occurred infrequently, with the most common event being progression of end-stage kidney disease, which developed in approximately 10% of patients in each group, with the contributing events occurring in patients with NDD-CKD. TEAEs leading to death happened at a numerically lower frequency and IR/100 patient-exposure years in patients randomized to roxadustat compared to ESA. Drug-related TEAEs, and drug-related TEAEs leading to discontinuation of the study drug, occurred more frequently with roxadustat; however, this difference is likely influenced by investigator awareness of treatment assignment in these open-label studies. Adverse events may be more likely to be attributed to the new treatment in open-label studies when the comparator has been the standard of care for some time.
The times to first MACE, MACE+ , and all-cause mortality were comparable for roxadustat and ESA, demonstrating no evidence of increased cardiovascular risk for the entire pooled population; outcomes were consistent for each individual study. These findings are similar to those described for the ID-DD population in a recent four-study pooled analysis comparing roxadustat to ESA, in which the cardiovascular and mortality risk for patients treated with roxadustat was estimated to be comparable to the cardiovascular and mortality risk for patients treated with ESA therapy, based on data from direct comparison of both therapies [25]. An exception exists for MACE+ , suggesting the possibility of a favorable risk profile with roxadustat compared with ESA when hospitalization for congestive heart failure or unstable angina is also considered, though this finding is considered hypothesis-generating. Additionally, the point estimates and upper limit of the 95% CI for MACE and MACE+ were higher in a pooled analysis of three studies evaluating roxadustat in patients with NDD-CKD; however, those results may have been biased because of differential dropout and the potential for informative censoring favoring the placebo group [28]. Although the results in subgroup analyses for MACE and MACE+ were consistent with the overall findings, future investigation into subgroups of interest could be warranted.
A numerically greater percentage of patients with NDD or ID-DD CKD randomized to roxadustat discontinued treatment because of a TEAE compared with ESA. The IR of TEAEs leading to study drug discontinuation was < 1/100 patient-exposure years for all organ classes except for cardiac disorders and infections and infestations, and only infections and infestations had numerically greater IR/100 patient-exposure years for roxadustat compared with ESA. However, the frequencies of serious or grade ≥ 3 TEAEs in these categories were numerically similar between roxadustat and ESA groups, which may suggest that any potential difference in TEAE leading to discontinuation of roxadustat or ESA may be due to chance or the result of individualized patient factors [28]. The most frequent TEAEs among patients randomized to roxadustat or ESA included hypertension, progression of end-stage kidney disease, diarrhea, and hyperkalemia. Apart from end-stage kidney disease, which had the same incidence in both groups, the other three TEAEs had minimal or no presence in the serious or grade ≥ 3 TEAE lists, suggesting these TEAEs were predominately concerns of tolerability rather than safety.
Pneumonia development and arteriovenous fistula thrombosis were the most commonly occurring serious TEAEs in this analysis, which may not have been influenced by the assigned treatment as they are well-established complications in patients with ID-DD CKD that are independently affected by other patient- and treatment-specific factors [29,30,31]. However, because variables such as the type of line access, fistula maturation, and time on use were not available for comparison between patients who received roxadustat or ESA, the arteriovenous fistula thrombosis rate must be interpreted with caution. As expected, based on the most frequent serious or grade ≥ 3 TEAEs, the three categories of TEAEs that comprised the majority of TEAEs leading to death in this study were cardiac disorders, infections and infestations, and general disorders and administration site condition, which were numerically similar between groups. Although these TEAEs leading to death were not classified as specifically related to the study drug and are common complications of end-stage kidney disease, critical evaluation for these TEAEs should be considered in patients treated with roxadustat or an ESA.
The heterogeneous cohort in this analysis is representative of a high-risk population of patients with anemia of CKD, who are either ESA-naïve or have been treated minimally with ESA prior to randomization. Nevertheless, this is a mixed population for which results may not necessarily be fully generalizable to either subgroup in the cohort. However, the high-risk nature of the subgroups in this population, the comparison of roxadustat to an active ESA comparator, and the recognition of this population in clinical practice are strengths of the analysis. The focus of this analysis was on safety, as comparable efficacy of roxadustat versus ESA has already been confirmed in previous publications [2, 21,22,23]. Although this pooled, post hoc analysis was not powered for non-inferiority testing, the upper bound limits for MACE and MACE+ evaluations are below the pre-specified, established non-inferiority margin. If the upper bound limit for the 95% CI were 1.25, as was used in studies evaluating the cardiovascular safety of vadadustat and daprodustat, the interpretation of these findings would be unaffected because the upper limits did not exceed 1.05 [29, 32]. Additionally, while most patients received care in Europe, multiple other continents were well represented, increasing the global application of these results. The safety outcomes evaluated in these four studies were comparable, which improves this analysis’s internal validity. Outcomes may have been affected by study drug dose adjustment frequency, as roxadustat-treated patients were permitted a dose adjustment every 4 weeks to maintain hemoglobin between 10 and 12 g/dL whereas ESA-treated patients followed local label, and dose adjustments may not have been as frequent in all patients. While multiple explanations may exist for study drug discontinuation, TEAE development, a potential bias from the open-label study design, and a comparison between an investigational drug versus standard of care may have impacted study drug discontinuation, regardless of the actual concerns surrounding efficacy, safety, or tolerability. TEAEs were classified at the discretion of the investigator, not definitively associated with study drug, and could have been affected by patient-specific factors that were not evaluated. Additionally, some specific TEAEs (e.g., arteriovenous fistula complications) affected patients mainly in one subgroup (i.e., ID-DD) and may not be generalizable to patients in the other subgroup. While some numerical differences in the IR of TEAEs may also be statistically significant, this pooled analysis was not designed to statistically test those hypotheses.
Conclusions
There was no evidence of an increased risk of cardiovascular events or mortality with roxadustat compared with ESA in patients with NDD or ID-DD CKD and anemia. Although TEAEs were common in both groups, patients infrequently discontinued the study drug because of an AE. Further investigation into patient-specific factors that affect TEAE development with roxadustat and ESAs is needed to meet patient needs in the NDD and ID-DD CKD population. Roxadustat represents an efficacious and well-tolerated oral alternative to ESAs for correcting and maintaining a target hemoglobin level for anemia of CKD in patients who are NDD or ID-DD.
References
Wingard RL, Chan KE, Lazarus JM, Hakim RM. The “right” of passage: surviving the first year of dialysis. Clin J Am Soc Nephrol. 2009;4(Suppl 1):S114–20.
Provenzano R, Shutov E, Eremeeva L, et al. Roxadustat for anemia in patients with end-stage renal disease incident to dialysis. Nephrol Dial Transplant. 2021;36:1717–30.
Johnson DW, Pollock CA, Macdougall IC. Erythropoiesis-stimulating agent hyporesponsiveness. Nephrology (Carlton). 2007;12:321–30.
Besarab A, Bolton WK, Browne JK, et al. The effects of normal as compared with low hematocrit values in patients with cardiac disease who are receiving hemodialysis and epoetin. N Engl J Med. 1998;339:584–90.
Pfeffer MA, Burdmann EA, Chen CY, et al. A trial of darbepoetin alfa in type 2 diabetes and chronic kidney disease. N Engl J Med. 2009;361:2019–32.
Singh AK, Szczech L, Tang KL, et al. Correction of anemia with epoetin alfa in chronic kidney disease. N Engl J Med. 2006;355:2085–98.
Fishbane S, Schiller B, Locatelli F, EMERALD Study Groups, et al. Peginesatide in patients with anemia undergoing hemodialysis. N Engl J Med. 2013;368:307–19.
Szczech LA, Barnhart HX, Inrig JK, et al. Secondary analysis of the CHOIR trial epoetin-alpha dose and achieved hemoglobin outcomes. Kidney Int. 2008;74:791–8.
Zhang Y, Thamer M, Stefanik K, Kaufman J, Cotter DJ. Epoetin requirements predict mortality in hemodialysis patients. Am J Kidney Dis. 2004;44:866–76.
Kilpatrick RD, Critchlow CW, Fishbane S, et al. Greater epoetin alfa responsiveness is associated with improved survival in hemodialysis patients. Clin J Am Soc Nephrol. 2008;3:1077–83.
Zhao L, Hu C, Cheng J, Zhang P, Jiang H, Chen J. Haemoglobin variability and all-cause mortality in haemodialysis patients: a systematic review and meta-analysis. Nephrology (Carlton). 2019;24:1265–72.
Bae MN, Kim SH, Kim YO, et al. Association of erythropoietin-stimulating agent responsiveness with mortality in hemodialysis and peritoneal dialysis patients. PLoS One. 2015;10: e0143348.
Chen N, Hao C, Liu BC, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med. 2019;381:1011–22.
Chen N, Hao C, Peng X, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381:1001–10.
Akizawa T, Iwasaki M, Otsuka T, Yamaguchi Y, Reusch M. Phase 3 study of roxadustat to treat anemia in non-dialysis-dependant CKD. Kidney Int Rep. 2021;6:1810–28.
Akizawa T, Iwasaki M, Yamaguchi Y, Majikawa Y, Reusch M. Phase 3, randomized, double-blind, active-comparator (darbepoetin alfa) study of oral roxadustat in CKD patients with anemia on hemodialysis in Japan. J Am Soc Nephrol. 2020;31:1628–39.
Shutov E, Sulowicz W, Esposito C, et al. Roxadustat for the treatment of anemia in chronic kidney disease patients not on dialysis: a phase 3, randomized, double-blind, placebo-controlled study (ALPS). Nephrol Dial Transplant. 2021;36:1629–39.
Coyne DW, Roger SD, Shin SK, et al. Roxadustat for CKD-related anemia in non-dialysis patients. Kidney Int Rep. 2021;6:624–35.
Fishbane S, El-Shahawy MA, Pecoits-Filho R, et al. Roxadustat for treating anemia in patients with CKD not on dialysis: results from a randomized phase 3 study. J Am Soc Nephrol. 2021;32:737–55.
Csiky B, Schomig M, Esposito C, et al. Roxadustat for the maintenance treatment of anemia in patients with end-stage kidney disease on stable dialysis: a European phase 3, randomized, open-label, active-controlled study (PYRENEES). Adv Ther. 2021;38:5361–80.
Charytan C, Manllo-Karim R, Martin ER, et al. A randomized trial of roxadustat in anemia of kidney failure: SIERRAS study. Kidney Int Rep. 2021;6:1829–39.
Fishbane S, Pollock CA, El-Shahawy M, et al. Roxadustat versus epoetin alfa for treating anemia in patients with chronic kidney disease on dialysis: results from the randomized phase 3 ROCKIES study. J Am Soc Nephrol. 2022;33:850–66.
Barratt J, Andric B, Tataradze A, et al. Roxadustat for the treatment of anaemia in chronic kidney disease patients not on dialysis: a phase 3, randomized, open-label, active-controlled study (DOLOMITES). Nephrol Dial Transplant. 2021;36:1616–28.
Hicks KA, Tcheng JE, Bozkurt B, et al. 2014 ACC/AHA Key Data Elements and Definitions for Cardiovascular Endpoint Events in Clinical Trials: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Data Standards (Writing Committee to Develop Cardiovascular Endpoints Data Standards). J Am Coll Cardiol. 2015;66:403–69.
Barratt J, Sulowicz W, Schomig M, et al. Efficacy and cardiovascular safety of roxadustat in dialysis-dependent chronic kidney disease: pooled analysis of four phase 3 studies. Adv Ther. 2021;38:5345–60.
US Department of Health and Human Services, Food and Drug Administration. Guidance for Industry Diabetes Mellitus—Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes. December 2008. https://www.fda.gov/media/71297/download. Accessed 22 Apr 2022.
European Medicines Agency Committee for Medicinal Products for Human Use (CHMP). Reflection paper on assessment of cardiovascular safety profile of medicinal products. 25 Feb 2016. https://www.ema.europa.eu/en/documents/scientific-guideline/reflection-paper-assessment-cardiovascular-safety-profile-medicinal-products_en.pdf. Accessed 22 Apr 2022.
Provenzano R, Szczech L, Leong R, et al. Efficacy and cardiovascular safety of roxadustat for treatment of anemia in patients with non-dialysis-dependent CKD: pooled results of three randomized clinical trials. Clin J Am Soc Nephrol. 2021;16:1190–200.
Eckardt KU, Agarwal R, Aswad A, ASCEND-ND Study Group, et al. Safety and efficacy of vadadustat for anemia in patients undergoing dialysis. N Engl J Med. 2021;384:1601–12.
Lilly MP, Lynch JR, Wish JB, et al. Prevalence of arteriovenous fistulas in incident hemodialysis patients: correlation with patient factors that may be associated with maturation failure. Am J Kidney Dis. 2012;59:541–9.
Guo H, Liu J, Collins AJ, Foley RN. Pneumonia in incident dialysis patients–the United States Renal Data System. Nephrol Dial Transplant. 2008;23:680–6.
Singh AK, Carroll K, McMurray JJV, ASCEND-ND Study Group, et al. Daprodustat for the treatment of anemia in patients not undergoing dialysis. N Engl J Med. 2021;385:2313–24.
Acknowledgements
Funding
Funding for this study and its publication, including the journal’s Rapid Service and Open Access fees, was provided by Astellas Pharma, Inc. The sponsor was involved in the trial design and collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors. Roxadustat is being developed by FibroGen, AstraZeneca, and Astellas. The studies were funded by Astellas Pharma, Inc. (DOLOMITES: NCT02021318), FibroGen, Inc. (SIERRAS: NCT02273726 and HIMALAYAS: NCT02052310), and AstraZeneca (ROCKIES: NCT02174731). We thank the participants and clinical investigator teams of the studies.
Medical Writing/Editorial Support
Medical writing/editorial support was provided by Callie Grimes, PhD, Drayton Hammond, PharmD, and Carol Cadmus, ELS from Peloton Advantage, LLC, an OPEN Health company, Parsippany, NJ, USA, and was funded by the study sponsor, Astellas Pharma, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Specific contributions for each author are as follows: Jonathan Barratt contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. Frank Dellanna contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. Jose Portoles contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. Gabriel Choukroun contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. Luca De Nicola contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. James Young contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. Nada Dimković contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript. Michael Reusch contributed to the conception or design of the manuscript, interpretation of the data, drafting and critically revising the manuscript for important intellectual content, and final approval of the manuscript.
Disclosures
Michael Reusch was an employee of Astellas Pharma Europe B.V. until December 2021 and is currently an employee of Guard Therapeutics International AB. James Young is an employee of Astellas Pharma, Inc., Northbrook, IL, USA. Jose Portoles has received lecture and advisory fees from Astellas Pharma, Inc., Otsuka Pharmaceutical Co., and GlaxoSmithKline Pharmaceutical, Ltd. Luca De Nicola has received fees for lectures and scientific consultation from Astellas Pharma, Inc. and AstraZeneca Pharmaceuticals LP. Jonathan Barratt and Gabriel Choukroun have received honoraria from Astellas Pharma, Inc. for advisory board and training events. Frank Dellanna and Nada Dimković have no conflicts of interest to declare.
Compliance with Ethics Guidelines
All study protocols for the previously published studies were approved by institutional review boards and/or ethics committees for the study sites, and were conducted in accordance with the tenets of the Declaration of Helsinki of 1964 and its later amendments, the International Council for Harmonization guidelines for Good Clinical Practice, and any other applicable local health and regulatory requirements. Individual study sites are available at https://clinicaltrials.gov/ct2/show/NCT02021318 (DOLOMITES), https://clinicaltrials.gov/ct2/show/NCT02273726 (SIERRAS), https://clinicaltrials.gov/ct2/show/NCT02052310 (HIMALAYAS), and https://clinicaltrials.gov/ct2/show/NCT02174731 (ROCKIES). As this was a pooled post hoc analysis, approval by an ethics committee was not necessary. Consent to participate: All patients provided written informed consent before enrollment.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Barratt, J., Dellanna, F., Portoles, J. et al. Safety of Roxadustat Versus Erythropoiesis-Stimulating Agents in Patients with Anemia of Non-dialysis-Dependent or Incident-to-Dialysis Chronic Kidney Disease: Pooled Analysis of Four Phase 3 Studies. Adv Ther 40, 1546–1559 (2023). https://doi.org/10.1007/s12325-023-02433-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-023-02433-0