
Abstract
Introduction
PF-06881894 is a proposed biosimilar to pegfilgrastim (Neulasta®). This study evaluated the pharmacodynamic/pharmacokinetic (PD/PK) equivalence, immunogenicity, and safety of PF-06881894 vs pegfilgrastim reference products (US- and EU-Neulasta®) in healthy volunteers.
Methods
A phase 1, open-label, randomized, crossover study was conducted to assess the pharmacologic equivalence and safety of a single 6-mg dose of PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU. The primary PD endpoints were area under the effect-versus-time curve for absolute neutrophil count (ANC) from dose administration to 288 h postdose, and maximum observed ANC value among subjects confirmed negative for anti-pegfilgrastim antibodies. Primary PK variables included area under the serum pegfilgrastim-versus-time curve from the time of dose administration to time infinity and maximum observed serum pegfilgrastim concentration. A second phase 1, open-label, randomized (1:1), parallel-group, non-inferiority study was conducted to assess the immunogenicity and safety of multiple 6-mg doses of PF-06881894 versus pegfilgrastim-US. The primary endpoint for the immunogenicity study was the proportion of subjects with both negative baseline and confirmed positive postdose anti-pegfilgrastim antibodies at any time during the study.
Results
Across the single- and multiple-dose studies (N = 153 and N = 420 treated subjects, respectively), demographics for age (18–65 years), male gender (n = 264/573), and white race (n = 423/573) were similar. Three-way PD/PK equivalence of PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU was demonstrated with the primary PD endpoints and primary PK variables being completely contained within the predefined 90% confidence interval acceptance limits (80–125%). The non-inferiority of PF-06881894 versus pegfilgrastim-US in terms of immunogenicity was established according to the prespecified non-inferiority margin (≤10%). Overall, there were no clinically meaningful differences in safety profiles among or between study groups.
Conclusions
Single-dose PF-06881894 demonstrated PD/PK equivalence and comparable safety with US- and EU-pegfilgrastim reference products. Multiple-dose PF-06881894 demonstrated immunogenicity non-inferiority to pegfilgrastim-US with comparable safety. Both studies contributed to the totality of evidence supporting biosimilarity.
Trial Registration
ClinicalTrials.gov identifiers: NCT02629289 (C1221001); NCT03273842 (C1221005).
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Why carry out this study? |
The treatment of neutropenia with a biosimilar of pegfilgrastim could provide an additional therapeutic option for those patients requiring a more affordable and long-acting hematopoietic stimulant |
This study assessed PF-06881894, a proposed biosimilar to pegfilgrastim, and the US-licensed and EU-approved pegfilgrastim reference products (Neulasta®) in healthy volunteers to determine if there were any clinically meaningful differences among the study drugs |
What has been learned from the study? |
Single-dose pharmacodynamic and pharmacokinetic equivalence was established with values contained within the predefined limit of 80–125% when PF-06881894 was compared with US-licensed and EU-approved reference products in study C1221001 |
PF-06881894 was non-inferior to US-licensed pegfilgrastim with respect to immunogenicity after multiple dosing; the upper bound of the 90% confidence interval for risk difference was contained within the prespecified non-inferiority margin of ≤ 10% in study C1221005 |
These studies demonstrated an overall comparable safety profile and supported the biosimilarity of PF-06881894 to Neulasta® |
Introduction
Neutropenia is a decrease in circulating neutrophils in the non-marginal pool and is a major dose-limiting toxicity of several chemotherapy regimens, including myelosuppressive chemotherapy [1,2,3]. Human granulocyte-colony stimulating factor (G-CSF) is a hematopoietic stimulant that promotes proliferation, differentiation, and maturation of neutrophil precursors and is used to treat neutropenia [4].
Filgrastim is a recombinant methionyl human G-CSF (rhG-CSF) that has been shown to enhance neutrophil production and mobilize hematopoietic stem cells from bone marrow to the blood [5]. It is rapidly cleared from systemic circulation—elimination half-life is ~ 3.5 h due to its small size (4 nm diameter and ~ 19 kDa molecular weight), which necessitates daily administration to augment neutrophil recovery [5, 6]. Pegfilgrastim (Neulasta®; Amgen, Thousand Oaks, CA) is a covalent conjugate of filgrastim and a 20-kDa monomethoxypolyethylene glycol (PEG) polymer [7, 8]. The hydrodynamic diameter of filgrastim expands to ~ 6 nm with the addition of PEG (PEGylation) to reduce renal clearance [6] and is contingent upon neutrophil-mediated clearance. At a molecular weight of 39 kDa, the half-life of pegfilgrastim after subcutaneous (SC) injection ranges from 15 to 80 h, which corresponds with the start of neutrophil recovery [6, 8]. The administration of pegfilgrastim is less frequent than for filgrastim, with one dose administered subcutaneously per chemotherapy cycle for patients with cancer receiving myelosuppressive chemotherapy [9].
A biosimilar is a therapeutic protein that is highly similar to a licensed originator (or reference) biologic product—notwithstanding minor differences in clinically inactive components—and for which there are no clinically meaningful differences in terms of safety, purity, or potency [10]. PF-06881894 is a proposed biosimilar to reference pegfilgrastim sourced from the USA (pegfilgrastim-US) and from the European Union (pegfilgrastim-EU). The assessment of similarity between a proposed biosimilar and its reference product involves rigorous analysis of its structural and functional activity conducted in a stepwise manner, as well as non-clinical studies (if required by regulatory authorities), clinical pharmacology assessments, and confirmatory comparative clinical trials [10]. Together, data from these studies comprise the “totality of evidence” required for the regulatory licensure of a biosimilar [10]. Regulatory guidance on clinical pharmacology studies to support the demonstration of biosimilarity outside of the USA recommends that, when possible, healthy individuals should be investigated to evaluate pharmacokinetics (PK) and pharmacodynamics (PD) to identify any product-specific differences that might exist [11, 12]. This approach aligns with US Food and Drug Administration (FDA) guidance, which suggests that healthy subjects are generally considered a more sensitive and homogeneous population with less likelihood of PK and/or PD variability and reduced potential for confounding factors compared with subjects who may have an unsuspected and/or concomitant disease, be heavily medicated, and/or myelosuppressed [13, 14]. Furthermore, the choice of a single well-established, highly sensitive dose-dependent PD surrogate biomarker for clinical efficacy outcomes, or multiple PD measurements (e.g., area under the effect-versus-time curve [AUEC] for absolute neutrophil count [AUECANC] for pegfilgrastim with a simultaneous measurement of the time of maximum value [ANC_Tmax]) of absolute neutrophil count (ANC) are recognized by the FDA as a streamlined method for reducing any residual uncertainty about clinically meaningful differences in the safety, purity, and potency between a proposed biosimilar and its reference product, albeit with the expectation that immunogenicity will still need to be adequately assessed [13, 15]. The most recently (2018) revised draft regulatory guidelines from the European Medicines Agency’s Committee for Medicinal Product for Human Use (CHMP) propose that product quality and clinical pharmacology comparisons can be sufficient to demonstrate similarity between a proposed pegfilgrastim biosimilar and its reference product without the need for dedicated comparative efficacy trial data [16]. In Europe, ANC PD endpoints are favored surrogate biomarkers for efficacy, and no further justification of comparability limits is considered necessary by the CHMP when a 95% confidence interval (CI) with a bioequivalence range of 90–111% is employed for PD measurements made after the last dose has been administered [16, 17].
Here we describe the findings from a phase 1, single-dose, PD/PK equivalence study of PF-06881894 versus pegfilgrastim-US and pegfilgrastim-EU, and a phase 1, multiple-dose, immunogenicity study of PF-06881894 versus pegfilgrastim-US. Safety outcomes were also evaluated in both studies and immunogenicity was an exploratory assessment in the PD/PK study.
Methods
Study Design
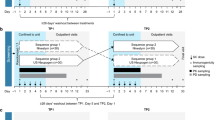
C1221001 (ClinicalTrials.gov identifier NCT02629289) was an open-label, randomized, single-dose, comparator-controlled, three-treatment, three-period, six-sequence, crossover study to assess the PD, PK, immunogenicity, and safety of PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU in healthy volunteers. Each subject was randomly assigned to one of six sequence groups and received each study drug by SC dosing over three study periods, with a washout of at least 56 days between each drug (Fig. 1).

Clinical development program for PF-06881894 in healthy volunteers. a C1221001: a single-dose PD/PK study of PF-06881894 versus pegfilgrastim-US and pegfilgrastim-EUa. b C1221005: a multiple-dose comparative immunogenicity study of PF-06881894 versus pegfilgrastim-USb. a For the PD evaluations, blood samples (4.0 mL) for ANC were collected by either intravenous catheter or venipuncture into evacuated collection tubes within 1 h before dose administration and at 12, 24, 48, 72, 96, 120, 144, 168, 192, 216, 240, 264, and 288 h postdose. A total of 14 samples were planned for PD analysis in each period. For PK evaluations, blood samples (5.0 mL) for the pegfilgrastim assay were collected within 1 h before dosing and at 1, 2, 4, 6, 8, 10, 12, 16, 24, 48, 72, 96, 120, 144, 168, 192, 216, 240, 264, and 288 h postdose. Blood samples (7 mL) for ADA testing were collected within 1 h before dosing on days 1 and 13 of each period, at day 30 of the follow-up visit of each period, or early termination (if applicable). b Identical dosing procedures were performed on day 1 of phase 1 (first drug dose) and phase 2 (second drug dose) in this parallel-design study. Blood samples (7.5 mL) for ADAs were collected before dosing (day 1, phase 1 only) and after fasting for at least 4 h (on days 13 and 30 ± 2, phase 1 and on days 13, 30 ± 2, 60 ± 5 or at early withdrawal, phase 2). Early termination from the study required a subject to have completed the day 30 ± 2, phase 2 assessments. ISR data collection occurred up to 90 min postdose (days 1, 3, and 5 of phases 1 and 2); other ISR data were captured outside the ISR schedule during AE monitoring. ADA anti-drug antibody, AE adverse event, ANC absolute neutrophil count, ISR injection-site reaction, NAb neutralizing antibody, PD pharmacodynamic, pegfilgrastim-EU pegfilgrastim sourced from the European Union (reference product), pegfilgrastim-US pegfilgrastim sourced from the USA (reference product), PF-06881894 proposed biosimilar of pegfilgrastim (test product), PK pharmacokinetic, SC subcutaneous
C1221005 (NCT03273842) was an open-label, randomized, multiple-dose, parallel-design study to assess the immunogenicity and safety of PF-06881894 and pegfilgrastim-US administered SC to healthy volunteers. Each subject received a total of two injections of the study drug to which they had been randomized (PF-06881894 or pegfilgrastim-US), once on day 1 in each treatment phase, with ~ 1 month between each 6-mg dose (Fig. 1). The protocol and all amendments were approved by the institutional review board at each of the investigational centers (Bellberry Limited, MidLands Independent Review Board, Advarra d/b/a Schulman Institutional Review Board and Schulmam IRB) participating in studies C1221001 and C1221005. Both studies were conducted in compliance with the protocol, International Conference on Harmonisation guidelines, all applicable regulatory requirements, and the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from all individual subjects prior to the performance of any study-specific procedures, and they were permitted to withdraw from the study at any time.
Participants
The study inclusion and exclusion criteria were generally consistent across both studies (Online Resources 1 and 2). In both studies, eligible participants comprised healthy, non-smoking male or female volunteers aged 18–65 years, with a body mass index of 19–30 kg/m2. Subjects with any active systemic or immunologic disease or condition (e.g., cardiovascular/pulmonary, hepatorenal, systemic infection, lactation), or history of or current malignancy—except for adequately treated squamous or basal cell carcinoma of the skin or cervical carcinoma in situ within 5 years—were excluded from both studies. Subjects with any disease or condition that might interfere with the absorption, distribution, metabolism, or excretion of the study drug or place them at increased risk were also excluded.
Objectives, Endpoints, and Assessments
The primary objective of study C1221001 was to assess the PD equivalence of PF-06881894 with pegfilgrastim-US and pegfilgrastim-EU. The primary endpoint was the AUECANC from the time of dose administration to 288 h postdose, and the maximum observed value for ANC (ANC_Cmax). ANC_Tmax was assessed as a secondary PD parameter. PD and PK sampling times are depicted in Fig. 1. The measurement of ANC using a hematology analyzer (Sysmex XN-3000; Japan) consisted of a cytochemical reaction of the cells with a reagent set, followed by fluorescence flow-cytometric analysis. Like PK, the PD variables were calculated utilizing non-compartmental methods (Phoenix™ for WinNonlin, v6.4; Pharsight, CA, USA). Subjects who were confirmed positive for anti-pegfilgrastim antibodies at any time were not included in the primary PD/PK population.
The secondary objectives in study C1221001 were to assess the PK equivalence of PF-06881894 with pegfilgrastim-US and pegfilgrastim-EU, and the PD/PK equivalence of pegfilgrastim-US and pegfilgrastim-EU as a single SC dose—to support the use of a single comparator in study C1221005—as well as the comparable safety of PF-06881894. Serum pegfilgrastim concentrations (range, 100–5000 pg/mL) were determined utilizing a validated enzyme-linked immunosorbent assay (ELISA) procedure employing the double-antibody sandwich method [18].
Exploratory testing for anti-drug antibodies (ADAs) in study C1221001 was performed predose and postbaseline, concomitant with PD/PK analyses and safety evaluations, according to a prespecified sampling schedule to establish the clinical relevance of ADAs [10, 19]. Immunogenicity was assessed using a multi-tiered approach consistent with FDA Guidance for Industry [10, 19, 20]. Samples that tested anti-pegfilgrastim antibody positive at screening using a validated electrochemiluminescent (ECL) bridging assay format (tier 1) were further tested using the same ECL assay format for confirmation and to determine ADA specificity for the filgrastim moiety (tier 2). Measurement of antibody titer in the ECL assay was tested (tier 3) before further characterization of confirmed positive samples to measure neutralizing antibodies (NAbs) using a cell-based assay. NAb-positive samples were further analyzed for filgrastim NAb (in an amendment to study C1221001). Anti-PEG antibody screening was performed for all ADA samples, regardless of the positivity score in the anti-pegfilgrastim antibody ECL assay. Samples were screened via the aforementioned process using a validated ELISA-based assay (tier 1). PEG specificity was assessed (tier 2) before the measurement of antibody titer in confirmed antibody-positive samples (tier 3). Confirmatory cut-points for ADA assay methodologies used a 99% CI to generate an ~ 1% false-positive rate. Additional immunogenicity methodology is described in Online Resource 3.
Subjects who were confirmed positive for anti-pegfilgrastim antibodies at any time were excluded from the PD and PK populations for primary analysis in study C1221001. Sensitivity analyses were performed to evaluate the effect of confirmed positive anti-pegfilgrastim antibodies on PD/PK.
The primary objective of study C1221005 was to assess the non-inferiority of PF-06881894 versus pegfilgrastim-US with respect to immunogenicity. The primary endpoint was the proportion of subjects with a negative baseline anti-pegfilgrastim antibody test result who had a confirmed postdose positive anti-pegfilgrastim antibody test result at any time during the study. The secondary endpoint was the proportion of subjects with a negative baseline anti-pegfilgrastim antibody test and a postdose positive NAb result during the study. Immunogenicity testing followed the same multi-tiered approach described for study C1221001, except the original ADA assay methodology validation parameters were updated for study C1221005 to remain aligned with the evolution of FDA guidance since study C1221001 (Online Resource 3) [20, 21].
Assessments of safety in these studies included adverse events (AEs) as well as AEs of special interest (AESIs) coded using the Medical Dictionary for Regulatory Activities (v19.1 and v21.1 for studies C1221001 and C1221005, respectively). AESIs were identified prospectively, based on the known safety profile of US- and EU-approved pegfilgrastim reference products [7]. Laboratory variables, vital signs, 12-lead electrocardiograms (ECGs), and physical examinations were also performed; local injection-site reactions (ISRs) were a secondary endpoint in study C1221005.
Statistical Analysis and Determination of Sample Size
C1221001
PD equivalence was assessed using analysis of variance to obtain geometric mean ratios (test/reference) and CIs for AUECANC and ANC_Cmax. Sequence, period, and treatment were factors for log-transformed data, and baseline ANC was used as a covariate for PD parameters. AUECANC was calculated via the linear trapezoidal rule from predose to the last measurable positive value. PD equivalence was concluded if the 90% CIs for both AUECANC and ANC_Cmax were completely contained within the prespecified acceptance limits of 80–125%.
The study was powered for the assessment of key secondary PK parameters (area under the serum–time curve [AUC] from time of dose administration to infinity [AUC0–∞] and Cmax). Assuming a within-subject coefficient of variation of 68% and an equivalence range of 0.80–1.25 of the test/reference mean ratio for AUC0–∞ and Cmax, a total of 150 enrolled subjects were required to demonstrate PK equivalence with at least 90% power at a one-sided alpha level of 0.05.
Step-down statistical testing was used to control the family-wise type 1 error for the PD and PK analyses. PF-06881894 was tested against pegfilgrastim-US, and whenever this test was positive, PF-06881894 was tested against pegfilgrastim-EU. Whenever the latter test was positive, pegfilgrastim-US was also tested against pegfilgrastim-EU. Results from the PD and PK population analyses were separately assessed for consistency and robustness via sensitivity analyses, which, in addition to the respective PD and PK populations, included subjects confirmed positive for anti-pegfilgrastim antibodies.
C1221005
The non-inferiority of PF-06881894 versus pegfilgrastim-US was statistically demonstrated if the upper bound of the 90% CI was ≤ 0.10 for the difference in anti-pegfilgrastim antibody rates in subjects who had a negative baseline anti-pegfilgrastim antibody status and confirmed postdose positive anti-pegfilgrastim antibody test at any time during the study. Assuming that this proportion is 1–5% in each group, a sample size of 210 randomized subjects per group provided at least 90% power to demonstrate non-inferiority. The CI was constructed using the exact method of Chan and Zhang [22]. An additional analysis was performed that included subjects with a positive baseline anti-pegfilgrastim result and higher-titer ADAs after baseline, and who were counted as positive.
Results
Subject Disposition, Baseline Demographics, and Other Characteristics
C1221001
Altogether, 153 subjects were enrolled and randomized to one of six treatment sequences (mean age, 30.4 [range, 18–65] years; 52.9% were male; 86.3% were white; Online Resource 4). Baseline demographic characteristics were similar among sequences. Of the enrolled subjects, 142 (92.8%) completed the study; 11 (7.2%) prematurely discontinued because of withdrawal by subject (n = 7), AEs (n = 2), physician’s decision not to initiate study drug in period 2 due to elevated alanine aminotransferase in period 1, which subsequently resolved (n = 1), or other reasons related to the subject leaving the country (n = 1).
C1221005
Altogether, 422 subjects were randomized to one of two treatments (n = 212 to PF-06881894 and n = 210 to pegfilgrastim-US) and 210 subjects in each group were treated (mean age, 36.6 [range, 18–65] years; 56.4% were female; 69.3% were white; Online Resource 4). Baseline demographic characteristics were balanced between treatment groups. Of the enrolled subjects, 376 (89.1%; 87.7% in the PF-06881894 group and 90.5% in the pegfilgrastim-US group) completed the study and 46 (10.9%; 12.3% and 9.5%, respectively) discontinued. Reasons for premature discontinuation were withdrawal by subject (n = 14) or loss to follow-up (n = 18). Other reasons were all-causality AEs (n = 2), protocol deviations (n = 2), unspecified reasons (n = 9), and no longer meeting the eligibility criteria (n = 1).
Pharmacodynamics (PD)
C1221001
The geometric values for the primary PD endpoints of AUECANC and ANC_Cmax were similar for PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU (Table 1). The 90% and 95% CIs for AUECANC and ANC_Cmax were completely contained within the predefined limit of 80–125% for all study drug comparisons. The sensitivity analysis showed similar PD results for the PD population plus subjects confirmed positive for anti-pegfilgrastim antibodies. The mean ANC-over-time profiles were similar between PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU (Fig. 2).

Mean (SD) serum absolute neutrophil count concentration over time in the single-dose study (C1221001) (PD population). ANC absolute neutrophil count, PD pharmacodynamic, pegfilgrastim-EU pegfilgrastim sourced from the European Union (reference product), pegfilgrastim-US pegfilgrastim sourced from the USA (reference product), PF-06881894 proposed biosimilar of pegfilgrastim (test product), SD standard deviation
Pharmacokinetics (PK)
C1221001
The geometric means for the primary PK endpoints were similar among PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU (Table 2). The 90% CIs for AUC0–∞ and Cmax were completely contained within the predefined limit of 80–125% for all study drug comparisons. The sensitivity analysis showed similar PK results for the PK population plus subjects confirmed positive for anti-pegfilgrastim antibodies. The mean serum pegfilgrastim concentration–time profiles were similar between three treatments (Fig. 3).

Mean (SD) serum pegfilgrastim concentration over time in the single-dose study (C1221001) (PK population). Pegfilgrastim-EU pegfilgrastim sourced from the European Union (reference product), pegfilgrastim-US pegfilgrastim sourced from the USA (reference product), PF-06881894 proposed biosimilar of pegfilgrastim (test product), PK pharmacokinetic, SD standard deviation
Mean values for the secondary PK endpoints of AUC from time of dose administration to time of last measurable concentration (AUC0–t), time to maximum concentration (Tmax), and elimination rate constant (λz) were similar across study drugs (data not shown). The mean elimination half-life ranged from 49.4 to 54.7 h for PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU.
Immunogenicity
C1221001
Ten subjects had anti-pegfilgrastim antibody-positive results confirmed during the study for the exploratory immunogenicity assessment (Table 3). Five subjects (n = 4, PF-06881894 and n = 1, pegfilgrastim-US groups) had a single positive result with low titers (< 20–40) in the absence of NAbs. The remaining five subjects (n = 2 in each of the PF-06881894 and pegfilgrastim-EU groups, n = 1 in the pegfilgrastim-US group) had more than one anti-pegfilgrastim antibody-positive sample confirmed during the study, but not at baseline, and three of these subjects were negative for NAbs (Online Resource 5). Seven of the 10 subjects confirmed as anti-pegfilgrastim antibody-positive had responses specific for the filgrastim protein moiety (n = 4, PF-06881894; n = 1, pegfilgrastim-US; n = 2, pegfilgrastim-EU groups). At the final visit, eight of the 10 subjects with confirmed anti-pegfilgrastim antibody-positive samples during the study tested negative for anti-pegfilgrastim antibodies.
Of 153 (72.5%) subjects in the safety analysis set, 111 tested positive for anti-PEG antibodies at least once during the study, and the incidences and titers were balanced across the three study drugs. Anti-PEG antibodies were predominantly pre-existing or detected on day 13 of period 1 (Table 3). Over the duration of the study, the incidence of anti-PEG antibodies decreased to 43/148 subjects (29.1%) by the final visit. There was no evidence of boosting of the anti-PEG antibody response upon administration of the second or third study drug.
Among the 10 subjects with an anti-pegfilgrastim antibody test result, two (both in the PF-06881894 group) were positive for NAbs detected on day 13 of period 1, but not thereafter (Online Resource 6). Both subjects with NAbs were negative for filgrastim NAb. Among all study subjects, including those with more than one anti-pegfilgrastim antibody test result, one—who was NAb-positive and tested de novo anti-PEG antibody-positive on day 13 of period 1—exhibited PD/PK profiles that demonstrated a notably diminished treatment effect. This was most pronounced during period 2 (pegfilgrastim-US) and still markedly evident during period 3 (pegfilgrastim-EU; Online Resource 7).
Among the subjects positive for ADAs, no specific trends in AEs were identified by the presence of anti-pegfilgrastim antibody, with the exception of the single PEG-related NAb-positive subject who developed a treatment-related (PF-06881894) urticarial rash on the left deltoid injection site on day 11 of period 1, along with a second, less-extensive urticarial rash on day 2 of period 2 (Table 3). An additional three subjects who were positive for anti-pegfilgrastim antibody reported AEs of vessel puncture-site rash (from a tape reaction), rhinitis allergic (from concurrent illness), and injection-site erythema.
C1221005
The upper bound of the 90% CI of the risk difference between PF-06881894 and pegfilgrastim-US (− 5.915% to 2.675%) was within the prespecified non-inferiority margin of ≤ 10%. This result met the primary endpoint by demonstrating the non-inferiority of PF-06881894 versus pegfilgrastim-US with respect to immunogenicity in healthy volunteers with negative baseline anti-pegfilgrastim, and confirmed postdose positive anti-pegfilgrastim antibody at any visit (Table 4).
Twelve subjects (5.9%) in the PF-06881894 group and 15 (7.5%) in the pegfilgrastim-US group had a confirmed postdose positive anti-pegfilgrastim antibody test at any visit beyond baseline; 16 and 23 subjects, respectively, were confirmed postdose anti-pegfilgrastim antibody-positive at any visit, including at baseline. Overall, anti-pegfilgrastim titers in subjects assessed at any visit were higher on day 13 of phase 1 and waned over the course of phase 2.
One subject in the PF-06881894 group, who was anti-pegfilgrastim antibody-negative at baseline, was anti-pegfilgrastim antibody-positive postdose, at study completion, but was lost to follow-up after the final prespecified visit. No subject who was anti-pegfilgrastim negative at baseline was NAb-positive at any visit (Table 4). A subject in the PF-06881894 group who tested anti-pegfilgrastim positive at baseline had two anti-pegfilgrastim antibody samples that tested positive for NAbs during phase 1, on day 13 (titer 16) and on day 30 (titer 8), and one sample in phase 1 tested positive for anti-PEG antibodies on day 13 (titer 12,800). Throughout phase 1, the positive anti-pegfilgrastim antibody samples for this subject were consistently negative when tested for specificity to the filgrastim protein moiety, suggesting that the NAb response was the result of the PEG moiety. No discernible effect of NAbs was observed on this subject’s ANC trends compared with subjects without anti-pegfilgrastim antibodies, but this subject did experience a treatment-emergent adverse event (TEAE) of ISR related to study drug, and discontinued by day 5 of phase 2. This subject was not included in the primary and secondary endpoint calculations because of their positive anti-pegfilgrastim antibody result at baseline (titer 80).
No clinically meaningful differences were observed in the anti-PEG antibody status at baseline (21.9% and 22.9% of subjects were confirmed anti-PEG antibody-positive) between the PF-06881894 (n = 210) and pegfilgrastim-US (n = 210) groups, respectively, or throughout the duration of the study. Confirmed positive anti-PEG antibodies peaked on day 13 of phase 1 for 56.0% and 59.9% of the subjects, respectively, and declined to 39.9% and 44.5% by the last postbaseline visit.
Safety
C1221001 and C1221005
Overall, the safety profiles of PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU in study C1221001 were similar, as were those for PF-06881894 and pegfilgrastim-US in study C1221005; the percentages of subjects with all-causality TEAEs (C1221001: 99.3%, 95.2%, and 95.3%, respectively; C1221005: 92.4% and 96.2%, respectively) were comparable across groups within studies (Table 5).
The most frequently reported all-causality TEAEs in study C1221001 were musculoskeletal pain for 118 (79.7%) subjects in the PF-06881894 group, 116 (79.5%) in the pegfilgrastim-US group, and 111 (75.0%) in the pegfilgrastim-EU group, and headache for 101 (68.2%), 99 (67.8%), and 105 (70.9%) subjects, respectively. In study C1221005, the TEAE incidence was highest for back pain, which was reported for 123 (58.6%) subjects in the PF-06881894 group and for 126 (60.0%) in the pegfilgrastim-US group, and next for headache, which was reported for 111 (52.9%) and 107 (51.0%) subjects, respectively.
Four severe TEAEs (skin abrasion and skin laceration in one subject in the PF-06881894 group, arthralgia in one subject in the PF-06881894 group, and renal colic unrelated to the study drug in one subject in the pegfilgrastim-EU group) were reported in study C1221001, and no TEAEs were severe in either treatment group in study C1221005. There were no deaths in either study.
In study C1221001, three subjects (2.0%) discontinued study drug administration: one subject in the PF-06881894 group following a non-drug-related spontaneous abortion, and two subjects in the pegfilgrastim-US group following TEAEs of alanine aminotransferase increased and rash generalized. Two of these three subjects also discontinued from the study as a result of these events (spontaneous abortion and rash generalized). In study C1221005, two subjects (0.5%) permanently discontinued from the study as a result of a TEAE: one in the PF-06881894 group, with a moderately serious urinary tract infection considered unrelated to the study drug; the other one in the pegfilgrastim-US group, with a moderate, non-serious angioedema event of moderate severity considered related to the study drug.
A similar percentage of subjects experienced treatment-emergent AESIs across groups in both studies, and the most frequently reported AESI was potential allergic reactions (C1221001: 5.4%, 7.5%, and 6.8% in PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU, respectively; C1221005: 2.9% and 2.4% in PF-068818894 and pegfilgrastim-US, respectively; Table 5). One subject (0.7%) each in the PF-06881894 and pegfilgrastim-US groups in study C1221001 and no subjects in study C1221005 had an AESI in the splenomegaly category. The rates of AESI of thrombocytopenia were similar between the PF-06881894 and pegfilgrastim-US groups (2.4% and 1.9%, respectively) in study C1221005, and not present in study C1221001.
The ISR assessment in study C1221005 showed that 11 subjects (5.2%) in the PF-06881894 group and eight (3.8%) in the pegfilgrastim-US group reported at least one event during the study. In the PF-06881894 group, injection-site pain was reported for eight (3.8%) subjects, injection-site ecchymosis for five subjects (2.4%), and injection-site erythema or pruritus for one subject (0.5%) each. In the pegfilgrastim-US group, injection-site ecchymosis was reported for six subjects (2.9%) and injection-site pain by three (1.4%). All ISRs, and ISRs captured during AE monitoring, were mild in severity and none were clinically significant.
In both studies, there were no clinically significant findings for vital signs, ECG, physical examination, or clinical laboratory results.
Discussion
Studies C1221001 and C1221005 were conducted to support the totality of evidence for demonstrating the biosimilarity of PF-06881894 to its US-licensed and EU-approved reference products. Both studies met their primary and secondary endpoints and collectively resulted in the demonstration of PD/PK equivalence among PF-06881894, pegfilgrastim-US, and pegfilgrastim-EU, the non-inferior immunogenicity of PF-06881894 versus pegfilgrastim-US, and no clinically meaningful differences in the safety profile of PF-06881894 when considered with respect to the safety and tolerability of the reference products in healthy volunteers.
Study C1221001 demonstrated the pharmacologic equivalence of PF-06881894 with pegfilgrastim-US and pegfilgrastim-EU for the primary PD endpoints (AUECANC and ANC_Cmax) and primary PK variable (AUC0–∞ and Cmax) on the basis of the 90% CIs being completely contained within the 80–125% acceptance limits. Study C1221001 also demonstrated robustness, with equivalence extending to the results of the PD/PK sensitivity analyses, as well as to the PD results and sensitivity analysis (AUECANC and ANC_Cmax) on the basis of the construction of a 95% CI. In addition, in study C1221001 it was important to support bridging between local and foreign pegfilgrastim products to satisfy regulatory filing requirements in major global jurisdictions [23].
The crossover design of study C1221001 was not suitable or powered for comparing immunogenicity among the three study treatments. Although immunogenicity testing in study C1221001 was strictly exploratory, the results aided an early approximation of the incidence and timing of ADAs, and paved the way for the subsequent and more robust parallel, multiple-dose, comparative immunogenicity study (C1221005) that was appropriately designed and powered for immunogenicity determinations.
Although PEGylation extends the circulating kinetics of filgrastim and other biotherapeutics, the PEG moiety is not immunologically inert [24, 25]. The ubiquitous presence of PEG in a variety of manufactured goods has likely induced strong memory immune responses to PEGylated polymers in some individuals [25]. Approximately 72% of the general population test positive for anti-PEG antibodies, which is similar to the 72.5% of subjects who were anti-PEG antibody positive at least once during study C1221001 [26]. Anti-PEG antibodies can potentially reduce the circulation half-life of PEGylated agents between two- to tenfold on average and have been associated with reduced therapeutic efficacy via enhanced clearance due to neutralizing capacity [25, 27]. However, patients who develop ADAs that are transient or not clinically relevant can remain responsive to treatment. All 10 subjects identified as anti-pegfilgrastim antibody-positive during study C1221001 were confirmed anti-pegfilgrastim antibody-negative by the end of the study, with no impact on PD/PK and safety, except in one subject. That single subject was one of two who had sustained high titer anti-PEG responses and demonstrated transient NAbs that were not specific for the filgrastim protein moiety, but rather related to the PEG moiety.
It should be noted that assay development and validations related to immunogenicity testing for study C1221001 were aligned with the FDA’s 2009 draft guidance for industry, which was available at the time, whereas updated FDA regulatory guidance for immunogenicity testing in 2016 was applied to study C1221005 [20, 21]. Therefore, in study C1221005, updated partial validations for the anti-pegfilgrastim ADA ECL method, the anti-PEG ADA ELISA, and the cell-based NAb assay methods were applied in response to evolving guidance. As a result, a lower positive control was subsequently implemented that represented a 1% assay failure rate in confirmatory cut-point assays to ensure that the minimum false-positive rates were met for the anti-pegfilgrastim ADA determination. Normal serum predose samples from study C1221005 were used for the screening and confirmatory cut-point validations of ELISAs and the screening cut-point validations of cell-based NAb assays.
The results for comparative immunogenicity in study C1221005—which confirmed the non-inferiority of PF-06881894 versus pegfilgrastim-US (12 [5.9%] vs 15 [7.5%] subjects with treatment-emergent anti-pegfilgrastim antibodies, respectively)—were balanced and generally consistent with the exploratory findings in study C1221001 in terms of ADAs and time profiles, and supported the conclusion of biosimilarity. The detection of ADAs consistently peaked on day 13 of period/phase 1.
In study C1221005, on day 13 of phase 1, anti-pegfilgrastim antibodies were detected in 4.9% of subjects who received PF-06881894 and in 4.5% of subjects who received pegfilgrastim-US, whereas anti-PEG antibodies were detected in 56.0% and 59.9% of subjects, respectively. The proportions of subjects who were anti-PEG antibody positive reflect the conservative approach adopted in relation to this standalone assay, in which every randomized subject was tested, regardless of positivity in the anti-pegfilgrastim antibody assay. In addition, the ELISA used to determine anti-PEG specificity was found to provide the highest sensitivity and specificity to all anti-PEG antibodies tested during methodology development assessments when ELISA and ECL formats were compared. No NAbs were detected among subjects who were negative for anti-pegfilgrastim antibodies at baseline in study C1221005. However, NAbs were detected twice in one subject in study C1221005 who had pre-existing anti-pegfilgrastim antibodies at baseline, but no discernible clinical effect of the NAbs was observed.
Beyond baseline detection of ADAs, many previous clinical studies have measured ADAs when assessing pegfilgrastim and/or proposed biosimilars of pegfilgrastim at day 21, 28, or later after dosing has commenced [2, 28,29,30,31,32,33], or at unspecified intervals [34, 35]. Among earlier timed immunogenicity assessments was a phase 1, randomized, double-blind, three-way, three-period, crossover study in which proposed pegfilgrastim biosimilar MYL-1401H (pegfilgrastim-jmdb; Fulphila®, Mylan GmbH, Zurich, Switzerland) was compared with pegfilgrastim-US and -EU reference products in healthy volunteers [36]. ADAs were assessed on days 7–9 of period 1. The incidence of treatment-emergent ADAs ranged from 22% to 30% among treatment groups. Two subjects in the MYL-1401H group and one subject each in the pegfilgrastim-US and -EU reference groups developed treatment-emergent NAbs that, in the MYL-1401H group, were characterized by very low titers (8–10 ng/mL). Among them, one subject each in the MYL-1401H group and the pegfilgrastim-US group was anti-PEG positive, and the remaining two subjects were both anti-pegfilgrastim positive and anti-PEG positive. No loss of efficacy and no safety issues were associated with the detected NAbs in subjects.
ADA binding location and affinities are known to variously impact drug clearance or metabolism, efficacy, and/or safety [37, 38]. In addition, the incidence of ADAs, including NAbs, as a result of exposure to biologic products of the same sequence or structural homology, tend to be disparately influenced by a range of intrinsic and extrinsic factors, including (but not limited to) assay methodology and parameters (despite the use of fully validated assays), sample collection and handling methods, concomitant medications, and disease conditions [20]. Importantly, the FDA advises that the immunogenicity of homologous biologic products can only ever be appropriately compared via head-to-head clinical studies that employ standardized assays under the same sensitivity and specificity conditions [20].
Limitations in this study are intertwined with the overall biosimilar development program for which the latitude is limited. We acknowledge that the PD/PK in healthy volunteers is potentially different than that of patients; a small pilot study was conducted in a patient population to determine that the PD response and PK exposure would be relatable (data not shown). Our studies did not generate long-term safety data, although the use of pegfilgrastim is typically not chronic, and most treatment-emergent AEs have a rapid onset. Rather, a robust program of long-term toxicology data was generated. The methodology employed in these studies used highly sensitive bioanalytical methods to detect antibodies to pegfilgrastim as well as their specificity for the component molecules filgrastim and PEG. Not all prior studies of pegfilgrastim (including biosimilars) immunogenicity have included specificity testing, particularly by testing for anti-PEG antibody, the presence of which was found to result in PK and PD changes in at least one subject in our study. The timing of antibody samples was also relevant, as the highest anti-PEG response was observed at or before day 13. As a result, it is not possible to compare immunogenicity results across programs or products.
Overall, the safety results observed in clinical studies C1221001 and C1221005 were consistent with the known safety profiles of US- and EU-approved pegfilgrastim, as described in the US Prescribing Information and Summary of Product Characteristics [7, 9]. PF-06881894 was generally as well tolerated as the reference products in each study, with no clinically meaningful differences reported for TEAEs, SAEs, and AESIs, and no trends were identified related to TEAEs, contingent upon the presence of anti-pegfilgrastim antibodies. The clinical laboratory results and patterns observed with PF-06881894 were also consistent with the known therapeutic response and the safety profiles for the reference products [7, 9].
Conclusions
PF-06881894, a proposed biosimilar of pegfilgrastim, demonstrated PD and PK equivalence to the US- and EU-approved pegfilgrastim reference products (Neulasta®) in a single-dose, three-period, crossover study. In a multiple-dose, comparative immunogenicity study, PF-06881894 was non-inferior to US-approved pegfilgrastim. Across both studies, there were no clinically meaningful differences in terms of safety. Together, these studies demonstrate support for biosimilarity of PF-06881894 to Neulasta®.
References
Crawford J, Dale DC, Lyman GH. Chemotherapy-induced neutropenia: risks, consequences, and new directions for its management. Cancer. 2004;100(2):228–37. https://doi.org/10.1002/cncr.11882.
Green MD, Koelbl H, Baselga J, et al. A randomized double-blind multicenter phase III study of fixed-dose single-administration pegfilgrastim versus daily filgrastim in patients receiving myelosuppressive chemotherapy. Ann Oncol. 2003;14(1):29–35. https://doi.org/10.1093/annonc/mdg019.
Sureda A, Domingo-Domenech E, Gautam A. Neutropenia during frontline treatment of advanced Hodgkin lymphoma: incidence, risk factors, and management. Crit Rev Oncol Hematol. 2019;138:1–5. https://doi.org/10.1016/j.critrevonc.2019.03.016.
Metcalf D. The colony-stimulating factors and cancer. Cancer Immunol Res. 2013;1(6):351–6. https://doi.org/10.1158/2326-6066.CIR-13-0151.
Amgen. NEUPOGEN® (filgrastim) prescribing information. Amgen. 2018 (last update June 2018). https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/neupogen/neupogen_pi_hcp_english.pdf. Accessed 17 Jan 2019.
Arvedson T, O’Kelly J, Yang B-B. Design rationale and development approach for pegfilgrastim as a long-acting granulocyte colony-stimulating factor. BioDrugs. 2015;29(3):185–98. https://doi.org/10.1007/s40259-015-0127-4.
Amgen. NEULASTA® (pegfilgrastim) summary of product characteristics. Amgen. 2018 (last update September 2018). https://www.medicines.org.uk/emc/product/6770/smpc#DOCREVISION. Accessed 7 Mar 2019.
Ho R, Gibaldi M. Biotechnology and biopharmaceuticals: transforming proteins and genes into drugs. In: Ho R, Gibaldi M, editors. Therapeutics based on biotechnology. Hematopoietic growth factors and coagulation factors. Hoboken: Wiley; 2003. p. 157–158.
Amgen. NEULASTA® (pegfilgrastim) prescribing infomation. 2020. https://www.pi.amgen.com/~/media/amgen/repositorysites/pi-amgen-com/neulasta/neulasta_pi_hcp_english.pdf. Accessed 27 May 2020.
US Food and Drug Administration. Scientific considerations in demonstrating biosimilarity to a reference product. Guidance for industry. US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Silver Spring, MD. 2015. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM291128.pdf. Accessed 9 Mar 2020.
European Medicines Agency. Biosimilars in the EU: Information guide for healthcare professionals. 2017. https://www.medicinesforeurope.com/wp-content/uploads/2017/05/HCP-guide-on-Biosimilars.pdf. Accessed 17 Jun 2019.
World Health Organization. Annex 2, Guidelines on evaluation of similar biotherapeutic products (SBPs): WHO Technical Report Series No. 977. 2013. https://www.who.int/biologicals/publications/trs/areas/biological_therapeutics/TRS_977_Annex_2.pdf?ua=. Accessed 27 May 2020.
US Food and Drug Administration. Clinical pharmacology data to support a demonstration of biosimilarity to a reference product. Guidance for industry. US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Silver Spring, MD. 2016. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM397017.pdf. Accessed 5 Mar 2019.
Gascon P, Fuhr U, Sorgel F, et al. Development of a new G-CSF product based on biosimilarity assessment. Ann Oncol. 2010;21(7):1419–29. https://doi.org/10.1093/annonc/mdp574.
Li J, Florian J, Campbell E, et al. Advancing biosimilar development using pharmacodynamic biomarkers in clinical pharmacology studies. Clin Pharmacol Ther. 2020;107(1):40–2. https://doi.org/10.1002/cpt.1653.
European Medicines Agency. Guideline on similar biological medicinal products containing recombinant granulocyte-colony stimulating factor (rG-CSF), EMEA/CHMP/BMWP/31329/2005 Rev 1 2018. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-similar-biological-medicinal-products-containing-recombinant-granulocyte-colony_en.pdf Accessed 10 Jan 2020.
Wolff-Holz E, Tiitso K, Vleminckx C, Weise M. Evolution of the EU biosimilar framework: past and future. BioDrugs. 2019;33(6):621–34. https://doi.org/10.1007/s40259-019-00377-y.
US Food and Drug Administration. Bioanalytical method validation. Guidance for industry. US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM), Silver Spring, MD. 2018. https://wayback.archive-it.org/7993/20190916051913/https:/www.fda.gov/media/70858/download. Accessed 20 Nov 2019.
US Food and Drug Administration. Immunogenicity assessment for therapeutic protein products. Guidance for industry. US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Silver Spring, MD. 2014. https://www.fda.gov/media/85017/download. Accessed 1 Nov 2019.
US Food and Drug Administration. Assay development and validation for immunogenicity testing of therapeutic protein products. Guidance for industry (draft guidance). US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Center for Devices and Radiological Health (CDRH), Silver Spring, MD. 2016. https://www.fda.gov/media/77796/download. Accessed 3 Nov 2019.
US Food and Drug Administration. Guidance for industry assay development for immunogenicty testing of therapeutic proteins: Guidance. US Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Biologics Evaluation and Research (CBER), Silver Spring, MD. 2009. https://www.regulations.gov/document?D=FDA-2009-D-0539-0002. Accessed 13 Jan 2020.
Chan IS, Zhang Z. Test-based exact confidence intervals for the difference of two binomial proportions. Biometrics. 1999;55(4):1202–9. https://doi.org/10.1111/j.0006-341x.1999.01202.x.
Webster CJ, Woollett GR. A 'global reference' comparator for biosimilar development. BioDrugs. 2017;31(4):279–86. https://doi.org/10.1007/s40259-017-0227-4.
Hamidi M, Azadi A, Rafiei P. Pharmacokinetic consequences of pegylation. Drug Deliv. 2006;13(6):399–409. https://doi.org/10.1080/10717540600814402.
Yang Q, Lai SK. Anti-PEG immunity: emergence, characteristics, and unaddressed questions. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2015;7(5):655–77. https://doi.org/10.1002/wnan.1339.
Yang Q, Jacobs TM, McCallen JD, et al. Analysis of pre-existing IgG and IgM antibodies against polyethylene glycol (PEG) in the general population. Anal Chem. 2016;88(23):11804–12. https://doi.org/10.1021/acs.analchem.6b03437.
Ishida T, Kiwada H. Anti-polyethyleneglycol antibody response to PEGylated substances. Biol Pharm Bull. 2013;36(6):889–91. https://doi.org/10.1248/bpb.b13-00107.
Blackwell K, Donskih R, Jones CM, et al. A comparison of proposed biosimilar LA-EP2006 and reference pegfilgrastim for the prevention of neutropenia in patients with early-stage breast cancer receiving myelosuppressive adjuvant or neoadjuvant chemotherapy: pegfilgrastim randomized oncology (supportive care) trial to evaluate comparative treatment (PROTECT-2), a phase III, randomized, double-blind trial. Oncologist. 2016;21(7):789–94. https://doi.org/10.1634/theoncologist.2016-0011.
Desai K, Catalano T, Rai G, Misra P, Shah N. Confirmation of biosimilarity in a pharmacokinetic/pharmacodynamic study in healthy volunteers for an analytically highly similar pegfilgrastim. Clin Pharmacol Drug Dev. 2016;5(5):354–63. https://doi.org/10.1002/cpdd.269.
Zou L, Buchner A, Roberge M, Liu PM. Immunogenicity assessment of lipegfilgrastim in patients with breast cancer receiving chemotherapy. J Immunol Res. 2016;2016:9248061. https://doi.org/10.1155/2016/9248061.
Gladkov O, Moiseyenko V, Bondarenko IN, et al. A phase III study of balugrastim versus pegfilgrastim in breast cancer patients receiving chemotherapy with doxorubicin and docetaxel. Oncologist. 2016;21(1):7–15. https://doi.org/10.1634/theoncologist.2015-0152.
Yang BB, Morrow PK, Wu X, Moxness M, Padhi D. Comparison of pharmacokinetics and safety of pegfilgrastim administered by two delivery methods: on-body injector and manual injection with a prefilled syringe. Cancer Chemother Pharmacol. 2015;75(6):1199–206. https://doi.org/10.1007/s00280-015-2731-x.
Harbeck N, Lipatov O, Frolova M, et al. Randomized, double-blind study comparing proposed biosimilar LA-EP2006 with reference pegfilgrastim in breast cancer. Future Oncol. 2016;12(11):1359–67. https://doi.org/10.2217/fon-2016-0016.
Holmes FA, O'Shaughnessy JA, Vukelja S, et al. Blinded, randomized, multicenter study to evaluate single administration pegfilgrastim once per cycle versus daily filgrastim as an adjunct to chemotherapy in patients with high-risk stage II or stage III/IV breast cancer. J Clin Oncol. 2002;20(3):727–31. https://doi.org/10.1200/jco.2002.20.3.727.
Holmes FA, Jones SE, O'Shaughnessy J, et al. Comparable efficacy and safety profiles of once-per-cycle pegfilgrastim and daily injection filgrastim in chemotherapy-induced neutropenia: a multicenter dose-finding study in women with breast cancer. Ann Oncol. 2002;13(6):903–9. https://doi.org/10.1093/annonc/mdf130.
Waller CF, Tiessen RG, Lawrence TE, et al. A pharmacokinetics and pharmacodynamics equivalence trial of the proposed pegfilgrastim biosimilar, MYL-1401H, versus reference pegfilgrastim. J Cancer Res Clin Oncol. 2018;144(6):1087–95. https://doi.org/10.1007/s00432-018-2643-3.
Shankar G, Arkin S, Cocea L, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides-harmonized terminology and tactical recommendations. AAPS J. 2014;16(4):658–73. https://doi.org/10.1208/s12248-014-9599-2.
van Brummelen EM, Ros W, Wolbink G, Beijnen JH, Schellens JH. Antidrug antibody formation in oncology: clinical relevance and challenges. Oncologist. 2016;21(10):1260–8. https://doi.org/10.1634/theoncologist.2016-0061.
Acknowledgements
The authors thank Amy Freyman, PhD, Gina Buckley, MS, and Sarah Ruta Jones, BS, of Pfizer Inc for their assistance with providing and interpreting the clinical trial data.
Funding
Study C1221001 was funded by Hospira Inc, which was acquired by Pfizer Inc in September 2015, and by Pfizer Inc. Study C1221005 was funded by Pfizer Inc. The journal’s Rapid Service and Open Access Fees were funded by Pfizer Inc.
Medical Writing Assistance
Medical writing support was provided by Sue Reinwald, PhD, and Neel Misra, MSc, of Engage Scientific Solutions and funded by Pfizer Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work, and have given their approval for this version to be published.
Authorship Contributions
TBM, H-MY, and JZ contributed to the conception and design of the work and TB and RE were involved in data acquisition. TBM and JST (immunogenicity data), SM (safety data), H-MY (pharmacology data), and JZ (statistical analyses) were involved in data analysis. All authors were involved in data interpretation and developing the manuscript and/or revising it critically for important intellectual content and provided final approval of the submitted version of the work. H-MY is the guarantor for the overall content.
Disclosures
Shahrzad Moosavi, Martin Summers, Jeffrey S. Thomas, Hsuan-Ming Yao, and Jeffrey Zhang are full-time employees of and own stock or options in Pfizer Inc. Thomas B. May and Reginald Ewesuedo were full-time employees of Pfizer Inc at the time of study conduct and hold stock or options in Pfizer Inc. Thomas B. May is now retired. Reginald Ewesuedo is now an employee of Tesaro Inc. Stuart Harris was, and Troy Borema and Jeffrey Levy are employees of Quotient Sciences, a Contract Research Organization, which received funding from Pfizer Inc to perform the studies described in the manuscript. Stuart Harris is now retired.
Compliance with Ethics Guidelines
The protocol and all amendments were approved by the institutional review board at each of the investigational centers (Bellberry Limited, MidLands Independent Review Board, Advarra d/b/a Schulman Institutional Review Board and Schulmam IRB) participating in studies C1221001 and C1221005. Both studies were conducted in compliance with the protocol, International Conference on Harmonisation guidelines, all applicable regulatory requirements, and the 1964 Declaration of Helsinki and its later amendments. Written informed consent was obtained from all individual subjects prior to the performance of any study-specific procedures, and they were permitted to withdraw from the study at any time.
Data Availability
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the USA and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12280886.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Moosavi, S., Borema, T., Ewesuedo, R. et al. PF-06881894, a Proposed Biosimilar to Pegfilgrastim, Versus US-Licensed and EU-Approved Pegfilgrastim Reference Products (Neulasta®): Pharmacodynamics, Pharmacokinetics, Immunogenicity, and Safety of Single or Multiple Subcutaneous Doses in Healthy Volunteers. Adv Ther 37, 3370–3391 (2020). https://doi.org/10.1007/s12325-020-01387-x
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01387-x