
Abstract
Introduction
HyQvia (Immune Globulin Infusion 10% [Human] with Recombinant Human Hyaluronidase) was developed to combine the advantages of intravenous and subcutaneous immune globulin (SCIG), allowing administration of larger volumes at a single subcutaneous site with less frequent dosing when compared to other SCIG products. Current US prescribing guidelines for HyQvia are limited to adults and do not encompass the flexibility required to achieve success in all patients with primary immunodeficiency (PID).
Methods
This retrospective study was designed to evaluate the clinical experience of treating patients with PID with HyQvia regimens outside of package insert recommendations as well as in pediatric patients. Data were abstracted from 38 patient records (317 HyQvia infusions), including five patients less than 16 years of age, from seven US immunology clinics.
Results
Among 37 patients receiving HyQvia regimens differing from prescribing guidelines, the most notable variations included shorter ramp-up periods, use of two rather than one infusion site, and slower than maximal infusion rates to mitigate local adverse events (AEs). The medication volume infused for single site doses ranged from 75 to 200 mL and doses split between two sites ranged from 100 to 750 mL. The most common type of regimen variation was a condensed ramp-up phase (shorter schedule, higher doses), and 96% (24/25) of patients managed in this way completed ramp-up. The most common ramp-up schedule was three infusions (one at 25–45%, another at 50–75%, and the final at 100% of target dose) spread over 2–4 weeks.
Conclusions
A shorter ramp-up schedule did not appear to increase the number of AEs compared to standard ramp-up schedules. For patients with AEs, slower infusion rates and the use of two sites may improve medication tolerability. Four of five pediatric patients reported no AEs, and only one discontinued, stating a fear of needles. HyQvia may be tailored to adults requiring alternative rates, ramp-up, and/or dosing regimens and may be especially well-suited to children.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
HyQvia offers distinct advantages for the treatment of PID, but the current prescribing guidelines do not specify the range of administration options needed to optimize care in all patients, and little data is published on its use in pediatrics. |
A need exists to examine clinical practice experience with administration of HyQvia at variance with current guidelines and use in children. |
The current analysis sought to identify and characterize real-world use of alternative HyQvia regimens in adults as well as HyQvia use in pediatric patients. |
What was learned from the study? |
HyQvia may be used in adults requiring alternative ramp-up and dosing schedules and may be especially well suited to use in children. |
The most notable variations included shorter ramp-up periods, use of two rather than one infusion site, and slower than maximum permitted infusion rates to mitigate local adverse events (AEs). |
Four of the five pediatric patients reported no AEs, and only one discontinued therapy. |
Introduction
Immunoglobulin (IG) replacement therapy, in use for over six decades [1,2,3,4,5,6,7], is well established as an effective treatment for patients with primary immunodeficiency (PID), a group of diseases in which part of the body’s immune system is missing or functioning improperly. IG can be administered either intravenously (IVIG) or subcutaneously (SCIG) and is usually a treatment that continues over the life span of the patient. Although each route of administration has inherent advantages and disadvantages, subcutaneous (SC) treatment has been shown to be as effective as intravenous (IV) treatment and is associated with fewer systemic adverse reactions [8,9,10]. It is also particularly beneficial for patients with limited venous access, as well as for patients who seek to lessen their burden of care by self-administration in a home setting [12,13,14,15]. However, as a result of the smaller volume of IG that can be infused into a single SC site and its lower IG bioavailability compared to IV administration, conventional SC treatments require more frequent dosing (typically weekly as opposed to monthly with IVIG), and multiple injection sites and needle sticks per infusion [16, 17].
HyQvia (Shire US Inc., Lexington, MA, USA), an IG replacement product indicated for the treatment of PID in adults, was developed to combine the advantages of IVIG and SCIG administration. HyQvia is supplied in a dual vial unit containing one vial of immune globulin infusion 10% (human) and one vial of recombinant human hyaluronidase. Hyaluronidase is an enzyme that increases tissue permeability and bulk fluid flow by catalyzing the hydrolysis of hyaluronan, a component of the extracellular matrix. Thus, the hyaluronidase component, administered SC just prior to the IG component, allows for administration of larger volumes of IG at a single SC site and, therefore, less frequent dosing than other SCIG products (i.e., every 3–4 weeks as opposed to weekly). Several studies have confirmed the safety and efficacy of recombinant human hyaluronidase in facilitating the absorption and dispersion of medications in human SC tissues [18,19,20,21].
Clinical trials have demonstrated a high degree of safety and efficacy with the use of HyQvia in patients with PID, including pediatric patients [11, 22, 23]. HyQvia is particularly well suited for patients whose current IVIG or conventional SCIG therapy is unsatisfactory. This includes patients with systemic side effects from IVIG, IVIG patients who require shorter infusion times, patients with difficult IV access or aversion to IV administration, patients who prefer SC administration with the reduced burden of dosing every 3–4 weeks or who prefer receiving SC infusions in the office/infusion center, patients who desire SC administration with fewer needle sticks, and high-risk patients (kidney disease, heart disease, thrombotic risk). HyQvia dosing guidelines for initiation of therapy include a 7-week ramp-up period designed to improve patient tolerability of the large SC doses and facilitate patient training. During this time, the HyQvia dose and rate of infusion, as well as the length of time between infusions, are gradually increased as tolerated until the patient’s target dose and schedule are attained [24]. Following ramp-up, HyQvia is recommended at doses of 300–600 mg/kg at 3- or 4-week intervals for those switching from SCIG, or at the same IG dose and frequency as previous treatments for those switching from IVIG [24] (see Appendix II in the supplementary material for complete dosing recommendations).
While HyQvia offers distinct treatment advantages, its introduction into the PID market is relatively recent (late 2014), and current prescribing guidelines do not encompass the range of administration options needed to optimize care in all patients. Recommendations in the USA are also mainly limited to adults, with little information regarding its use in children. To date, there are two retrospective studies addressing the use of HyQvia in adults and pediatric patients in a real-world setting, but these studies included fewer than 20 patients each and provide little detail on tailored HyQvia therapy [25, 26]. Thus, a need exists to further examine real-world clinical experience with administration of HyQvia at variance with current guidelines and use in children.
The HyQvia Experience Study was a chart review study designed to gather data on HyQvia usage as part of routine medical practice. The current analysis sought to identify and characterize real-world use of alternative HyQvia ramp-up and/or maintenance regimens that differed from those recommended in the package insert. As a secondary objective, use of HyQvia in pediatric patients was evaluated, regardless of dosing regimen.
Methods
Study Design
This was a retrospective chart review of patient data pertaining to HyQvia infusions performed on or before November 16, 2016, from member sites of the Consortium of Independent Immunology Clinics (CIIC). The CIIC is an independent organization comprising board-certified immunologists in private practice who specialize in treating patients with PID (see Appendix I in the supplementary material for a listing of CIIC investigators). Given the retrospective nature of the study, an exemption from full Institutional Review Board review was granted for the study protocol by the North Texas Institutional Review Board for all participating sites. All subject data were de-identified and kept confidential in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice [27]. The investigators and/or clinical staff at each site identified potential candidates for the study whose eligibility was confirmed through a screening process by the lead investigator at the site.
Patient Population
Patients of any age were included if they had a diagnosis of PID requiring IG replacement therapy (identified by relevant ICD-10 codes) and were treated with HyQvia using a regimen that varied from package insert recommendations, i.e., used a medication dose or rate not specified in the prescribing guidelines. Patients less than 16 years old (per Federal Drug Administration [FDA] guidance of pediatric as under 16) were included regardless of HyQvia regimen. Subjects were excluded if they used HyQvia for a diagnosis other than PID.
Data Collection
For each patient, data were collected retrospectively, starting from the time of HyQvia therapy initiation through the end of the data collection period or HyQvia treatment discontinuation, whichever came first. For the purposes of this study, discontinuations of interest were those made for clinical reasons, such as medication intolerance/AEs, and all subsequent mention of “discontinuations” should be interpreted as such. Discontinuations due to insurance and/or financial issues were deemed non-clinician and were not included in this category. Data were hand-recorded onto case report forms on-site and subsequently entered into a secure, electronic, centralized database. Data sources included patient charts (paper and/or electronic medical records), laboratory reports, infusion logs, pharmacy logs, and appointment logs. Data collected included patient demographics (age, gender), relevant medical history (primary diagnosis, concomitant illnesses, comorbid conditions, medications), previous IG treatments (regimen, product, AEs), and details on each HyQvia treatment (regimen and rationale, duration of therapy, dates/number of infusions, site and volume of infusion, dosage and flow rates, AEs, and nonclinical reasons for discontinuation). All reported AEs were categorized as either local or systemic. Coding and relation of AEs to treatment with HyQvia were limited to what was recorded in the patient’s chart and AEs were not interpreted or coded outside of the information directly available upon retrospective assessment.
Data Analysis
A sample size of 20 patients who received a HyQvia regimen at variance with the package insert was planned. For analyses, variations occurring during ramp-up dosing were separated from variations occurring during maintenance dosing. Variations were further divided into categories based on whether or not the variation in treatment was a change in dose, rate, schedule, or a combination of the three from the recommendations found in the package insert (see Appendices II and III in the supplementary material for official HyQvia ramp-up and dosing guidelines and definitions of variations). Most outcomes were analyzed by age group, at least 16 years of age (hereafter, “adults”) and less than 16 years of age. Descriptive statistics were used to characterize endpoints of interest. Patients were categorized according to whether or not they discontinued HyQvia for a clinical reason. The one-tailed Fisher’s exact test was used to compare the percentages of patients who discontinued therapy with those who did not discontinue with respect to reported AEs (by category: any AE, local AE, systemic AE).
Results
Patient Population
A total of 38 patients from seven CIIC sites met the criteria for this analysis, 37 of whom had some type of regimen variation from the HyQvia package insert, and one who was included only for being less than 16 years old (no regimen variation). The majority of patients were female (71.1%) and the average (standard deviation [SD]) age was 45 (21.6) years (min, max: 8, 78 years) (Table 1). There were five (13.2%) patients under the age of 16. One patient was pregnant while using HyQvia. The most common diagnosis was some form of common variable immune deficiency (CVID), which was recorded for 27 (71.1%) patients, followed by antibody deficiency with near normal immunoglobulins recorded in 5 (13.2%) patients. Prior to treatment with HyQvia, 14 (36.9%) patients had received IVIG treatment, 19 (50.0%) had used SCIG, and 5 (13.2%) were naïve to IG treatment.
Infusions
Dose and administration details were available for a total of 317 individual HyQvia infusions among the 38 patients. Of these 317 infusions, 311 occurred in the physician’s office, three occurred at a freestanding infusion center, and three were done by the patient at home. A total of 224 doses (70.7%) were administered using two infusion sites, 73 (23.0%) were administered using one site, and 3 (0.9%) were administered using three sites (number of sites not recorded for 17 infusions). For doses infused at one site, the mean (SD) was 13.8 (8.8) g (138 [88] mL) and ranged from 7.5 to 20 g (75–200 mL). For doses infused at two sites, the mean (SD) total dose was 42.2 (12.9) g (422 [129] mL) and ranged from 10.0 to 75.0 g (100–750 mL).
Most of the patients (n = 23; 60.5%) started out using one infusion site and later switched to two infusion sites. Six (15.8%) patients used two sites for all infusions; three patients used one site for all infusions, two of whom were pediatric (both 10 years old). One patient started using two infusion sites and went down to one site before discontinuing. One patient varied from one to three sites per infusion. Four patients, including two pediatric patients, did not have sufficient information to allow for characterization of infusion site patterns.
HyQvia Regimen Variations: Ramp-Up
Figure 1a illustrates the nature of HyQvia regimen variations during ramp-up. All of the 35 patients with regimen variations had some type of variation during ramp-up. The most common variation (n = 25) was a condensed ramp-up regimen, characterized by higher doses and a shorter ramp-up schedule as compared to those recommended in the HyQvia package insert. For the patients who used a shorter ramp-up period, the most common dosing schedule was three ramp-up infusions spread over 2–4 weeks, with the patient receiving 25–45% of their target dose for the first infusion, 50–75% of their target dose for the second infusion, and 100% of their target dose for the third and final ramp-up infusion. Four patients used a two-dose ramp-up regimen spread over 1–3 weeks, receiving 30–50% of their target dose at the first infusion and the full target dose at the second and final ramp-up infusion. One patient skipped ramp-up and went straight to full maintenance dosing. Six of the patients using a condensed ramp-up regimen also used a slower infusion rate than recommended in the package insert, while three patients used a faster than recommended infusion rate.

Nature of HyQvia ramp-up (a) and maintenance (b) variations by discontinuationa status in each phase. a Discontinuations for clinical reasons. b Two non-discontinuation patients in this category used a faster infusion rate than recommended; six non-discontinuation patients in this category used a slower infusion rate than recommended. c All four patients in this category used a slower infusion rate than recommended. d One non-discontinuation patient in this category also used a slower infusion rate. e One patient in this category also infused at a slower rate
Four patients used a longer than recommended ramp-up schedule, with lower doses per infusion over a longer period of time; none of these patients discontinued during ramp-up. One patient used a longer than recommended ramp-up schedule with higher than recommended doses and did not discontinue. There were six patients whose only variation during ramp-up was a slower rate of HyQvia infusion per dose. One patient underwent ramp-up with recommended doses and infusion rates, but according to a more gradual schedule with longer intervals between doses.
Three patients discontinued during ramp-up. One followed a condensed ramp-up regimen and discontinued due to headaches, which this patient had also reported during prior IVIG therapy. The ramp-up variations of the other two patients who discontinued treatment were characterized by slower infusion rates only, and these patients discontinued because of AEs (anaphylactic-type reaction; headache/vomiting).
HyQvia Regimen Variations: Maintenance Phase
Thirty-five of the 38 patients (92.1%) reached the HyQvia maintenance phase, 18 of whom received maintenance regimens consistent with package insert recommendations. Only one of these 18 (5.6%) patients discontinued during the maintenance phase (infusion site swelling was unacceptable).
Fifteen patients received HyQvia maintenance therapy using a regimen that varied in some way from package insert recommendations (Fig. 1b). In nine patients, the only variation from package insert recommendations was a slower HyQvia infusion rate, and five of these patients discontinued during maintenance therapy. An additional three patients used a slower infusion rate along with higher than recommended doses, and each of these patients discontinued. Three patients used lower doses than recommended in the package insert; one of these patients discontinued. One of the patients using a lower dose also used slower infusion rates (did not discontinue). Two patients used a regimen that varied only in the use of shorter intervals between doses; both of these patients discontinued.
All slower rates were due to AEs; infusion site discomfort or leakage or headaches while using faster infusion rates. Upon slowing the rate, all patients reported a decrease in AEs and continued at this slower rate for all subsequent HyQvia infusions.
Drug-Related Adverse Events
Overall, 29 (76.3%) patients experienced one or more AE (Table 2). Of the 15 patients who discontinued HyQvia treatment, a total of 14 (93.3%), reported AEs, while 15 (65.2%) of the 23 patients who did not discontinue reported AEs.
Local AEs were reported by 23 (60.5%) patients. The most common local AE was erythema (42.1%), followed by pain/burning during infusion (18.4%), itching (15.8%), excessive swelling (13.2%), and pain at the infusion site (13.2%). Systemic AEs were reported by 63.2% of all patients, and 86.7% of those who discontinued. The most common systemic AEs were headache (47.4%), malaise (26.3%), and gastrointestinal disorders (e.g., nausea/vomiting/diarrhea) (15.8%).
Fourteen of 29 patients who experienced any AE discontinued therapy, while one of nine patients who experienced no AEs discontinued therapy (48.3% vs 11.1%; P = 0.0501). Ten of 23 patients who experienced local AEs discontinued therapy as compared to five of 15 patients who did not experience local AEs (43.5% vs 33.3%; P = 0.3897). Thirteen of 24 patients who experienced a systemic AE discontinued therapy, as compared to two of 14 patients who did not experience a systemic AE (54.2% vs 14.3%; P = 0.0165).
Of the 29 patients reporting an AE during treatment with HyQvia, 17 had also experienced AEs while on previous IG therapy. Eleven of these patients experienced HyQvia-related AEs (most commonly headache) that were similar to the AEs associated with previous IVIG treatment.
Pediatric Patient Summary
This study included five patients under the age of 16 years, four of whom received HyQvia using a dosing regimen that varied from the recommendations in the package insert. One pediatric patient was administered HyQvia ramp-up and maintenance at recommended doses and intervals, but at a slower infusion rate (150 mL/h due to discomfort at 300 mL/h); this patient reported pain/burning during the hyaluronidase infusion, pain at the site, and discomfort at the site. This patient was the only pediatric patient to discontinue treatment with HyQvia, stating fear of needles/anxiety over self-administration as the main reason.
Three pediatric patients were administered HyQvia ramp-up at higher doses than recommended in the package insert and over a shorter timeframe. One of these also had faster than recommended infusion rates during ramp-up. All three received maintenance regimens consistent with the package insert, none of them reported any AEs, and none of them discontinued HyQvia. The fifth pediatric patient received HyQvia ramp-up and maintenance according to package insert recommendations, reported no AEs, and did not discontinue treatment.
Discussion
In the pivotal clinical trial [11] and long-term extension [23] for HyQvia, all patients followed the recommended ramp-up period, and the majority of patients (97.8%) received HyQvia without changes in administration (including no flow rate reductions, interruptions in treatment, or discontinuations related to tolerability or AEs) [28]. In real-world practice, there may be situations or preferences that lead to decisions regarding modified dosing regimens of HyQvia. Yet, there is little published data describing the use of HyQvia outside of the official prescribing guidelines. Further, while the clinical studies of HyQvia included 26 patients under the age of 18 years, including 13 patients between the ages of 4 and 12 [22], real-world pediatric data always has clinical value, particularly in rare diseases. The findings of the current observational study contribute to the growing body of knowledge on the individualized use of HyQvia to treat PID in real-world situations in both adult and pediatric patients.
The most common type of regimen variation noted in this study was a condensed ramp-up phase (shorter schedule with higher doses), and 96% (24/25) of patients managed in this way completed ramp-up. The most common ramp-up schedule followed was that of three infusions (one at 25–45% of target dose, another at 50–75% of target dose, and the final at 100% of target dose) spread over 2–4 weeks. Several patients had a successful two-dose ramp-up (50% of target dose, followed by 100% of target dose); one patient skipped ramp-up entirely and went straight to target dose (30 g) with no apparent adverse consequences. Overall, the patients following a shorter ramp-up schedule did not appear to have an increased number of AEs when compared to those who followed the standard ramp-up schedule. In fact, only one patient out of 25 who underwent a condensed ramp-up phase discontinued during ramp-up, and this was related to headaches, which had also been reported by this patient during prior IVIG therapy. The 7-week ramp-up period recommended in the HyQvia package insert was designed to improve patient satisfaction with HyQvia treatment. By gradually increasing the HyQvia dose and lengthening the time between infusions in a stepwise approach, patients are given time to adjust to the large SC doses while simultaneously gaining comfort in HyQvia administration. When considering that the current PID recommendations for HyQvia ramp-up require a substantial time commitment from patients and include multiple provider visits, use of a shorter schedule carries the potential to broaden the pool of patients willing to start HyQvia. A condensed ramp-up may be especially useful for patients transitioning from conventional SCIG because they are already comfortable with subcutaneously self-administered IG.
Prescribing guidelines recommend the use of one infusion site, with use of a second site at the discretion of the physician and patient on the basis of tolerability and total volume [24]. The real-world data collected in this study suggest a trend for using two infusion sites as the dose of HyQvia increased (usually by the third infusion for the majority of patients). The volume of medication infused for single site doses ranged from 75 to 200 mL and doses split between two sites ranged from 100 to 750 mL. Only three patients, two of whom were pediatric, used one site for every infusion. Most patients (61%) progressed from one to two infusion sites, while 16% used two sites for every infusion. In another real-world study, only 62% of patients using HyQvia initiated treatment using a single infusion site, seven of whom increased the number of infusion sites after completing dose ramp-up to lessen the volume per infusion site [25]. A preference to use two sites for larger HyQvia doses may also reflect a desire for shorter infusion duration, improved patient comfort, or both. Pediatric patients, however, may be better suited for a single infusion site, as they generally require lower doses as compared to adults and are more likely to suffer from anxiety related to multiple needle sticks. In clinical trials, 82.5% of all HyQvia infusions in children aged less than 18 years were completed using a single infusion site [22]. Among the three pediatric patients in the current study with infusion site information, only one (15 years old) used more than a single infusion site; this patient switched from one to two sites only after the target dose of 30 g was reached.
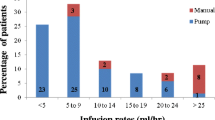
After the first two HyQvia infusions, the official prescribing guidelines recommend increasing the flow rate, as tolerated, to 300 mL/h per site for subjects weighing at least 40 kg and 160 mL/h per site in patients weighing less than 40 kg [24]. The infusion rates used in the HyQvia phase 3 trial followed these same weight-based recommendations. Accordingly, by age group, the median maximum infusion rates used in that study were 300 mL/h (range 10–300 mL/h) in subjects aged at least 12 years and 160 mL/h (range 80–300 mL/h) in patients aged 2–12 years, with few variations in flow rate throughout treatment [11]. The flow rate was reduced, interrupted, or stopped in only 1.7%, 0.4%, and 0.2% of infusions, respectively, in response to patient tolerability or AE issues [11].
In the current study, 17 patients (51.2%) aged 16 and older and one patient (20%) aged less than 16 years received HyQvia at a rate that was slower than the recommendations in the package insert at some point in therapy. In all cases, the rate was lowered after initially receiving HyQvia at the recommended maximum rate (either during a previous infusion or during the current infusion) as a result of AEs, including headache, site discomfort, and site swelling. Following the slowing of the infusion, all adult patients reported a decrease in their AE(s) (both immediately and post-infusion), and these patients continued to receive treatment during subsequent visits at a slower rate with a noticeable reduction in their reported AEs. This observation may be beneficial in guiding the treatment of patients who are experiencing AEs during treatment with HyQvia. For these patients, individualization of the maximum rate reached per site may improve comfort, tolerability, and treatment adherence. It should be noted, however, that when a slower infusion rate was the sole variation in the HyQvia regimen, about half of such patients discontinued therapy, whether during ramp-up (two of four patients) or maintenance (five of nine patients). Further, all three patients who used a slower infusion rate along with a higher than recommended dose during maintenance discontinued therapy.
It is worth noting that 18 patients used maintenance regimens that were consistent with the package insert, and only one of those patients discontinued (6%), suggesting that the package insert maintenance recommendations are acceptable for most patients. Of the 17 patients who had some type of maintenance variation, 11 (65%) discontinued. It is likely that the patients who opted for some type of maintenance variation had some preexisting concerns about treatment that may have made them more likely to discontinue therapy.
The patient group in this study included some of the earliest users following FDA approval of HyQvia. Given that HyQvia was designed with the intent to minimize common systemic reactions to traditional IVIG treatment, it is likely that early adopting physicians may have chosen patients to receive HyQvia in part because of systemic tolerability problems with previous treatments. This could have thereby increased the probability of systemic AEs among the patients in this cohort. In fact, 17 (60.7%) patients in this study reporting an AE during treatment with HyQvia reported AEs while receiving their previous treatment, and 11 (39.3%) reported similar AEs while receiving HyQvia as compared to their previous treatment. On the basis of the available data on treatment with HyQvia in this study, clinical trials, and extension studies, it is likely that strategies focused on the alleviation of local side effects (such as the use of two infusion sites to reduce site swelling, slower infusion rates, the use of heating/cooling packs, etc.) may be useful in increasing patient tolerability and adherence to treatment with HyQvia. Analysis of AE data suggested that patients who experienced systemic AEs were statistically more likely to have discontinued therapy; however, it should be noted that this study was not designed or powered to robustly evaluate the influence of AEs on discontinuation.
The experiences of the five patients under the age of 16 years included in this study support the safety and tolerability of HyQvia in pediatric patients under the age of 16 years, only one of whom discontinued treatment because of anxiety over needles. At the time of manuscript preparation, four patients were actively using HyQvia with no reports of efficacy, safety, or tolerability problems to date. These findings parallel those described previously by Wasserman et al. [22], who reported low rates of infection, minimal local and systemic reactions, and well-tolerated infusions of HyQvia in 12 patients aged less than 18 years. The findings of this real-world study suggest that HyQvia may be especially well suited to both address the unique needs of pediatric patients with PID and reduce the treatment burden of patients and caregivers. Surveys of both patients and caregivers using HyQvia indicate that over half of all adults and all responding pediatric patient caregivers prefer HyQvia to IVIG or conventional SCIG treatment [22, 23]. Unlike conventional SCIG, HyQvia can be administered once a month, and, as the actual dose in g (mL) in children is lower than adults, most children can be treated with a single infusion site. This decreases the number of needle sticks and infusions per month for an age group in which needle phobia and frequent infusions may be a prominent deterrent to treatment adherence. Of the three pediatric patients with information regarding number of infusion sites, two 10-year-old patients in this study completed all infusions using a single site every 3–4 weeks, while a 15-year-old patient progressed from one to two sites over time. Prior studies have reported use of a single infusion site in 83% and 70% of pediatric patients using HyQvia [22, 26].
Given the study’s retrospective and real-world nature, all findings described herein are subject to several limitations and are not designed to serve as an evaluation of drug efficacy or safety. These limitations include a small sample size, non-randomization (allowing for possible selection bias), lack of a direct control group (data gathering was limited only to those patients either receiving HyQvia outside of prescribing guidelines or younger than the age of 16), incompleteness of chart data for outcomes of interest, and data restricted to objective findings, thereby precluding evaluations of patient satisfaction. Collection of safety data was limited to information previously recorded during routine patient follow-up visits, and, as such, the coding and drug reaction probability score of AEs was not captured. Additionally, as there was no control group, it is impossible to determine if the AEs and discontinuations experienced by the reported cohort are in any way correlated to their alternative HyQvia regimens.
Conclusions
Despite the noted limitations, the experience shared in this small, clinical practice, observational study has clinical value, suggesting that HyQvia ramp-up and administration specifics can be tailored to individual patient needs and desires; however, as with any mode of IG administration, individual patients may choose to discontinue regardless of efforts to accommodate apparent patient needs. An alternative ramp-up schedule may be especially well suited to adults with limited availability while also addressing the special needs of the pediatric population and reducing the treatment burden of caregivers. While these data should not take precedence over those from controlled clinical studies or official prescribing recommendations, the findings may be useful for clinicians whose patients require individualized treatment strategies in real-world situations.
References
Eley B. An update of the primary antibody disorders. Curr Allergy Clin Immunol. 2008;21(1):13–9.
Wasserman RL. Progress in gammaglobulin therapy for immunodeficiency: from subcutaneous to intravenous infusions and back again. J Clin Immunol. 2012;32(6):1153–64.
McCormack PL. Immune globulin subcutaneous (human) 20%: in primary immunodeficiency disorders. Drugs. 2012;72(8):1087–97.
McCormack PL. Immune globulin (human) 10% liquid: a review of its use in primary immunodeficiency disorders. BioDrugs. 2013;27(4):303–400.
Lucas M, Lee M, Lortan J, Lopez-Granados E, Misbah S, Chapel H. Infection outcomes in patients with common variable immunodeficiency disorders: relationship to immunoglobulin therapy over 22 years. J Allergy Clin Immunol. 2010;125(6):1354.e4–1.
Bruton OC. Agammaglobulinemia. Pediatrics. 1952;9(6):722–8.
Perez EE, Orange JS, Bonilla F, et al. Update on the use of immunoglobulin in human disease: a review of evidence. J Allergy Clin Immunol. 2017;139(3S):S1–46.
Gardulf A, Hammarström L. Subcutaneous administration of immunoglobulins. Clin Immunother. 1996;6:108–16.
Berger M. Subcutaneous immunoglobulin replacement in primary immunodeficiencies. Clin Immunol. 2004;112(1):1–7.
Wasserman RL, Melamed I, Kobrynski L, et al. Efficacy, safety, and pharmacokinetics of a 10% liquid immune globulin preparation (GAMMAGARD LIQUID, 10%) administered subcutaneously in subjects with primary immunodeficiency disease. J Clin Immunol. 2011;31(3):323–31.
Wasserman RL, Melamed I, Stein MR, et al. Recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulins for primary immunodeficiency. J Allergy Clin Immunol. 2012;130(4):951–7.e11.
Garcia-Lloret M, McGhee S, Chatila TA. Immunoglobulin replacement therapy in children. Immunol Allergy Clin North Am. 2008;28(4):833–49.
Gardulf A, Hammarström L, Smith CI. Home treatment of hypogammaglobulinaemia with subcutaneous gammaglobulin by rapid infusion. Lancet. 1991;338(8760):162–6.
Fasth A, Nyström J. Quality of life and health-care resource utilization among children with primary immunodeficiency receiving home treatment with subcutaneous human immunoglobulin. J Clin Immunol. 2008;28(4):370–8.
Nicolay U, Kiessling P, Berger M, et al. Health-related quality of life and treatment satisfaction in North American patients with primary immunedeficiency diseases receiving subcutaneous IgG self-infusions at home. J Clin Immunol. 2006;26(1):65–72.
Misbah S, Sturzenegger MH, Borte M, et al. Subcutaneous immunoglobulin: opportunities and outlook. Clin Exp Immunol. 2009;158(Suppl 1):51–9.
Ochs HD, Gupta S, Kiessling P, Nicolay U, Berger M, Subcutaneous IgG Study Group. Safety and efficacy of self-administered subcutaneous immunoglobulin in patients with primary immunodeficiency diseases. J Clin Immunol. 2006;26(3):265–73.
Yocum RC, Kennard D, Heiner LS. Assessment and implication of the allergic sensitivity to a single dose of recombinant human hyaluronidase injection: a double-blind, placebo-controlled clinical trial. J Infus Nurs. 2007;30(5):293–9.
Harb G, Lebel F, Battikha J, Thackara JW. Safety and pharmacokinetics of subcutaneous ceftriaxone administered with or without recombinant human hyaluronidase (rHuPH20) versus intravenous ceftriaxone administration in adult volunteers. Curr Med Res Opin. 2010;26(2):279–88.
Allen CH, Etzwiler LS, Miller MK, et al. Recombinant human hyaluronidase-enabled subcutaneous pediatric rehydration. Pediatrics. 2009;124(5):e858–67.
Thomas JR, Yocum RC, Haller MF, von Gunten CF. Assessing the role of human recombinant hyaluronidase in gravity-driven subcutaneous hydration: the INFUSE-LR study. J Palliat Med. 2007;10(6):1312–20.
Wasserman RL, Melamed I, Kobrynski L, et al. Recombinant human hyaluronidase facilitated subcutaneous immunoglobulin treatment in pediatric patients with primary immunodeficiencies: long-term efficacy, safety and tolerability. Immunotherapy. 2016;8(10):1175–86.
Wasserman RL, Melamed I, Stein MR, et al. Long-term tolerability, safety, and efficacy of recombinant human hyaluronidase-facilitated subcutaneous infusion of human immunoglobulin for primary immunodeficiency. J Clin Immunol. 2016;36(6):571–82.
HYQVIA [prescribing information]. Westlake Village, CA: Baxalta US Inc. https://www.shirecontent.com/PI/PDFs/HYQVIA_USA_ENG.pdf.
Rosenbach KP, Rabbat CJ, Rozen L, Hughes SM. Real-world use of recombinant human hyaluronidase-facilitated subcutaneous infusion of immunoglobulin (IGHy; HYQVIA) in patients with primary immunodeficiency disorders (PID). Presented at: Clinical Immunology Society. 2016 April 14–17, Boston, MA.
Gruenemeier P, Ernst C, White C, Duff K. Real-world pediatric experience with hyaluronidase-facilitated subcutaneous immunoglobulin (IGHY) infusion parameters [Abstract]. Ann Allergy Asthma Immunol. 2016;117(5):S98.
International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. ICH Harmonised Tripartite Guideline. Guideline for Good Clinical Practice E6(R1). June 1996. http://www.ich.org/products/guidelines/efficacy/efficacy-single/article/good-clinical-practice.html. Accessed Oct 8 2015.
Wasserman RL. Overview of recombinant human hyaluronidase-facilitated subcutaneous infusion of IgG in primary immunodeficiencies. Immunotherapy. 2014;6(5):553–67.
Acknowledgements
The full list of the CIIC study investigators can be found within the supplementary material.
Funding
This study and medical writing assistance was funded by an unrestricted research grant to CIIC from Takeda, who will also be funding the journal’s Rapid Service and Open Access fees.
Medical Writing and Editorial Assistance
Study design, protocol development, and data collection and analysis assistance was provided by Frank Rodino, MHS, PA and Rachel Hathcock, RN, BSN of Churchill Outcomes Research, LLC; medical writing and editorial assistance were provided by Rachel Hathcock, RN, BSN and Sandra Westra, Pharm.D., of Churchill Communications, LLC, Maplewood, NJ. This assistance was funded through an unrestricted research grant from the Baxalta, now part of the Takeda group of companies.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Dr. Richard Wasserman has served as a consultant for Takeda, EvolveBio, and Korean Green cross; he has served as a speaker for Takeda and CSL Behring; he has served as an investigator for CSL Behring, EvolveBio, Prometic, Kedrion, Korean Green Cross, and Takeda.
Compliance with Ethics Guidelines
Given the retrospective nature of the study, an exemption from full IRB review was granted for the study protocol by the North Texas Institutional Review Board for all participating sites. All subject data were de-identified and kept confidential in accordance with the International Conference on Harmonization Guidelines for Good Clinical Practice [27]. The investigators and/or clinical staff at each site identified potential candidates for the study whose eligibility was confirmed through a screening process by the lead investigator at the site.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.11847387.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Wasserman, R.L., HyQvia Experience Study Group. Clinical Practice Experience with HyQvia in Adults Using Alternative Dosing Regimens and Pediatric Patients: A Retrospective Study. Adv Ther 37, 1536–1549 (2020). https://doi.org/10.1007/s12325-020-01264-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-020-01264-7