
Abstract
Introduction
Tofacitinib is an oral Janus kinase inhibitor for the treatment of rheumatoid arthritis (RA). To provide additional clinical evidence in regulatory submissions for a modified-release (MR) once-daily (QD) tofacitinib formulation, we compared real-world adherence and effectiveness between patients initiating the MR QD formulation and patients initiating an immediate-release (IR) twice-daily (BID) formulation.
Methods
Two noninterventional cohort studies were conducted. First, adherence and two effectiveness proxies were compared between patients with RA who newly initiated tofacitinib MR 11 mg QD or IR 5 mg BID in the IBM® MarketScan® Commercial and Medicare Supplemental US insurance claims databases (March 2016–October 2018). Second, using data collected in the Corrona US RA Registry (February 2016–August 2019), two Clinical Disease Activity Index (CDAI)-based measures of effectiveness were compared between tofacitinib MR 11 mg QD and IR 5 mg BID, and against noninferiority criteria derived from placebo-controlled clinical trials of the tofacitinib IR formulation. Multiple sensitivity analyses of the registry data were conducted to reassure regulators of consistent results across different assumptions.
Results
In each study, approximately two-thirds of patients initiated the MR formulation. In the claims database study, improved adherence and at least comparable effectiveness were observed with tofacitinib MR vs IR over 12 months, particularly in patients without prior advanced therapy. In the registry study, the noninferiority of tofacitinib MR vs IR was demonstrated for both CDAI outcomes at ~6 months; this finding was robust across multiple sensitivity analyses.
Conclusion
These results demonstrate the value of real-world evidence from complementary data sources in understanding the impact of medication adherence with a QD formulation in clinical practice. These analyses were suitable for regulatory consideration as an important component of evidence for the comparability of tofacitinib MR 11 mg QD vs IR 5 mg BID in patients with RA.
Trial Registration
Claims database study: ClinicalTrials.gov identifier NCT04018001, retrospectively registered July 12, 2019. Corrona US RA Registry study: ClinicalTrials.gov identifier NCT04267380, retrospectively registered February 12, 2020.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Evidence of effectiveness from real-world data (RWD) analyses can complement data from randomized clinical trials and thereby further inform clinical decision-making on once-daily (QD) vs twice-daily (BID) formulations of medications. |
What was learned from the study? |
In US claims databases, tofacitinib modified-release (MR) 11 mg QD improved adherence and demonstrated at least comparable estimated effectiveness (per a claims-based algorithm and duration of therapy proxy measures) vs immediate-release (IR) 5 mg BID in patients with rheumatoid arthritis (RA). |
In the Corrona US RA Registry, the effectiveness of tofacitinib MR 11 mg QD was noninferior to that of tofacitinib IR 5 mg BID, assessed using two Clinical Disease Activity Index (CDAI)-based outcomes, augmenting previous randomized clinical trial results. |
Findings from these analyses of two complementary RWD sources indicated comparable outcomes with the tofacitinib MR 11 mg QD and IR 5 mg BID formulations in patients with RA. |
Digital Features
This article is published with digital features to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12918956.
Introduction
Real-world evidence (RWE) has a well-established role in meeting the pharmacovigilance requirements agreed with regulatory authorities. It is being increasingly leveraged to demonstrate treatment effectiveness to support, and potentially expedite, regulatory decisions, especially in settings of single-arm trials, off-label use, and limited safety data [1, 2]. Globally, regulatory agencies are implementing programs to drive the application of RWE [3,4,5,6,7]. In the US, the 21st Century Cures Act [8] and Prescription Drug User Fee Act Reauthorization (PDUFA VI) commitments [9] require the US Food and Drug Administration to develop draft guidance on incorporating RWE into the approval process (expected late 2021), consistent with the approach for medical devices [9, 10]. In the absence of accepted standards for RWE in the evolving regulatory environment, regulators may be reassured by consistent conclusions from analyses conducted across different data sources and under a variety of assumptions.
Rheumatoid arthritis (RA) is an inflammatory disease characterized by joint inflammation and destruction [11]; given its chronic nature, treatment adherence is often suboptimal [12]. Tofacitinib is an oral Janus kinase (JAK) inhibitor for the treatment of RA. An immediate-release (IR) 5 mg formulation for twice-daily (BID) dosing was approved in the US in November 2012, and in Europe in March 2017 [13]. A modified-release (MR) 11 mg tofacitinib formulation for once-daily (QD) dosing is approved in the US (termed “extended-release”; February 2016) [14] and Europe (termed “prolonged-release”; December 2019) [13]. Patient preference is a key aspect of shared decision-making between patients and physicians, and patient preferences for RA treatments often include dosing frequency [15]. Incorporating patient preferences, for example by granting the option of QD or BID dosing, may lead to improved adherence [16, 17] and thus better treatment outcomes.
Clinically meaningful improvements in RA were demonstrated with tofacitinib MR 11 mg QD in a global phase 3b/4 randomized controlled trial (RCT) [18] and a Japanese phase 3 double-blind RCT [19]. The latter is the only double-blind RCT that assessed noninferiority of tofacitinib MR 11 mg QD vs IR 5 mg BID. Noninferiority was not demonstrated per change from baseline (∆) in Disease Activity Score in 28 joints, C-reactive protein (DAS28-4[CRP]) at week 12 [19]. However, this analysis used a narrower noninferiority margin than would commonly be used, as a result of Japanese Pharmaceutical and Medical Devices Agency requests [19]. Furthermore, the real-world benefit of the MR QD administration is unlikely to be measured in a traditional RCT with mandated compliance and double-dummy trial design, which required that the patients assigned to the MR treatment took a pill BID, thus obscuring the potential convenience and adherence benefits of QD vs BID regimens [16, 20], which could improve effectiveness.
To support regulatory submissions, we aimed to generate RWE to supplement the limited RCT evidence comparing the efficacy of tofacitinib formulations, and to understand whether real-world medication adherence differed between patients receiving the formulations in clinical practice. Using US insurance claims databases, we compared adherence and two effectiveness proxies in patients with RA initiating either formulation. Because clinical outcome assessments are not collected in claims databases, the primary effectiveness comparison was determined using the longitudinal Corrona US RA Registry to conduct propensity score (PS)-matched analyses comparing two Clinical Disease Activity Index (CDAI)-based measures of effectiveness between initiators of tofacitinib 11 mg QD and 5 mg BID. The Corrona US RA Registry collects high-quality clinical data for the purposes of effectiveness research including validated disease activity endpoints [21]. Given the intended use of the analyses for regulatory consideration, multiple sensitivity analyses were conducted to evaluate whether analytic choices or assumptions modified the conclusions of the primary analysis of CDAI-based effectiveness in the Corrona US RA Registry.
Methods
Comparative Analysis of Treatment Adherence and Effectiveness Using US Claims Data (ClinicalTrials.gov Identifier NCT04018001)
Study Design
This was a retrospective, noninterventional cohort analysis of patients ≥ 18 years of age with RA (International Classification of Diseases [ICD]9: 714.xx; ICD10: M05.x or M06.x), newly initiating tofacitinib MR 11 mg QD or IR 5 mg BID between March 1, 2016 (following US approval of the MR formulation on February 24, 2016) and October 31, 2018, in the IBM® MarketScan® Commercial and Medicare Supplemental US insurance claims databases (Table 1). Patients were continuously enrolled for ≥ 12 months before and ≥ 12 months after index claim (first tofacitinib use during study period), with ≥ 1 prior claim for the conventional synthetic disease-modifying antirheumatic drug (csDMARD) methotrexate (MTX).
Patients were excluded if they had: claims during the 12-month pre-index period for other conditions for which advanced therapies (biologic DMARDs [bDMARDs] or JAK inhibitors other than tofacitinib) are used; evidence of index medication use during the 12-month pre-index period; or prescription claims for > 1 advanced therapy or both tofacitinib MR and IR formulations at index.
Compliance with Ethics Guidelines
Institutional Review Board (IRB) review was not required as the study used commercially available deidentified secondary data sources and was considered exempt from the requirements for “human subjects research” in the US. As the study did not involve data subject to privacy laws according to applicable legal requirements, obtaining informed consent from patients was not required. Patient consent for publication was not required. The study was conducted in accordance with legal and regulatory requirements.
Treatment Adherence
The primary focus of this study was treatment adherence, which was evaluated for the 6- and 12-month post-index periods using proportion of days covered (PDC) and medication possession ratio (MPR). PDC was the number of days covered by arrays for each tofacitinib prescription fill, adjusted for a < 15-day overlap in days’ supply (numerator) during the 6- or 12-month post-index period (denominator). MPR was the total tofacitinib days’ supply filled, divided by the number of days between first and last tofacitinib prescription in the 6- or 12-month post-index period. MPR was capped at 1.0. A PDC/MPR ≥ 0.8 represented high adherence.
Treatment Effectiveness Proxies
Patients were considered effectively treated if they met all six criteria in a validated claims-based algorithm [22] during the 12-month post-index period: adherence (PDC ≥ 0.8); no index medication dose escalation; no advanced therapy switch; no new csDMARD; no new/increased oral glucocorticoid dose; and < 2 days’ intra-articular glucocorticoid injections.
Duration of therapy, which has been proposed as a measure of real-world effectiveness [23], was assessed over the 6- and 12-month post-index periods. The 12-month evaluation was the primary focus, in line with the claims-based algorithm and continuous enrollment requirement. Duration of therapy was defined as the number of days between tofacitinib initiation and the first of the following: date of last tofacitinib prescription followed by a 60-day gap after days’ supply expiration without evidence of another advanced therapy; day before receipt of another advanced therapy; or end of follow-up period. Persistence (i.e., no gap in tofacitinib refills or advanced therapy switch) was also evaluated over the 6- and 12-month post-index periods.
Statistical Analyses
Data are presented overall and for patients with no prior advanced therapies (bDMARD, tofacitinib, or baricitinib) any time pre-index (censored at January 1, 2013 based on licensed data availability). Simple t tests (continuous variables) and chi-squared tests (categorical variables) were used to examine differences between tofacitinib MR 11 mg QD and IR 5 mg BID; p values < 0.05 were considered significant.
To control for possible confounders, adjusted analyses used Cox (duration of therapy) and logistic regression (adherence, algorithm-based effectiveness and its criteria, and persistence) models (see the supplementary material) [24]. The 95% confidence intervals (CIs) and p values (significant at < 0.05) were calculated for adjusted odds ratios (adjORs) and hazard ratios (adjHRs).
Noninferiority Analysis Using US Registry Data (ClinicalTrials.gov Identifier NCT04267380)
Study Design
This was a noninterventional cohort analysis of patients ≥ 18 years of age initiating tofacitinib MR 11 mg QD or IR 5 mg BID (“initiators”) between February 1, 2016 and September 30, 2018 in the Corrona US RA Registry [21].
The initiation formulation was defined as the formulation on which the patient first initiated tofacitinib treatment; patients were not excluded from the analysis for switching formulations during the analysis period.
For the primary analysis, the data cut was September 30, 2018, and initiators were required to have a follow-up visit at 6 (± 3) months post-tofacitinib initiation, and valid CDAI scores at initiation and each follow-up visit (Table 1).
Post hoc sensitivity analyses (Table 1) were conducted to determine whether results of the primary analysis were robust to different analytic approaches, including different follow-up eligibility criteria (± 3 months, ± 6 months, and no requirement), and use of a later data cut (August 31, 2019).
A separate exploratory analysis (data cut September 30, 2018) considered changes in disease activity among patients who initiated tofacitinib IR 5 mg BID during or after November 2012 and switched to MR 11 mg QD (“switchers”) between February 2016 (approval of new formulation) and September 2018.
Compliance with Ethics Guidelines
All participating investigators in the Corrona US RA Registry were required to obtain IRB approval. All patients were required to provide written informed consent prior to participating in the study. Patient consent for publication was not required. The Corrona registry study was conducted in accordance with the Helsinki Declaration.
Outcomes
Two endpoints were considered: achievement of minimum clinically important difference (MCID) in CDAI from initiation to the 6-month visit (primary, dichotomous endpoint; see the supplementary material) and ΔCDAI (secondary, continuous endpoint). MCID was dependent on baseline (at initiation) disease activity: improvement of ≥ 2 in CDAI if low CDAI (≤ 10) [25]; improvement of ≥ 6 in CDAI if moderate CDAI (> 10 to ≤ 22) [26]; and improvement of ≥ 11 in CDAI if high CDAI (> 22) [26].
Noninferiority margins of tofacitinib MR 11 mg QD vs IR 5 mg BID were determined for CDAI outcomes, based on the lower value of the CI boundary for the difference between tofacitinib IR and placebo observed in the tofacitinib clinical program (see the supplementary material). Margins were evaluated for unmatched and PS-matched populations, to evaluate the sensitivity of noninferiority conclusions to the analytic approach (see the supplementary material).
The noninferiority margin for the dichotomous endpoint was 0.59 (see the supplementary material); noninferiority was declared if the 95% CI lower bound of the odds ratio (OR; tofacitinib MR 11 mg QD/tofacitinib IR 5 mg BID) was > 0.59. For the continuous endpoint, the noninferiority margin was 4.04 (see the supplementary material; noninferiority was declared if the 95% CI upper bound for treatment difference [tofacitinib MR 11 mg QD − tofacitinib IR 5 mg BID] was < 4.04).
Statistical Analyses
Patient demographics and baseline disease characteristics were summarized descriptively, with standardized differences calculated. An imbalance in baseline characteristics between groups, defined as the absolute value of the standardized difference > 0.2, was identified for the following characteristics in the primary analysis, which were included as covariates in a PS model to derive PS-matched populations: prior tumor necrosis factor inhibitor (TNFi) use (0, 1, 2+); prior non-TNFi use (0, 1, 2+); current concomitant therapy (monotherapy, with MTX, with other csDMARD, with MTX plus other csDMARD); and line of therapy (1st, 2nd, 3rd, 4th). Imbalanced baseline characteristics not included in the model (and the rationale) were: prior bDMARD use (as it was a linear combination of prior TNFi and non-TNFi use); prednisone dose (only measured for a subset of patients using prednisone); and DAS28-4 (CRP) (not routinely collected, and missing in a large proportion of the population). Patients were matched 1:1 based on the PS model using a matching algorithm and a caliper of 0.01, to ensure the MR 11 mg QD and IR 5 mg BID groups were generally balanced.
The primary, dichotomous endpoint was compared between PS-matched groups using a chi-squared test of the difference in proportions of patients achieving MCID in CDAI between the MR 11 mg QD and IR 5 mg BID initiators. The primary endpoint was also compared using adjusted multivariable logistic regression models (ORs and 95% CIs calculated). This model was adjusted for age, gender, duration of RA, baseline CDAI (selected a priori as key potential confounders), and baseline swollen joint count (included due to residual imbalance between groups post-matching). For the dichotomous endpoint, patients who discontinued tofacitinib or switched to another therapy were considered as nonresponders. This was considered a conservative approach, as reasons for discontinuation may be due to adverse events (AEs) or otherwise unrelated to patient responses to treatment. Patients who switched to another therapy during the 6 months of follow-up were considered for the continuous outcomes at the time of discontinuation. Based on the sample size of the PS-matched population (N = 298; N = 149 per group) in the primary analysis, a difference of ≥ 16 percentage points between groups in the proportion of patients achieving an MCID improvement in CDAI was expected to be detected, with 80% power for the primary outcome.
The secondary continuous endpoint was compared between PS-matched groups using a linear regression model adjusted for the baseline value of the outcome, age, gender, duration of RA, baseline CDAI, and baseline swollen joint count.
The PS-matching procedure was reassessed in sensitivity analyses 1 and 2, so that imbalanced characteristics (absolute value of the standardized difference > 0.2) in each sensitivity analysis were included as covariates to derive PS-matched populations. The primary dichotomous endpoint and secondary continuous endpoint were compared between PS-matched populations, as per the primary analysis. The multivariable logistic regression model, used to compare the primary endpoint, and the linear regression model, used to compare the secondary endpoint, were adjusted for age, gender, duration of RA, and baseline CDAI.
Additionally, the primary dichotomous endpoint and the secondary continuous endpoint were compared in unmatched populations in the primary analysis and in sensitivity analyses 1–3.
Exploratory Analysis
In the exploratory analysis of patients who switched from tofacitinib IR 5 mg BID to MR 11 mg QD, effectiveness of tofacitinib MR 11 mg QD was assessed by comparing endpoint measures at the time of switch vs 6 (± 3) months post-switch. Mean change in CDAI from the time of the switch to 6 months post-switch was assessed as a continuous outcome, and the proportions of patients at 6 months post-switch with CDAI worsening, improving, or remaining steady were assessed as binary outcomes.
Results
Comparative Analysis of Treatment Adherence and Effectiveness Using US Claims Data
Patients
In the US claims analysis, 1057 tofacitinib initiators met inclusion criteria; 678 (64.1%) and 379 (35.9%) initiated MR 11 mg QD and IR 5 mg BID, respectively (Fig. 1). Demographics, clinical characteristics, and total healthcare expenditures during the 12-month pre-index period were generally similar between groups (Table 2). A greater proportion of tofacitinib MR 11 mg QD vs IR 5 mg BID initiators received ≥ 1 advanced therapy during the 12-month pre-index period.

US claims databases: cohort selection. BID twice daily, IR immediate-release, MR modified-release, MTX methotrexate, N number of patients, QD once daily, RA rheumatoid arthritis
Overall, 235 (22.2%) patients received no prior advanced therapies any time pre-index; 142 (60.4%) and 93 (39.6%) initiated tofacitinib MR 11 mg QD and IR 5 mg BID, respectively (p = 0.18) (Table 2). Approximately 89% of patients received a csDMARD during the 12-month pre-index period. Similar proportions of tofacitinib MR 11 mg QD and IR 5 mg BID initiators received a csDMARD (i.e., combination therapy) within 90 days post-index (54.6% and 56.5%, respectively; p = 0.55).
Treatment Adherence
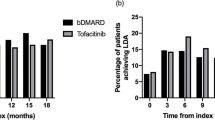
Through 12 months, 48.2% and 37.7% of tofacitinib MR 11 mg QD and IR 5 mg BID initiators, respectively, were highly adherent per the PDC ≥ 0.8 definition (p = 0.001; Fig. 2a), with an adjOR of 1.52 (p = 0.003; Fig. 2b). Per the MPR ≥ 0.8 definition, 80.1% and 69.9% of tofacitinib MR 11 mg QD and IR 5 mg BID initiators, respectively, were highly adherent (p = 0.0002; Fig. 2c), with an adjOR of 1.79 (p = 0.0002; Fig. 2d). Differences between tofacitinib formulations were greater in patients without prior advanced therapy vs overall (Fig. 2).

US claims databases: unadjusted proportion of patients and adjORs of patients who were highly adherent with tofacitinib treatment through 6 and 12 months: a, b PDCa ≥ 0.80; c, d MPRb ≥ 0.80. Data from the IBM® MarketScan® Commercial and Medicare Supplemental US insurance claims databases. adjOR adjusted odds ratio, bDMARD biologic disease-modifying antirheumatic drug, BID twice daily, CI confidence interval, IR immediate-release, MPR medication possession ratio, MR modified-release, N number of patients, OR odds ratio, PDC proportion of days covered, QD once daily, RA rheumatoid arthritis. *p < 0.05; **p < 0.01; ***p < 0.001 tofacitinib MR 11 mg QD vs IR 5 mg BID. aPDC was defined as number of days covered by arrays for each tofacitinib prescription fill, adjusted for a < 15-day overlap in days’ supply (numerator), during the 6- or 12-month post-index period (denominator). bMPR was defined as total tofacitinib days’ supply filled, divided by the number of days between first and last tofacitinib prescription in the 6- or 12-month post-index period. MPR was capped at 1.0. cNo bDMARDs, tofacitinib, or baricitinib. dAdjusted analyses were performed using logistic regression models, which included index medication (tofacitinib MR 11 mg QD or IR 5 mg BID), type of therapy (combination or monotherapy), insurance type, region, age, sex, index year, use of nonsteroidal anti-inflammatory drugs in the 12-month pre-index period (yes/no), and number of prior advanced therapies any time pre-index as covariates. Initially, the models included an interaction term of index medication by type of therapy; if the p value was > 0.20, it was removed and the simpler model was fitted instead
Through 6 months, adjORs for PDC ≥ 0.8 and MPR ≥ 0.8 were 1.11 (p = 0.45) and 1.41 (p = 0.04) overall; and 1.83 (p = 0.04) and 1.81 (p = 0.10) in those without prior advanced therapy (Fig. 2b, d).
Treatment Effectiveness Proxy: Algorithm-Based
In unadjusted analyses (Fig. 3a, c), significantly higher proportions of tofacitinib MR 11 mg QD vs IR 5 mg BID initiators were effectively treated through 12 months per the claims-based algorithm (p = 0.01 overall; p = 0.007 without prior advanced therapy). The adjORs were 1.41 (p = 0.02) and 1.99 (p = 0.04) overall and in patients without prior advanced therapy, respectively (Fig. 3b, d). Differences were driven by significant differences in adherence, which were more pronounced in those without prior advanced therapy (Fig. 3).

US claims databases: unadjusted proportion of patients and adjORs of patients meeting effectiveness (proxy) criteriaa: a, b overall; c, d no prior advanced therapy. Data from the IBM® MarketScan® Commercial and Medicare Supplemental insurance US claims databases. adjOR adjusted odds ratio, bDMARD biologic disease-modifying antirheumatic drug, BID twice daily, CI confidence interval, csDMARD conventional synthetic disease-modifying antirheumatic drug, IR immediate-release, MR modified-release, N number of patients, NE not estimable, OR odds ratio, PDC proportion of days covered, QD once daily. *p < 0.05; **p < 0.01; ***p < 0.001 tofacitinib MR 11 mg QD vs IR 5 mg BID. aAn algorithm-based proxy measure of effectiveness during the 12-month post-index period was based on the six criteria shown in a, c; patients meeting all criteria were considered effectively treated [22]. bORs were NE for “No dose escalation”, owing to rates at or close to 100%. c≤ 30 days of oral glucocorticoid between months 3 and 12 post-index in patients with no glucocorticoid prescriptions for 6 months pre-index. dNo increase in oral glucocorticoid dose ≥ 20% during months 6–12 post-index (for those with 6 months of pre-index glucocorticoid use). eAdjusted analyses were performed using logistic models, which included index medication (tofacitinib MR 11 mg QD or IR 5 mg BID), type of therapy (combination or monotherapy), insurance type, region, age, sex, index year, use of nonsteroidal anti-inflammatory drugs in the 12-month pre-index period (yes/no), and number of prior advanced therapies any time pre-index as covariates. Initially, the models included an interaction term of index medication by type of therapy; if the p value was > 0.20, it was removed and the simpler model was fitted instead. fNo bDMARDs, tofacitinib, or baricitinib
Treatment Effectiveness Proxy: Duration of Therapy
Over 12 months, mean duration of therapy was 243.4 and 235.7 days for tofacitinib MR 11 mg QD and IR 5 mg BID, respectively (p = 0.36; Fig. 4a). The adjHRs for tofacitinib discontinuation were 0.90 (p = 0.26) overall and 0.64 (p = 0.03) in those without prior advanced therapy (Fig. 4b). Persistence was 51.6% and 45.7% with tofacitinib MR 11 mg QD and IR 5 mg BID, respectively (p = 0.06; Fig. 4c), with adjORs of 1.23 (p = 0.13) overall and 2.04 (p = 0.02) in those without prior advanced therapy (Fig. 4d).

US claims databases: a mean duration of tofacitinib treatmenta over 6 and 12 months (unadjusted); b risk for tofacitinib discontinuation over 6 and 12 months (adjustedb); c proportion of patients who were persistentc with tofacitinib treatment through 6 and 12 months (unadjusted); d ORs of persistent tofacitinib treatment through 6 and 12 months (adjustedb). Data from the IBM® MarketScan® Commercial and Medicare Supplemental US insurance claims databases. bDMARD biologic disease-modifying antirheumatic drug, BID twice daily, CI confidence interval, HR hazard ratio, IR immediate-release, MR modified-release, N number of patients, OR odds ratio, QD once daily, SD standard deviation. *p < 0.05; **p < 0.01 tofacitinib MR 11 mg QD vs IR 5 mg BID. aDuration of therapy was defined as the number of days between tofacitinib initiation and the first of the following: date of last tofacitinib prescription followed by a 60-day gap after days’ supply expiration without evidence of another advanced therapy; day before receipt of another advanced therapy; or end of follow-up period. bAdjusted analyses were performed using Cox (duration of therapy) and logistic regression (persistence) models, which included index medication (tofacitinib MR 11 mg QD or IR 5 mg BID), type of therapy (combination or monotherapy), insurance type, region, age, sex, index year, use of nonsteroidal anti-inflammatory drugs in the 12-month pre-index period (yes/no), and number of prior advanced therapies any time pre-index as covariates. Initially, the models included an interaction term of index medication by type of therapy; if the p value was > 0.20, it was removed and the simpler model was fitted instead. cDefined as the continuation of index medication without a ≥ 60-day gap after prior prescription days’ supply had run out or advanced therapy (bDMARD or baricitinib) switch, and associated duration of therapy. dNo bDMARDs, tofacitinib, or baricitinib
Similarly, differences over 6 months were not significant in unadjusted or adjusted analyses overall, nor in adjusted analyses for patients without prior advanced therapy.
Comparative Effectiveness Analysis Using US Registry Data
Patients
Of the 2100 patients who initiated tofacitinib within the Corrona US RA Registry up to September 2018, 1008 did so during or after February 2016 (MR 11 mg QD, N = 611; IR 5 mg BID, N = 258; other or missing dose, N = 139). Of these, 450 patients (MR 11 mg QD, N = 297; IR 5 mg BID, N = 153) had a 6-month follow-up visit and available CDAI data at both initiation and follow-up, and were therefore included in the primary analysis (Fig. 5).

US registry: patients included in the primary analysis and post hoc sensitivity analyses. Percentages are with respect to all tofacitinib initiators (i.e., patients included in sensitivity analysis 3). BID twice daily, CDAI Clinical Disease Activity Index, IR immediate-release, MR modified-release, N number of patients, PS propensity score, QD once daily, RA rheumatoid arthritis
The sample sizes for the sensitivity analyses are also shown in Fig. 5. Patient characteristics were generally similar between tofacitinib MR 11 mg QD and IR 5 mg BID initiators, in PS-matched populations in the primary analysis and in sensitivity analyses 1 and 2 (Table 3).
Outcomes
In the PS-matched primary analysis, the proportion of patients with CDAI improvements ≥ MCID (primary outcome) was similar between MR 11 mg QD and IR 5 mg BID initiators without adjustment (25.5 vs 22.1%, respectively; p = 0.73) or with adjustment for covariates (OR 1.42; 95% CI 0.79, 2.53; adjusted p = 0.24). The 95% CI lower bounds for the OR were > 0.59 (0.79 [PS-matched population; Table 4] and 0.73 [unmatched population; see Table S1 in the supplementary material]), demonstrating noninferiority of tofacitinib MR 11 mg QD vs IR 5 mg BID.
Noninferiority of the MR vs IR formulation was also demonstrated for ∆CDAI, as 95% CI upper bounds were < 4.04 (1.43 [PS-matched population; Table 4] and 1.05 [unmatched population; see Table S1 in the supplementary material]).
Across the sensitivity analyses using the later data cut and different follow-up eligibility criteria, results were consistent with those in the primary analysis, with noninferiority criteria met when comparing tofacitinib MR 11 mg QD vs IR 5 mg BID for both dichotomous and continuous CDAI endpoints in PS-matched populations (Table 4) and unmatched populations (see Table S1 in the supplementary material).
Exploratory Analysis
Of 152 patients in the primary analysis who switched from tofacitinib IR 5 mg BID to MR 11 mg QD during their course of therapy between February 2016 and September 2018, 99 (65.1%) had a 6-month follow-up visit post-switch where effectiveness data (CDAI) could be obtained, and were included in the exploratory analysis. None of these patients had switched back to tofacitinib IR 5 mg BID before the 6-month follow-up visit. At 6 months post-switch, most patients had steady (64.6%) or improving (22.1%) CDAI scores (see Table S2 in the supplementary material).
Only 10 of the 297 tofacitinib MR 11 mg QD initiators included in the primary analysis switched to IR 5 mg BID by their 6-month visit, too few for separate analyses.
Discussion
RCT data comparing the tofacitinib MR and IR formulations are limited to a single Japanese RCT with a double-dummy trial design and, as such, patients randomized to receive tofacitinib MR 11 mg took two pills each day (one tofacitinib pill and one placebo pill); therefore, these data are limited with respect to understanding the potential convenience benefit of QD dosing [19]. As such, RWE of effectiveness may complement RCT findings in the totality of evidence concerning a potential improved adherence benefit related to administration burden and patient preference for a certain dosing regimen [15, 16, 20]. In real-world usage, where adherence benefit is not obscured by blinding/dosing regimen forced upon patients by randomization or close monitoring of patients in a highly selected RCT population, we demonstrated an adherence benefit, as well as clinical noninferiority in a high-quality data source across multiple sensitivity analyses, for tofacitinib MR 11 mg QD relative to IR 5 mg BID.
In the US insurance claims databases, the primary focus was on 12-month outcomes following initiation of tofacitinib MR 11 mg QD vs IR 5 mg BID. Adherence based on PDC/MPR ≥ 0.8 and effectiveness based on a claims-based algorithm [22] were improved with the MR vs IR formulation. Overall, duration of therapy, risk for discontinuation, and odds of persistence were not significantly different between formulations, although directionality favored MR 11 mg QD. The observed uptake of and increased adherence to the MR formulation may reflect ease of administration alongside physician/patient preference for QD formulations, especially in patients transitioning to their first advanced therapy. The improved adherence and persistence were most pronounced among patients without prior advanced therapy. In all outcomes at 12 months, differences favoring tofacitinib MR 11 mg QD were found in patients without prior advanced therapy. This is particularly relevant as JAK inhibitors are recommended by the European League Against Rheumatism following csDMARD failure [30], and prior research has indicated lower adherence and persistence with bDMARDs in patients with RA without prior bDMARD experience [31].
Given that effectiveness endpoints are not directly reported in claims data, the primary evidence of effectiveness was derived from the Corrona US RA Registry. Noninferiority criteria were established using margins derived from the tofacitinib clinical program. In the primary analysis, noninferiority criteria were met for dichotomous and continuous CDAI endpoints with tofacitinib MR 11 mg QD vs IR 5 mg BID. These results at 6-month follow-up in the registry are complemented by the persistence and adherence findings over 6 months in the US insurance claims databases; the effectiveness algorithm was not evaluated at 6 months, consistent with its validation and prior publications [22, 32]. However, it is also interesting to note that proportions of patients meeting criteria for effectiveness, and related ORs, were similar in the analysis of the six-item claims-based algorithm and in the registry analysis of MCID improvement in CDAI.
Furthermore, the conclusions of the Corrona noninferiority analyses were consistent across multiple sensitivity analyses. Using an updated data cut (August 31, 2019) improved the balance between the proportions of patients initiating the tofacitinib formulations with eligible 6-month data. In the primary analysis (data cut September 30, 2018), the proportions of eligible tofacitinib initiators were 48.6% (MR 11 mg QD) and 59.3% (IR 5 mg BID). With the later data cut used for the sensitivity analyses, proportions were 65.0% (MR 11 mg QD) and 65.9% (IR 5 mg BID). In an observational setting such as the Corrona US RA Registry, follow-up differences between groups could potentially be attributable to differential efficacy. However, these sensitivity analyses provide further assurance that findings relating to the noninferiority of tofacitinib MR 11 mg QD are not owing to bias resulting from nonrandom assignment or differential loss to follow-up.
Compared with an RCT, the non-interventional aspect of the Corrona analyses is potentially sensitive to important biases [33], overcome by the high quality of the Corrona US RA Registry, study design, and analytic methods. Confounding bias was addressed by PS-matching. Prevalent user bias was avoided by inclusion of only new users of tofacitinib. Incomplete follow-up bias was avoided by requiring a 6-month follow-up period for the primary analysis and using a 6-month change in CDAI, which is also reasonable from a real-world perspective in terms of timepoints for visits and being per usual clinical care guidelines. Channeling bias, whereby physicians direct a certain therapy to a certain patient type, was avoided in part by PS-matching. However, such bias was primarily avoided due to both treatment arms (11 mg QD and 5 mg BID) being prescribed the same medicine with equivalent labeling in the US market, allowing prescribers and patients to choose the preferred formulation. Reporting bias was avoided by employing the same data collection instrument and protocol for both contemporaneous cohorts. Attrition bias was demonstrated not to have occurred, as the proportion of patients with 6-month follow-up was 65% of patients prescribed 11 mg QD and 66% of patients prescribed 5 mg BID in sensitivity analysis 1.
On average, patients in the Corrona US RA Registry were older than patients in the claims databases. Furthermore, both of these populations were generally older than patients in the tofacitinib clinical program for RA [34], from which the noninferiority margins were derived. It should be noted that age may affect treatment outcomes [35], and this should be considered when applying noninferiority margins derived from clinical data to real-world data (RWD). In the general population [36] and in patients with RA [35, 37], the presence of comorbidities increases with increasing age. In clinical trials of advanced therapies in patients with RA, older patients (≥ 65 years of age) have been shown to have a numerically higher risk of serious AEs [35, 38] and discontinuations due to AEs [35] compared with younger patients ( < 65 years of age), thus adherence and persistence may be affected by patient age. However, older and younger patients experienced similar efficacy in terms of American College of Rheumatology 20/50 response rates and proportions achieving a Health Assessment Questionnaire-Disability Index improvement from baseline ≥ 0.22 (CDAI was not assessed in the analysis) [35].
For the analysis of US insurance claims databases, common limitations of claims data must be noted. Claims data collection is linked to payment processing; not all services provided may be billed and captured, and a prescription claim does not necessarily indicate that the patient took the medication. Furthermore, the diagnosis of RA and other autoimmune conditions may be potentially miscoded. Additionally, patients treated with an advanced therapy in the pre-index period were considered as prior users; however, patients with no bDMARD or JAK inhibitor treatment in this period may have been off therapy. Moreover, effectiveness was measured using a validated algorithm [22], which was restricted to commercially available bDMARDs at the time of development. The algorithm includes a “no dose escalation” criterion; results may be subject to bias, given that in the US there is only one approved tofacitinib dose for RA (i.e., MR 11 mg QD or IR 5 mg BID). Additionally, findings were generated in a commercially insured population and may not be representative of patients with other insurance types or no insurance. Finally, while adjusted analyses were conducted, differences in unobserved confounders may result in bias.
In contrast to claims analyses, the Corrona US RA Registry data are intended for effectiveness research due to the direct capture of disease activity effectiveness endpoints and treatment regimens. Unlike many RWD sources, Corrona data are collected explicitly for researching comparative safety and effectiveness, an attribute shared with RCT data and a key consideration when evaluating the validity of RWE [21]. The Corrona US RA Registry is the largest real-world, prospective-cohort study of patients with RA globally [39], and its patients have been shown to have good representativeness of the Medicare population [40]. However, as with RCTs, the patient sample is not necessarily generalizable to all adults with RA in the US. Particularly, these were patients with RA who attended clinical rheumatologist visits.
Future research could provide further insights into similarities and differences in outcomes with the two formulations of tofacitinib. We did not specifically evaluate the potential impact of longer-term (12-month) improved adherence on clinical effectiveness via clinical outcomes assessment tools or radiographic progressions. This is an area for future research and ideally requires linkages of clinical (i.e., registry) and claims data sources, as previously done with Corrona data and Medicare claims data by Curtis et al. [41]. Furthermore, understanding adherence beyond 12 months could be an area for future evaluation. Additionally, understanding the incidence and impact of AEs, while not the primary purpose of the studies reported herein, may be of interest in subsequent studies.
Conclusion
These results demonstrate the value of using RWE to contextualize RCT outcomes, and provide further evidence for the comparability of tofacitinib MR 11 mg QD vs IR 5 mg BID. Optimizing RWE generation in parallel with RCTs offers the potential to better understand drug benefit–risk profiles in settings more representative of clinical practice than a traditional RCT. As such, RWE can help inform regulatory decisions. Future research providing evidence for the real-world effectiveness of tofacitinib MR 11 mg QD vs other targeted therapies is warranted.
Change history
12 November 2020
In the original article, “Digital Features” link was not included. The link has been included to the Figshare page under "Additional Information".
References
Silverman B. A baker’s dozen of US FDA efficacy approvals using real world evidence. 2018. https://pink.pharmaintelligence.informa.com/PS123648/A-Bakers-Dozen-Of-US-FDA-Efficacy-Approvals-Using-Real-World-Evidence. Accessed 27 Feb 2020.
Dreyer NA. Advancing a framework for regulatory use of real-world evidence: when real is reliable. Ther Innov Regul Sci. 2018;52:362–8.
European Medicines Agency. Adaptive pathways. 2016. https://www.ema.europa.eu/en/human-regulatory/research-development/adaptive-pathways. Accessed 5 Feb 2020.
European Medicines Agency. HMA–EMA Joint Big Data Taskforce—summary report. 2019. https://www.ema.europa.eu/en/documents/minutes/hma/ema-joint-task-force-big-data-summary-report_en.pdf. Accessed 5 Feb 2020.
Health Canada. Optimizing the use of real world evidence to inform regulatory decision-making. 2019. https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/announcements/optimizing-real-world-evidence-regulatory-decisions.html. Accessed 5 Feb 2020.
Center for Drug Evaluation. Key considerations in using real-world evidence to support drug development (draft for public review). 2019. http://www.accestra.com/wp-content/uploads/2019/06/Key-Considerations-in-Using-Real-World-Evidence-to-Support-Drug-DevelopmentDraft-for-Public-Review.docx. Accessed 5 Feb 2020.
Uyama Y. Proceedings: DIA 2018 global annual meeting. Utilizing real world data: a PMDA perspective. 2018. https://globalforum.diaglobal.org/issue/august-2018/utilizing-real-world-data-a-pmda-perspective/. Accessed 27 Mar 2020.
US Food and Drug Administration. 21st Century Cures Act. 2016. https://www.fda.gov/regulatory-information/selected-amendments-fdc-act/21st-century-cures-act. Accessed 17 Mar 2020.
US Food and Drug Administration. Prescription Drug User Fee Act Reauthorization (PDUFA VI), Medical Device User Fee Act Reauthorization (MDUFA IV), Generic Drug User Fee Act Reauthorization (GDUFA II), and Biosimilar User Fee Act Reauthorization (BsUFA II). 2017. https://www.fda.gov/news-events/congressional-testimony/prescription-drug-user-fee-act-reauthorization-pdufa-vi-medical-device-user-fee-act-reauthorization. Accessed 17 Mar 2020.
US Food and Drug Administration. Use of real-world evidence to support regulatory decision-making for medical devices. 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/use-real-world-evidence-support-regulatory-decision-making-medical-devices. Accessed 17 Mar 2020.
Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388:2023–38.
Marengo MF, Suarez-Almazor ME. Improving treatment adherence in patients with rheumatoid arthritis: what are the options? Int J Clin Rheumtol. 2015;10:345–56.
European Medicines Agency. Tofacitinib (Xeljanz): summary of product characteristics. 2019. https://www.ema.europa.eu/en/documents/referral/xeljanz-article-20-procedure-annex-iii_en.pdf. Accessed 28 Feb 2020.
US Food and Drug Administration. XELJANZ® (tofacitinib): highlights of prescribing information. 2019. https://labeling.pfizer.com/ShowLabeling.aspx?id=959. Accessed 8 Apr 2020.
Taylor PC, Betteridge N, Brown TM, et al. Treatment mode preferences in rheumatoid arthritis: moving toward shared decision-making. Patient Prefer Adher. 2020;14:119–31.
Saini SD, Schoenfeld P, Kaulback K, Dubinsky MC. Effect of medication dosing frequency on adherence in chronic diseases. Am J Manag Care. 2009;15:e22–e33.
van Heuckelum M, Mathijssen EG, Vervloet M, et al. Preferences of patients with rheumatoid arthritis regarding disease-modifying antirheumatic drugs: a discrete choice experiment. Patient Prefer Adher. 2019;13:1199–211.
Cohen SB, Pope J, Haraoui B, et al. Methotrexate withdrawal in patients with rheumatoid arthritis who achieve low disease activity with tofacitinib modified-release 11 mg once daily plus methotrexate (ORAL Shift): a randomised, phase 3b/4, non-inferiority trial. Lancet Rheumatol. 2019;1:E23–E34.
Tanaka Y, Sugiyama N, Toyoizumi S, et al. Modified- versus immediate-release tofacitinib in Japanese rheumatoid arthritis patients: a randomized, phase III, non-inferiority study. Rheumatology (Oxford). 2019;58:70–9.
Coleman CI, Limone B, Sobieraj DM, et al. Dosing frequency and medication adherence in chronic disease. J Manag Care Pharm. 2012;18:527–39.
Kremer JM. The Corrona US registry of rheumatic and autoimmune diseases. Clin Exp Rheumatol. 2016;34:S96–9.
Curtis JR, Baddley JW, Yang S, et al. Derivation and preliminary validation of an administrative claims-based algorithm for the effectiveness of medications for rheumatoid arthritis. Arthritis Res Ther. 2011;13:R155.
Wolfe F. The epidemiology of drug treatment failure in rheumatoid arthritis. Baillieres Clin Rheumatol. 1995;9:619–32.
Harrell FE Jr. Regression modeling strategies: with applications to linear models, logistic regression, and survival analysis. Springer Series in Statistics. New York: Springer Science + Business Media; 2001.
Curtis JR, Yang S, Chen L, et al. Determining the minimally important difference in the clinical disease activity index for improvement and worsening in early rheumatoid arthritis patients. Arthritis Care Res (Hoboken). 2015;67:1345–53.
Kavanaugh A, Keystone E, Greenberg JD, et al. Benefit of biologics initiation in moderate versus severe rheumatoid arthritis: evidence from a United States registry. Rheumatology (Oxford). 2017;56:1095–101.
Deyo RA, Cherkin DC, Ciol MA. Adapting a clinical comorbidity index for use with ICD-9-CM administrative databases. J Clin Epidemiol. 1992;45:613–9.
Quan H, Sundararajan V, Halfon P, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Med Care. 2005;43:1130–9.
Ting G, Schneeweiss S, Scranton R, et al. Development of a health care utilisation data-based index for rheumatoid arthritis severity: a preliminary study. Arthritis Res Ther. 2008;10:R95.
Smolen JS, Landewé RBM, Bijlsma JWJ, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 update. Ann Rheum Dis. 2020;79:685–99.
Murage MJ, Tongbram V, Feldman SR, et al. Medication adherence and persistence in patients with rheumatoid arthritis, psoriasis, and psoriatic arthritis: a systematic literature review. Patient Prefer Adher. 2018;12:1483–503.
Bonafede MMK, Curtis JR, McMorrow D, Mahajan P, Chen C-I. Treatment effectiveness and treatment patterns among rheumatoid arthritis patients after switching from a tumor necrosis factor inhibitor to another medication. Clinicoecon Outcomes Res. 2016;8:707–15.
Sharma M, Nazareth I, Petersen I. Observational studies of treatment effectiveness: worthwhile or worthless? Clin Epidemiol. 2019;11:35–42.
Bird P, Bensen W, El-Zorkany B, et al. Tofacitinib 5 mg twice daily in patients with rheumatoid arthritis and inadequate response to disease-modifying antirheumatic drugs: a comprehensive review of phase 3 efficacy and safety. J Clin Rheumatol. 2019;25:115–26.
Curtis JR, Schulze-Koops H, Takiya L, et al. Efficacy and safety of tofacitinib in older and younger patients with rheumatoid arthritis. Clin Exp Rheumatol. 2017;35:390–400.
Fried LP, Ferrucci L, Darer J, Williamson JD, Anderson G. Untangling the concepts of disability, frailty, and comorbidity: implications for improved targeting and care. J Gerontol A Biol Sci Med Sci. 2004;59:255–63.
Widdifield J, Bernatsky S, Paterson JM, et al. Serious infections in a population-based cohort of 86,039 seniors with rheumatoid arthritis. Arthritis Care Res (Hoboken). 2013;65:353–61.
Bathon JM, Fleischmann RM, van der Heijde D, et al. Safety and efficacy of etanercept treatment in elderly subjects with rheumatoid arthritis. J Rheumatol. 2006;33:234–43.
CORRONA. Corrona® RA Registry. 2019. https://www.corrona.org/registry/rheumatoid-arthritis. Accessed 12 Mar 2020.
Curtis JR, Chen L, Bharat A, et al. Linkage of a de-identified United States rheumatoid arthritis registry with administrative data to facilitate comparative effectiveness research. Arthritis Care Res (Hoboken). 2014;66:1790–8.
Curtis JR, Chen L, Greenberg JD, et al. The clinical status and economic savings associated with remission among patients with rheumatoid arthritis: leveraging linked registry and claims data for synergistic insights. Pharmacoepidemiol Drug Saf. 2017;26:310–9.
Acknowledgments
Funding
These studies were sponsored and funded by Pfizer Inc. The journal’s Rapid Service and Open Access Fees for this article were funded by Pfizer Inc. The Corrona US RA Registry is sponsored by Corrona, LLC. Access to Corrona study data was limited to Corrona, LLC, and Corrona, LLC statisticians completed all of the analysis.
Medical Writing Assistance
Medical writing support, under the guidance of the authors, was provided by Karen Irving, PhD, and Sarah Piggott, MChem, CMC Connect, McCann Health Medical Communications, and Karleen Nicholson, PhD, on behalf of CMC Connect, and was funded by Pfizer Inc, New York, NY, USA in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163:461-4).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
JDG, JH, AM, TWS, DG, TL, JW, KJD, HJL, TB, CC, and JLR were involved in the study conception and design. JH, AM, DG, and TL were involved in acquisition of data. All authors contributed to analysis and interpretation of the results. All authors were involved in drafting the article or revising it critically for important intellectual content.
Disclosures
Stanley B. Cohen has received research grants and consulting fees from Pfizer Inc. Jeffrey D. Greenberg, Kimberly J. Dandreo, Heather J. Litman, and Taylor Blachley are employees of Corrona, LLC. Ann Madsen, David Gruben, Richard Zhang, Tatjana Lukic, John Woolcott, Anne Lenihan, Connie Chen, and Jose L. Rivas are employees and shareholders of Pfizer Inc. James Harnett was an employee and shareholder of Pfizer Inc at the time of analysis, and is currently an employee of Regeneron Pharmaceuticals. Timothy W. Smith was an employee and shareholder of Pfizer Inc at the time of analysis, and is currently an employee of Novartis. Maxime Dougados has received honoraria from AbbVie, Gilead, Lilly, and Pfizer Inc for his participation in advisory boards or symposia; moreover, his department has received research grants from AbbVie, Gilead, Lilly, and Pfizer Inc to conduct clinical trials. Corrona, LLC, has been supported through contracted subscriptions in the last 2 years by AbbVie, Amgen, Boehringer Ingelheim, Bristol-Myers Squibb, Celgene, Genentech, Gilead, Janssen, Lilly, Merck, Novartis, Ortho Dermatologics, Pfizer Inc, Regeneron, and Sun.
Compliance with Ethics Guidelines
NCT04018001: Institutional Review Board (IRB) review was not required as the study used commercially available deidentified secondary data sources and was considered exempt from the requirements for “human subjects research” in the US. As the study did not involve data subject to privacy laws according to applicable legal requirements, obtaining informed consent from patients was not required. Patient consent for publication was not required. The study was conducted in accordance with legal and regulatory requirements. NCT04267380: All participating investigators in the Corrona US RA Registry were required to obtain IRB approval. All patients were required to provide written informed consent prior to participating in the study. Patient consent for publication was not required. The Corrona registry study was conducted in accordance with the Helsinki Declaration.
Data Availability
Data used in this study from the MarketScan® Commercial and Medicare Supplemental US insurance claims databases were supplied by IBM®. Any analysis, interpretation, or conclusion based on these data is solely that of the authors and not IBM®. The Corrona data set is based on a large US multicenter study adhering to a number of IRBs, with complex logistics. Patients did not provide consent to raw-data sharing during the data collection for this purpose, and the Corrona data-sharing policies do not permit raw-data sharing for this purpose. An aggregated limited data set from the current analyses is available to qualified investigators with an approved protocol. Data requests may be sent to Corrona, represented by Dr. Jeffrey D. Greenberg, MD, MPH, Corrona, LLC, Waltham, MA, and NYU School of Medicine, New York, NY, USA. e-mail: jgreenberg@corrona.org.
Author information
Authors and Affiliations
Corresponding author
Additional information
James Harnett and Timothy W. Smith were employees of Pfizer Inc at the time of the analysis.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-Non-Commercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Cohen, S.B., Greenberg, J.D., Harnett, J. et al. Real-World Evidence to Contextualize Clinical Trial Results and Inform Regulatory Decisions: Tofacitinib Modified-Release Once-Daily vs Immediate-Release Twice-Daily for Rheumatoid Arthritis. Adv Ther 38, 226–248 (2021). https://doi.org/10.1007/s12325-020-01501-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01501-z