
Abstract
Introduction
The mineralocorticoid receptor (MR) blocker esaxerenone is a new treatment for hypertension in Japan and under development for treatment of diabetic nephropathy. Hepatic impairment is known to impact the pharmacokinetics (PKs) of other MR blocking drugs. The aim of the present study was to characterise the PKs and safety of a single oral dose of esaxerenone in Japanese subjects with mild–moderate hepatic impairment.
Methods
In this open-label, parallel-group study, subjects with mild (Child–Pugh grade A) or moderate (grade B) hepatic impairment, and healthy controls with normal hepatic function matched by age and BMI (all groups n = 6), received a single 2.5-mg oral dose of esaxerenone. Plasma concentrations were measured by liquid chromatography–tandem mass spectrometry, and PK parameters were calculated using non-compartmental analysis.
Results
Geometric least-squares mean (GLSM) ratios (90% confidence intervals [CIs]) for area under the plasma concentration–time curve (up to the last quantifiable time, up to infinity) in subjects with mild hepatic impairment versus normal hepatic function were 0.837 (0.637, 1.099) and 0.824 (0.622, 1.092), respectively. Corresponding values for moderate hepatic impairment versus normal hepatic function were 1.078 (0.820, 1.415) and 1.098 (0.829, 1.454). GLSM ratios (90% CIs) for peak plasma concentration (Cmax) were 0.959 (0.778, 1.182) for mild hepatic impairment versus normal hepatic function and 0.804 (0.653, 0.992) for moderate hepatic impairment versus normal hepatic function. Time to Cmax and clearance values were comparable between groups. The incidence of adverse events (AEs) was 16.7% in the moderate hepatic impairment and normal hepatic function groups. One serious AE (hepatic encephalopathy) occurred in one subject with moderate hepatic impairment.
Conclusions
Mild to moderate hepatic impairment had no clinically relevant effect on esaxerenone exposure. Esaxerenone dosage adjustment based on PKs is unlikely to be needed in patients with mild to moderate hepatic impairment.
Trial Registration
JapicCTI-163339.
Funding
Daiichi Sankyo Co., Ltd.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Safety concerns related to increased exposure to eplerenone, a mineralocorticoid receptor (MR) blocker used to treat hypertension and heart failure, have been identified in patients with mild to severe hepatic impairment, which has led to advised caution or limitation of its use in these patients. |
Esaxerenone, a novel non-steroidal MR blocker, has a favourable pharmacokinetic (PK) profile for daily oral treatment in healthy subjects. |
This study aimed to examine the PK and safety profile of a single oral dose of esaxerenone in Japanese subjects with mild to moderate hepatic impairment. |
What was learned from the study? |
Mild to moderate hepatic impairment did not have any clinically relevant effect on esaxerenone exposure and the drug was safe in this subject population. |
Esaxerenone can be administered at unadjusted dosages to subjects with mild to moderate hepatic impairment. |
Because of this, side effects observed with other drugs in this class of MR blockers can likely be avoided during treatment. |
Introduction
Mineralocorticoid receptor (MR) blockers are used to treat hypertension and oedematous conditions such as heart failure and hepatic cirrhosis [1, 2]. Currently available MR agents include spironolactone and eplerenone, both of which improve prognoses in patients with heart failure [3,4,5]. However, use of these MR agents is limited by adverse events (AEs) such as hyperkalaemia [6, 7], suggesting a need for new treatment options.
Esaxerenone (CS-3150) is a novel, oral, non-steroidal MR blocker approved for the treatment of hypertension and currently in development for the treatment of diabetic nephropathy in Japan [8]. In a pharmacokinetic (PK) study in healthy Japanese subjects [9, 10], esaxerenone plasma concentrations were generally proportional to dose, with steady-state levels achieved at day 4 of once-daily dosing. The time (tmax) to peak plasma concentration (Cmax) of esaxerenone was approximately 2.5–3.5 h, and the half-life (t1/2) of esaxerenone was approximately 20 h. Phase 1 studies have shown that esaxerenone is suitable for daily dosing without safety and tolerability concerns [9, 10].
Optimal functioning of the liver is important for drug elimination, and chronic liver disease is linked to various factors that lower drug-metabolising activity [11]. Hepatic impairment reduces hepatic blood flow, leading to reduced plasma clearance of drugs and altered plasma protein binding, which in turn influences the PKs of drugs [12]. Portal systemic shunting, as seen in advanced hepatic impairment, can considerably increase drug exposure by reducing the first-pass effect [12]. Changes in metabolising enzyme activity can also influence drug clearance and cause PK variations when hepatic function is impaired [12].
Several studies have examined the effect of hepatic impairment on the PKs of existing MR blockers [13,14,15]. When a single oral 200-mg dose of spironolactone was administered to five patients with chronic liver disease, the mean t1/2 of the active metabolite canrenone was 59.4 h, which was about three times longer than the t1/2 in healthy adults [15]. In individuals with moderate hepatic impairment, the area under the plasma concentration–time curve (AUC0–24) for eplerenone at steady state was 42% higher than in healthy adults [14]. Therefore, the prescribing information for eplerenone advises caution in patients with mild or moderate hepatic impairment [16]. A previous study has shown that esaxerenone has good bioavailability (approximately 90%) [10] and is primarily excreted as metabolites into the urine and faeces [17]. The route of esaxerenone clearance is primarily via metabolism involving oxidation by cytochrome P450 (CYP) 3A4/3A5, glucuronidation by several isozymes of uridine diphosphate-glucuronosyltransferase (UGT), and hydrolysis [17]. On the basis of these data, it is important to evaluate the effect of hepatic impairment on esaxerenone clearance. Therefore, this study investigated the PKs and safety of a single oral 2.5-mg dose of esaxerenone in Japanese subjects with mild or moderate hepatic impairment.
Methods
Study Design and Treatments
This multicentre, single-arm, open-label, parallel-group study examined the effects of mild and moderate hepatic impairment on the PK profile of a single oral 2.5-mg dose of esaxerenone (2 × 1.25-mg tablets; Daiichi Sankyo Co., Ltd, Tokyo, Japan).
Eligible subjects were identified at a screening examination 2–30 days before esaxerenone administration. Subjects were admitted to hospital the day before esaxerenone administration and then remained at the study site for three additional days after dosing for a total of 5 days and 4 nights. Assessments were performed at admission, before esaxerenone administration, and after administration but before discharge. Esaxerenone was administered on the second day of hospitalisation. Subjects were required to fast for at least 10 h on the day of dosing, then consume a standard meal [18]. Thirty minutes after eating, each subject took a single oral dose of esaxerenone with 200 mL of water. No other beverages were permitted for 1 h before or 2 h after dosing. Caffeinated drinks were prohibited during hospitalisation, and subjects were only permitted food provided at a predetermined time and prepared at the study centre. Two subsequent assessments were performed, the first on day 5 at discharge, and the second 10–14 days after esaxerenone administration (post-study examination).
The study was conducted in accordance with Good Clinical Practice guidelines, as defined by the International Conference on Harmonisation, and ethical principles outlined in the Declaration of Helsinki. The study was registered with JAPIC Clinical Trials Information (http://www.clinicaltrials.jp; JapicCTI-163339). The study received institutional review board approvals from ETHIPRO, Montreal, QC, Canada (IORG0000689); the Clinical Trials Review Committee of Hakata Clinic (1464P3S-6); and the Clinical Trials Review Committee of Kurume Clinical Pharmacology Clinic (approved on 22 August 2016; reference number not stated). All subjects provided written informed consent before inclusion in the study.
Study Subjects
Japanese male and female subjects who met the following criteria were included in the study: age 20 years or more; body mass index (BMI) less than 30 kg/m2 at screening; ability of smokers to quit smoking during hospitalisation and at each study visit; and no confirmed pregnancy for female subjects. Subjects were classified into three groups based on hepatic function at the screening examination, as determined by aspartate transaminase (AST) and alanine transaminase (ALT) levels, and Child–Pugh classification [19]: normal hepatic function (AST and ALT less than two times the upper limit of normal [ULN]); mild hepatic impairment (Child–Pugh grade A [score 5–6]); or moderate hepatic impairment (Child–Pugh grade B [score 7–9]). Healthy adult subjects with normal hepatic function were matched by age and BMI to the patients with hepatic impairment.
Exclusion criteria included any subject who had undergone whole blood collection, plasmapheresis, or platelet apheresis after screening (except blood collection for retesting); who was breastfeeding; who could not take medically reliable contraception from the time of screening to 12 weeks after the last study visit; who had an estimated glomerular filtration rate (eGFR) less than 60 mL/min/1.73 m2 at admission; who had used or planned to use prohibited concomitant drugs; who had consumed grapefruit (juice or pulp) within 7 days before esaxerenone administration; who had any previous serious disease affecting thyroid, central nervous system, respiratory, blood/haematopoietic, gastrointestinal, pituitary, or adrenal function; who had laboratory values outside reference ranges (except for hepatic function) or clinically relevant symptoms; or who had an abnormal electrocardiogram on admission. Additional exclusion criteria for subjects with mild or moderate hepatic impairment included the presence of ascites that required invasive treatment, cholestatic liver disease, or any change in medication from admission to the post-study examination.
Subjects with normal hepatic function were prohibited from taking other drugs or supplements from 14 days before esaxerenone administration until completion of the post-study assessments. Subjects with mild or moderate hepatic impairment could not receive the following medications from 2 days before dosing until completion of the post-study assessments: K+-sparing diuretics, K+ preparations, insulin preparations, ion-exchange resins, and blood products. Additionally, drugs associated with inhibition or induction of CYP3A were contraindicated from 14 days before esaxerenone administration until after the post-study assessments. Other investigational drugs were prohibited from 120 days before dosing until completion of the post-study assessments.
PK Assessments
Full details of plasma esaxerenone concentration measurement have been described previously [9]. Blood samples were collected into a vacuum tube containing ethylenediaminetetraacetic acid dipotassium salt, before, and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 48, 72, and 120 h after esaxerenone administration. Drug concentrations were measured by liquid chromatography–tandem mass spectrometry (LC–MS/MS). Chromatographic separation was performed using a CAPCELL PAK C18 MGIII (Shiseido Co., Ltd.; Tokyo, Japan) column (2.0 × 150 mm, 5 μm). Detection was performed using an API 4000 (AB SCIEX, Framingham, Massachusetts, USA) tandem mass spectrometer with TurboIonSpray source by electrospray ionisation in the negative ion mode and multiple-reaction monitoring of esaxerenone (m/z 465–365) and its internal standard (m/z 472–370) as described previously [9]. For esaxerenone test samples of 0.3, 4.0, and 80.0 ng/mL, intra-study assay precision was 3.7%, 3.7%, and 3.5%, respectively. Accuracy of the assay ranged from 0.3% to 1.0%, with a lower limit of quantification of 0.1 ng/mL.
The primary endpoint of the study was esaxerenone PKs, based on plasma concentrations using a non-compartmental method: Cmax, AUC up to the last quantifiable time (AUClast), AUC up to infinity (AUCinf), tmax, t1/2, apparent volume of distribution based on the terminal phase (Vz/F), and apparent total body clearance (CL/F).
Phoenix® WinNonlin® (version 6.3, Certara USA Inc., Princeton, NJ, USA) was used for PK parameter calculation.
Safety Assessments
Safety assessments were performed at various time points throughout the study and included vital signs (blood pressure, pulse rate, and body temperature), laboratory tests, body weight, and electrocardiogram findings. AEs were categorised in accordance with the Medical Dictionary for Regulatory Activities (MedDRA/J Ver. 14.0) System Organ Class and Preferred Terms for the individual AEs. MedDRA® the Medical Dictionary for Regulatory Activities terminology is the international medical terminology developed under the auspices of the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
Statistical Analyses
The planned sample size of 18 subjects, six in each subgroup, was based on guidance from the United States Food and Drug Administration [20] and the European Medicines Agency [21]. The PK analysis set comprised subjects who received a single dose of the study drug, who had no serious study protocol violation, and who had samples and measurements available for each assessment. The safety analysis set comprised subjects who received a single dose of the study drug.
Baseline values were defined as the last non-missing measurement (including repeated and unscheduled measurements) before the study drug dose, unless otherwise specified. To assess the effect of hepatic impairment (mild or moderate) versus normal hepatic function, an analysis of variance (ANOVA) was used with hepatic impairment as a fixed effect. Geometric least-squares mean (GLSM) ratio and 90% confidence interval (CI) values for AUCinf, AUClast, and Cmax were calculated. Scatter plots were created for CL/F with respect to hepatic function (Child–Pugh score and albumin concentration).
Statistical analysis was performed using SAS (version 9.2, SAS Institute, Cary, NC, USA).
Results
Subject Disposition
A total of 18 subjects were enrolled in the study, six in each group. Except for hepatic function, baseline characteristics did not differ significantly between the study groups (Table 1). All subjects enrolled in the mild or moderate hepatic impairment groups had liver cirrhosis caused by hepatitis virus infection, hepatic steatosis, or other conditions. All subjects completed the study and were included in the PK analysis and safety analysis sets.
PKs
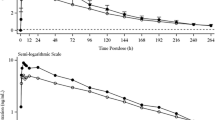
The time course of plasma esaxerenone concentrations is shown in Fig. 1a, b. The PK profile of esaxerenone did not differ across hepatic function groups; thus, PK parameters were relatively similar in subjects with different degrees of hepatic impairment (Table 2). Although mean Cmax values were similar in the mild hepatic impairment group (25.5 ng/mL) and normal hepatic function group (26.0 ng/mL), AUClast (514 vs 602 ng h/mL) and AUCinf (521 vs 620 ng h/mL) were lower in the mild hepatic impairment than normal hepatic function group. The value for mean Cmax was lower (21.2 vs 26.0 ng/mL) in the moderate hepatic impairment versus normal hepatic function group, whereas the mean value for AUClast was slightly greater (659 vs 602 ng h/mL), as was the mean value for AUCinf (692 vs 620 ng h/mL). No major differences were evident between the three hepatic function groups regarding tmax, t1/2 and Vz/F. Moreover, no trend was evident between the three study groups regarding differences in exposure to esaxerenone, and CL/F was similar between the three groups (Table 2).

Plasma concentration–time profiles of esaxerenone in Japanese subjects with normal or impaired hepatic function: a linear plot; b semi-log plot. LLOQ lower limit of quantification, SD standard deviation
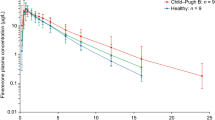
The GLSM ratios for Cmax, AUClast, and AUCinf are shown in Table 3. For subjects with mild hepatic impairment versus normal hepatic function, the GLSM ratios (90% CI) for AUClast and AUCinf were 0.837 (0.637, 1.099) and 0.824 (0.622, 1.092), respectively, and 0.959 (0.778, 1.182) for Cmax. For subjects with moderate hepatic impairment versus normal hepatic function, GLSM ratios (90% CI) for AUClast and AUCinf were 1.078 (0.820, 1.415) and 1.098 (0.829, 1.454), respectively, and 0.804 (0.653, 0.992) for Cmax. CL/F values were distributed within a fixed range (approximately 3–7 L/h) in all groups and did not show any clear trends as Child–Pugh score increased (Fig. 2a) or as baseline albumin levels decreased (Fig. 2b).

Scatter plots of a Child–Pugh score and b albumin at baseline versus apparent total body clearance of esaxerenone in Japanese subjects with normal or impaired hepatic function
Safety
Overall, two subjects experienced three AEs (Table 4). One patient with moderate hepatic impairment had severe hepatic encephalopathy, as judged by the investigator, 3 days after esaxerenone administration—this event was judged as a serious AE by the investigator—and mild headache; these events were considered possibly related to esaxerenone. The hepatic encephalopathy resolved after hospitalisation and pharmacological treatment, and the headache resolved after treatment. This subject had a history of hepatic encephalopathy and constipation; indeed, their blood ammonia level was high before and after administration, with 224 μg/dL 1 month before administration and 223 μg/dL after AE onset (normal range 30–80 μg/dL), and the subject had ongoing constipation. The subject underwent colonic hydrotherapy and their blood ammonia concentration decreased to 98 μg/dL 2 days after AE onset, and improvement of hepatic encephalopathy was observed. Therefore, the possibility that this event was worsened by constipation was considered. Esaxerenone PKs in this subject (Cmax 15.5 ng/mL, AUClast 783 ng h/mL) did not differ from that of other study subjects.
One subject with normal hepatic function had an increased AST level, considered by the investigator to be unrelated to esaxerenone. This change was transient and resolved without pharmacological treatment; at follow-up, the change was determined to have been owing to an altered living environment.
There were no deaths or discontinuations and no abnormal changes in vital signs, electrocardiogram, and other laboratory tests (including K+) during the study.
Discussion
This study evaluated the effect of hepatic impairment on esaxerenone PKs and safety after single-dose administration in Japanese subjects. The results indicate that esaxerenone PKs after a single 2.5-mg oral dose are not influenced by mild or moderate hepatic impairment. The scatter plot of Child–Pugh scores at baseline and esaxerenone CL/F values showed that the CL/F for each subject was distributed within a fixed range and that there was no clear trend of decreased clearance as Child–Pugh score increased, with considerable overlap between mean CL/F values in the different groups. When plasma albumin levels decrease owing to impaired hepatic function, the PKs of highly protein-bound drugs are affected [22]. Although esaxerenone has a high protein-binding rate (98.2–99.0%; unpublished data on file, Daiichi Sankyo, Tokyo, Japan), only two subjects with hepatic impairment had blood albumin levels below 35 g/L in our study, which may explain why no correlation was identified between blood albumin level and esaxerenone clearance (Fig. 2b). Thus, between-group differences in esaxerenone clearance indicate no consistent trend associated with the extent of hepatic impairment, and no clear influence of hepatic impairment on the PK profile of esaxerenone.
In a previous mass balance study of esaxerenone in healthy volunteers, excretion of unchanged esaxerenone in faeces and urine was 18.7% and 1.6%, respectively, of the administered dose (total excretion of radioactivity was 92.5% in faeces and urine); thus, clearance of esaxerenone was primarily via metabolism [17]. From the analysis of urinary and faecal metabolites after administration of radiolabelled esaxerenone to humans and from in vitro metabolic studies, esaxerenone metabolism is considered to involve oxidation by CYP3A4/3A5, glucuronidation by several isozymes of UGT, and hydrolysis [17]. Hepatic impairment reportedly influences various CYP enzymes, including CYP3A; however, UGT activity may be generally less susceptible to mild to moderate hepatic impairment than oxidation by CYP [12]. Because multiple pathways are involved in esaxerenone elimination, it seems likely that mild and moderate hepatic impairment will have only a minor influence on esaxerenone PKs.
The impact of hepatic function on the PKs of MR blockers was studied previously in patients receiving eplerenone [14]. Because the main route of eplerenone elimination is considered to be via CYP3A metabolism, non-Asian patients with moderate hepatic impairment who were receiving eplerenone had an AUC value approximately 1.4-fold higher than that in healthy adults [13], thus creating increased potential for AEs such as hyperkalaemia [6, 7]. In contrast, our study showed that esaxerenone AUC increased to a smaller extent by about 1.1-fold in moderate hepatic impairment versus normal hepatic function. Therefore, the PKs of esaxerenone are considered to be less affected by hepatic function than the PKs of eplerenone.
Esaxerenone was generally well tolerated in the current study, in agreement with existing data from two other phase 1 studies [9, 10]. A previous, multiple-dose study of esaxerenone safety in healthy Japanese men reported that all AEs were mild or moderate: two resolved with treatment and the others resolved without treatment [9].
Hyperkalaemia is a well-recognised AE of MR blockers, especially in patients with diabetes or kidney dysfunction. In a repeated-dose study of 10–100 mg esaxerenone for 10 days [9], serum K+ concentration increased with esaxerenone dosage. However, in our trial, no clinically significant changes in K+-related laboratory test values were found, and only one AST increase was observed in one subject. As our study was a small, single-dose study, we were unable to quantify the potential risk of hyperkalaemia in esaxerenone-treated patients with more advanced hepatic failure, such as liver cirrhosis complicated with ascites. A case of serious hepatic encephalopathy developed in the moderate hepatic impairment group in our study, but because blood ammonia concentration in this subject was high before esaxerenone administration, it is very likely that this event was due to chronic constipation; indeed, the event resolved with treatment and was considered unrelated to the overall safety profile of esaxerenone.
The limitations of this study include that it only investigated PK after a single esaxerenone dose and did not include a multiple-dose arm. Furthermore, the study sample size was only six subjects per group, which is the minimum required by FDA and EMA guidance [20, 21], and the study did not evaluate patients with severe hepatic impairment.
Conclusions
The current findings indicate that mild or moderate hepatic impairment has no clinically relevant effect on esaxerenone PKs and that dose adjustment is considered unnecessary from the PK viewpoint.
References
Bansal S, Lindenfeld J, Schrier RW. Sodium retention in heart failure and cirrhosis: potential role of natriuretic doses of mineralocorticoid antagonist? Circ Heart Fail. 2009;2:370–6.
Flatt DM, Brown MC, Mizeracki AM, King BJ, Weber KT. Mineralocorticoid receptor antagonists in the management of heart failure and resistant hypertension: a review. JAMA Cardiol. 2016;1:607–12.
The RALES Investigators. Effectiveness of spironolactone added to an angiotensin-converting enzyme inhibitor and a loop diuretic for severe chronic congestive heart failure (the Randomized Aldactone Evaluation Study [RALES]). Am J Cardiol. 1996; 78:902–7.
Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–21.
Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21.
Lainscak M, Pelliccia F, Rosano G, et al. Safety profile of mineralocorticoid receptor antagonists: spironolactone and eplerenone. Int J Cardiol. 2015;200:25–9.
Sica DA. Pharmacokinetics and pharmacodynamics of mineralocorticoid blocking agents and their effects on potassium homeostasis. Heart Fail Rev. 2005;10:23–9.
Arai K, Homma T, Morikawa Y, et al. Pharmacological profile of CS-3150, a novel, highly potent and selective non-steroidal mineralocorticoid receptor antagonist. Eur J Pharmacol. 2015;761:226–34.
Kato M, Furuie H, Shimizu T, Miyazaki A, Kobayashi F, Ishizuka H. Single- and multiple-dose escalation study to assess pharmacokinetics, pharmacodynamics and safety of oral esaxerenone in healthy Japanese subjects. Br J Clin Pharmacol. 2018;84:1821–9.
Kurata A, Furuie H, Ishizuka T, et al. Absolute bioavailability of esaxerenone and food effects on its pharmacokinetics after a single oral dose in healthy Japanese subjects. Adv Ther. 2019. https://doi.org/10.1007/s12325-019-00956-z.
Palatini P, De Martin S. Pharmacokinetic drug interactions in liver disease: an update. World J Gastroenterol. 2016;22:1260–78.
Verbeeck RK. Pharmacokinetics and dosage adjustment in patients with hepatic dysfunction. Eur J Clin Pharmacol. 2008;64:1147–61.
Brown NJ. Eplerenone: cardiovascular protection. Circulation. 2003;107:2512–8.
Jackson L, Branch R, Levine D, Ramsay L. Elimination of canrenone in congestive heart failure and chronic liver disease. Eur J Clin Pharmacol. 1977;11:177–9.
Sungaila I, Bartle WR, Walker SE, et al. Spironolactone pharmacokinetics and pharmacodynamics in patients with cirrhotic ascites. Gastroenterology. 1992;102:1680–5.
INSPRA® (eplerenone) US prescribing information. https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/021437s006lbl.pdf. Accessed 16 Aug 2018.
Yamada M, Mendell J, Takakusa H, Shimizu T, Ando O. Pharmacokinetics, metabolism, and excretion of [14C]esaxerenone, a novel mineralocorticoid receptor blocker in humans. Drug Metab Dispos. 2019;47:340–9.
Ministry of Health, Labor and Welfare. Japanese dietary intake standard (2010 edition). Report on the study meeting of ‘Japanese dietary intake standards’ study report (May 2009). https://www.mhlw.go.jp/shingi/2009/05/s0529-4.html. Accessed 25 Sept 2018.
Peng Y, Qi X, Guo X. Child–Pugh versus MELD score for the assessment of prognosis in liver cirrhosis: a systematic review and meta-analysis of observational studies. Medicine (Baltimore). 2016;95:e2877.
FDA. Guidance for Industry: Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. U.S. Department of Health and Human Services. Food and Drug Administration. May 2003. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072123.pdf. Accessed 18 Aug 2018.
EMA. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. CPMP/EWP/2339/02. February 2005. European Medicines Agency. Committee for Medicinal Products for Human Use. London: 2005. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003122.pdf. Accessed 18 Aug 2018.
Delcò F, Tchambaz L, Schlienger R, Drewe J, Krähenbühl S. Dose adjustment in patients with liver disease. Drug Saf. 2005;28:529–45.
Acknowledgements
The authors would like to express gratitude to all subjects who participated in this study.
Funding
Sponsorship for this study and the journal’s Rapid Service Fee and Open Access fees were funded by Daiichi Sankyo Co., Ltd. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Authorship
All authors contributed to drafting the manuscript, reviewing the manuscript content critically, and designing and implementation of the research. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing, Editorial, and Other Assistance
The authors wish to thank Tsunenori Nakazawa and Naoko Nakai for their help with the bioanalysis of samples in this study. The authors would also like to thank Nicola Ryan, David Murdoch and Sarah Bubeck, PhD of Edanz Medical Writing for providing medical writing support, which were funded by Daiichi Sankyo Co., Ltd. MedDRA® trademark is registered by IFPMA on behalf of ICH.
Disclosures
Akifumi Kurata is an employee of Daiichi Sankyo Co., Ltd. Tomoko Ishizuka is an employee of Daiichi Sankyo Co., Ltd. Takafumi Nakatsu is an employee of Daiichi Sankyo Co., Ltd. Takako Shimizu is an employee of Daiichi Sankyo Co., Ltd. Manabu Kato is an employee of Daiichi Sankyo Co., Ltd. Yasuhiro Nishikawa is an employee of Daiichi Sankyo Co., Ltd. Hitoshi Ishizuka is an employee of Daiichi Sankyo Co., Ltd. Takafumi Yoshida and Megumi Inoue have no potential conflicts of interest to disclose.
Compliance with Ethics Guidelines
The study received institutional review board approvals from ETHIPRO, Montreal, QC, Canada (IORG0000689); the Clinical Trials Review Committee of Hakata Clinic (1464P3S-6); and the Clinical Trials Review Committee of Kurume Clinical Pharmacology Clinic (approved on 22 August 2016; reference number not stated). The study was conducted in accordance with Good Clinical Practice guidelines, as defined by the International Conference on Harmonization, and ethical principles outlined in the Declaration of Helsinki. All subjects provided written informed consent before inclusion in the study.
Data Availability
Research data are not shared.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9929789.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Kurata, A., Yoshida, T., Inoue, M. et al. Pharmacokinetics and Safety of Single-Dose Esaxerenone in Japanese Subjects with Mild to Moderate Hepatic Impairment. Adv Ther 37, 253–264 (2020). https://doi.org/10.1007/s12325-019-01121-2
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-019-01121-2