
Abstract
Background and Objective
Zibotentan, a selective endothelin A receptor antagonist, is in development for chronic liver and kidney disease. The pharmacokinetics (PK) of zibotentan were previously investigated in patients with either renal impairment or hepatic impairment, but the impact of both pathologies on PK was not evaluated. This study evaluated the PK and tolerability of a single oral dose of zibotentan in participants with concurrent moderate renal impairment and moderate hepatic impairment versus control participants.
Methods
Twelve participants with moderate renal and hepatic impairment and 11 healthy matched control participants with no clinically significant liver or kidney disease were enrolled in an open-label, parallel-group study design. After administration of a single oral dose of zibotentan 5 mg, blood and urine sampling was performed. Pharmacokinetic parameters were determined for each of the two cohorts and compared. Comparisons between the cohorts were based on the geometric least squares mean ratio for the primary endpoints, which were area under the plasma concentration-time curve (AUC) from time zero to infinity (AUC∞) and from time zero to the time of the last measurable concentration (AUClast), and maximum plasma drug concentration (Cmax) on Day 1 through 120 h post-dose. Secondary endpoints included apparent total body clearance (CL/F) on Day 1 through 120 h post-dose. Safety endpoints were assessed up to discharge.
Results
In total, 11 participants with concurrent moderate renal and hepatic impairment, and 11 controls, completed the study. Zibotentan was generally well tolerated, and no new clinically significant safety findings were observed. Total exposure (AUC∞ and AUClast) was approximately 2.10-fold higher in participants with concurrent moderate renal and hepatic impairment versus controls, while Cmax and total nonrenal body clearance were similar among all groups. A regression-based post hoc analysis, comparing exposure and CL/F in patients with concurrent impairment to patients with either renal or hepatic impairment alone, showed that CL/F with concurrent impairment was approximately half of that in controls and was positively correlated with reduction of renal function. Inclusion of the data on concurrent moderate renal and hepatic impairment in the regression analysis led to a narrower confidence interval for the predicted mean CL/F in participants with moderate hepatic impairment.
Conclusion
The presented findings advance the understanding of the PK of zibotentan in both renal impairment and hepatic impairment, with and without overlapping pathologies, and will thus increase the confidence of dose selection in future studies, particularly in vulnerable patient populations with concurrent renal and hepatic impairment.
Trial Registration
ClinicalTrials.gov identifier: NCT05112419.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
After taking a single dose of the drug zibotentan, in patients with both kidney and liver disease, the body removed the drug approximately half as fast as in healthy people, so that approximately double the levels of the drug were found within their blood. |
These findings support the development of zibotentan in patients with both kidney and liver disease by providing important information that will help to decide whether some patients require a lower dose. |
1 Introduction
Liver cirrhosis represents a substantial health burden, with the number of deaths and disability-adjusted life years as well as the proportion of all global deaths due to cirrhosis increasing since 1990 [1]. Liver cirrhosis is initially described as ‘compensated’, which progressively worsens as portal pressure increases and liver function decreases. Clinically significant portal hypertension increases the risk for decompensation events including ascites, variceal bleeding, encephalopathy and associated complications, such as renal impairment (i.e., acute kidney injury/hepatorenal syndrome) [2, 3]. In addition to acute episodes of impaired renal function, there is also an increased risk of developing chronic kidney disease among patients with liver cirrhosis [4], which further increases the risk of morbidity and mortality [4,5,6]. Preventing cirrhosis progression by targeting pathological changes, such as fibrosis and portal hypertension, represents an attractive treatment strategy; however, this remains an unmet need in patients with cirrhosis [7].
Patients with liver cirrhosis have elevated circulating levels of endothelin-1 [8], a potent vasoconstrictor with effects mediated through binding to the endothelin A (ETA) receptor [9, 10]. A single oral dose of a selective ETA receptor antagonist has been shown to decrease the hepatic venous pressure gradient in patients with cirrhosis without affecting systemic haemodynamics [11], suggesting that ETA receptor antagonism reduces portal pressure by reducing hepatic vasoconstriction. Zibotentan is a selective ETA receptor antagonist originally studied as a treatment for cancer [12,13,14] and currently under development as a combination therapy with dapagliflozin for chronic kidney disease [15] and liver cirrhosis with features of clinically significant portal hypertension. Fluid retention remains a tolerability and safety concern with ETA receptor antagonists [16, 17], including zibotentan [12, 18]. Dapagliflozin, a sodium-glucose cotransporter 2 inhibitor, may mitigate the fluid retention risk associated with zibotentan [19,20,21]. Additionally, both dapagliflozin and ETA antagonists have evidence of fibrosis progression attenuation [22, 23].
The plasma clearance of zibotentan is impacted by both metabolism and renal excretion [24]. Zibotentan metabolism was found to be mainly mediated by the CYP3A4 isozyme [25]. Elimination of zibotentan and its metabolites occurs predominantly renally, with approximately 58% of the parent compound being eliminated unchanged in the urine [24]. The pharmacokinetics (PK) of zibotentan have previously been investigated in two separate studies in patients with either hepatic or renal impairment [26]. In these populations, exposure, as evaluated by the area under the plasma concentration curve [AUC] from time zero to infinity [AUC∞], was increased by up to 2.9-fold for hepatic impairment and 2.2-fold for renal impairment, compared with healthy participants. Exposure was greater in patients with higher degrees of hepatic or renal impairment due to slower clearance of zibotentan. No study to date has investigated the PK of zibotentan in patients with concurrent renal and hepatic impairment and the implications of PK in these overlying pathologies. Concurrent moderate renal and moderate hepatic impairment (as opposed to severe impairment) was selected for this study as it was anticipated to affect parts of the clinical target population, while patients with—e.g., concurrent severe renal and severe hepatic impairment—were regarded as too ill to be included into the clinical programme.
The aim of this study was to evaluate the PK and tolerability of zibotentan in patients with concurrent moderate renal and moderate hepatic impairment compared with a group of healthy matched controls. In addition, this study aimed to compare these findings with data from patients with either hepatic or renal impairment alone [26] in a post hoc analysis.
2 Methods
2.1 Study Design
This was an open-label, parallel-group study (NCT05112419) conducted at a single centre in Bulgaria from 10 November to 15 December 2021 in which participants received a single oral dose of zibotentan 5 mg (Fig. 1). This dose was selected to be lower than the 10 mg used in the previous separate renal impairment and hepatic impairment studies [26], since an exposure increase was to be expected, and creating particularly high exposures was not required for this PK analysis. Food intake has been found to decrease zibotentan Cmax, which is not considered clinically relevant but may when not standardised increase variability of PK; participants were therefore fasted from midnight prior to dosing until 4 h after dosing and not allowed fluids except water from 1 h prior to dosing until 2 h after dosing; meals were standardised and given at scheduled timepoints during the first 24 h after dosing.

Study design. n number of participants, PK pharmacokinetic
The study consisted of 2 cohorts: 12 participants with moderate renal and hepatic impairment in the first cohort and 11 healthy participants in the second cohort, who were matched for age (± 10 years), sex and body mass index (BMI) (± 20%) on a group level to participants with impairment. Following a screening period of ≤ 28 days, participants were admitted to the study centre for a residential period of 8 days, from 2 days before administration of zibotentan (Day − 2) to discharge after their final study assessments, 120 h post-dose (Day 6).
Pharmacokinetic blood sampling was performed pre-dose and at post-dose intervals ranging from 30 minutes to 120 h. Pharmacokinetic urine sampling was performed at 0–6 h, 6–12 h and subsequent 12-h intervals from 12 to 72 h post-dosing, with the sample volume recorded. Plasma and urine samples were received frozen and stored at − 60 to – 80 °C until analysis.
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, consistent with the International Conference on Harmonisation Guideline for Good Clinical Practice and applicable regulatory requirements, and all participants provided written informed consent prior to enrolment. The study protocol and informed consent documents were approved by the local Independent Ethics Committee.
2.2 Participants
2.2.1 Eligibility Criteria
Females of non-childbearing potential and males aged 18–80 years with a body weight ≥ 50 kg and BMI 18–35 kg/m2 were enrolled. Full inclusion and exclusion criteria are provided in the Supplementary Methods.
Specific inclusion criteria for participants with impairment were as follows: estimated glomerular filtration rate (eGFR) of 30–45 mL/min/1.73 m2 at screening (rather than 30–59 mL/min/1.73 m2 to prevent inclusion of patients with mild-to-moderate renal impairment), using the Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) formula; confirmed clinical diagnosis of cirrhosis with either ascites or moderate hepatic impairment, classified as Child–Pugh score of class B; and stable hepatic impairment within 28 days prior to screening.
Participants with impairment were excluded for presence of unstable medical or psychological conditions; fluctuating or rapidly deteriorating hepatic function, as indicated by clinical and/or laboratory signs, within 28 days prior to dosing; severe hepatic impairment (Child–Pugh score of class C), an isolated aspartate transaminase (AST) or alanine transaminase (ALT) > 5 × the upper limit of normal (ULN), total bilirubin >2 × ULN or a concurrent AST and ALT of > 3 × the ULN together with a total bilirubin of >2 × ULN; acute liver disease caused by drug toxicity or an infection; presence of hepatocellular carcinoma; planned liver or renal transplantation ≤ 3 months; receiving renal replacement therapy; recent acute or subacute deterioration of renal function; New York Heart Failure Association functional heart failure Class III/IV or unstable heart failure ≤ 6 months prior to screening; abnormal resting supine systolic or diastolic blood pressure; and change in dose of medically required medication ≤ 14 days prior to dosing.
Healthy controls included participants who were overtly healthy as determined by medical evaluation including medical history, physical examination and clinical laboratory tests, who had an eGFR of ≥ 90 mL/min/1.73 m2 using the CKD-EPI formula and no clinically significant liver or kidney disease as judged by the investigator. Those with a history of any clinically important disease or disorder were excluded.
2.3 Endpoints
2.3.1 Pharmacokinetic Endpoints
The primary endpoints were the PK parameters AUC∞, AUC from time zero to the time of last measurable concentration [AUClast] and maximum plasma drug concentration [Cmax] on Day 1 through 120 h post-dose in participants with renal and hepatic impairment compared with healthy matched controls.
Secondary endpoints included the following: plasma PK parameters (time to reach the maximum observed plasma concentration [tmax], AUC from time zero to 24 h [AUC0–24], concentration at 24 h post-dose [C24], half-life associated with terminal slope of a semi-logarithmic concentration-time curve [t½λz], terminal elimination rate constant [λz], apparent total body clearance of drug from plasma after extravascular administration [CL/F], apparent total nonrenal body clearance of drug from plasma after extravascular administration [CLNR/F] and apparent volume of distribution during the terminal phase after extravascular administration [Vz/F]) on Day 1 through 120 h post-dose in both cohorts and urine PK parameters (amount of unchanged drug excreted into urine [Ae], percentage of dose excreted unchanged in urine [fe] and renal clearance of drug from plasma [CLR]) on Day 1 through 72 h post-dose in both cohorts.
2.4 Tolerability and Safety Endpoints
Treatment-emergent adverse events (TEAEs), routine safety assessments and clinical laboratory evaluations were assessed up to the final tolerability and safety assessments at discharge (Day 6 or at 120 h post-dose).
2.5 Statistical Analyses
The PK analysis set included all participants with ≥ 1 quantifiable post-dose concentration and no important protocol deviations who received a single dose of zibotentan; the safety analysis set included all participants with any post-dose safety data who received a single dose of zibotentan. A sample size of 24 participants (12 participants per cohort) was selected to account for potential discontinuation, assuming an interparticipant coefficient of variation of 30%, with 10 participants in each cohort expected to give a relative precision of 1.8 (ratio between the upper and lower limits of the 90% confidence interval [CI]) with a probability of 80%.
Descriptive summary statistics are presented for all endpoints, with frequency counts for TEAEs. For the primary PK endpoints, differences between cohorts were analysed by analysis of variance using the natural logarithm of the parameters as the response variables and cohort as fixed effect; data are presented as geometric least squares mean ratios (90% CI).
Results were compared with data from participants with either renal impairment or hepatic impairment alone [26] in a post hoc analysis. Ratios of geometric means of AUC∞ and Cmax of each impairment group (concurrent moderate renal and hepatic impairment, hepatic impairment only [mild/moderate/severe] and renal impairment only [mild/moderate/severe]) compared with respective healthy controls and 90% CIs were calculated. A regression analysis of CL/F versus eGFR was performed to examine confidence in the data from the combined renal and hepatic impairment study. The analysis used model averaging with Akaike Information Criterion (AIC) based weights [27] and a model pool consisting of the following three linear regression models: (1) CL/F versus baseline eGFR independent of Child–Pugh class, (2) CL/F versus baseline eGFR with a Child–Pugh class-specific intercept and (3) CL/F versus baseline eGFR with a Child–Pugh class-specific intercept and slope.
Child–Pugh class as a categorical covariate (none, class A, class B, and class C) and eGFR as a continuous covariate were the only covariates in the regression model. The model pool was fit to a dataset containing data from participants in the renal impairment and the hepatic impairment study (n = 80) [26] (i.e., data available prior to conducting the combined renal and hepatic impairment study) as well as to a dataset containing data from participants in all three available studies, i.e., the renal, the hepatic and the combined impairment study (n = 103) (i.e., data available after conducting the combined renal and hepatic impairment study). Predictions for the mean CL/F versus baseline eGFR and associated 95% CIs were obtained using both fits for participants with no hepatic impairment as well as for participants with Child–Pugh class B, by averaging the predictions and standard errors from each model in the pool with weights proportional to exp(− 0.5 * ∆AIC), where ∆AIC is the difference to the lowest AIC in the pool.
All statistical analyses were performed using SAS® version 9.4 or later; data were plotted and regression analysis was performed in R version > 3.5.
3 Results
3.1 Participants
In total, 23 participants were enrolled in the study (participants with impairment, n = 12; healthy controls, n = 11) and 22 participants completed the study (participants with impairment, n = 11; healthy controls, n = 11); one participant was withdrawn from the cohort of participants with impairment and excluded from the PK analysis, as it was not possible to collect repeated blood and urine samples, resulting in protocol deviations.
Participants with renal and hepatic impairment and healthy controls were matched according to age, sex and BMI (Table 1). All participants were White. All participants with impairment had a Child–Pugh score of class B (range of 7–8), and 9 participants with impairment had clinically overt ascites. Mean (SD) eGFR was 37.1 (6.42) mL/min/1.73 m2 in participants with impairment versus 94.1 (4.61) mL/min/1.73 m2 in healthy controls. No prohibited concomitant medications were used by participants. Concomitant medication in the group of participants with impairment included one participant on angiotensin II receptor blockade (Valsartan) and four participants on paracetamol. In the matched control group, 4 participants were on paracetamol, and no other medications. No medical history events or concurrent illnesses were considered likely to affect the outcome of the study as judged by the investigator and sponsor.
3.2 Endpoints
3.2.1 Pharmacokinetic Endpoints
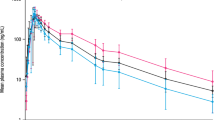
Geometric mean plasma concentration-time curves and PK parameters for each cohort are presented in Fig. 2 and Table 2.

Geometric mean plasma zibotentan concentration-time curves in linear (a) and semi-logarithmic (b) scale. Vertical lines represent geometric mean*/geometric standard deviation. h hour, N number of participants in the PK analysis set for each treatment, PK pharmacokinetic
3.2.1.1 Primary Endpoints
Total exposure, as measured by AUC∞ and AUClast, was approximately 2.10-fold higher in participants with renal and hepatic impairment compared with healthy controls (Table 2), with geometric least squares mean ratios (90% CI) of 211.0% (180.6, 246.5) and 210.6% (180.3, 246.1). Cmax was similar in both cohorts (Table 2).
3.2.1.2 Secondary Endpoints: Plasma Pharmacokinetic Parameters
The median (min–max) tmax was longer and had a wider range in participants with renal and hepatic impairment compared with healthy controls, 3.0 h (1.5–10.0) versus 1.5 h (0.5–4.0), respectively (Table 2). In addition, the geometric mean concentration at 24 h post-dose was approximately 4-fold higher and t½λz was longer (14.66 vs 9.82 h) in participants with impairment compared with healthy controls. The geometric mean CL/F in participants with impairment was approximately half of the geometric mean in healthy controls. In contrast, the geometric means of CLNR/F were comparable in both cohorts.
3.2.1.3 Secondary Endpoints: Urine Pharmacokinetic Parameters
Geometric means of Ae and CLR in participants with renal and hepatic impairment were 18.3 and 8.8% of the healthy control values, respectively (Table 2). The geometric mean (percentage coefficient of variation [CV%]) for fe was 10.01% (147.5) of the zibotentan dose in participants with impairment, ranging widely from 2.73 to 61.5%, which was 18.3% of the geometric mean for healthy controls (54.80% [15.45]).
3.2.1.4 Post Hoc Analysis: Comparison of Exposure and Clearance in Participants with Concurrent Renal and Hepatic Impairment Versus Those with Either Renal or Hepatic Impairment Alone
Total exposure of zibotentan, as measured by AUC∞, increased approximately 2.10-fold in participants with moderate renal and hepatic impairment; the AUC∞ in these participants could have been expected to increase by 2.31-fold, based on multiplying the expected AUC∞ of participants with moderate renal impairment by that of participants with moderate hepatic impairment (values listed below).
Area under the plasma concentration-time curve from zero to infinity in the previous renal impairment and hepatic impairment studies were estimated to increase by 1.27-, 1.59- and 1.78-fold in participants with mild, moderate and severe renal impairment, respectively, and 1.40-, 1.45- and 2.90-fold in participants with mild, moderate and severe hepatic impairment, respectively, compared with their respective healthy controls (Fig. 3a).

Comparison of zibotentan exposure in participants with renal or hepatic impairment and in those with renal and hepatic impairment versus healthy controls without impairment of a AUC∞, b Cmax and c regression analysis of CL/F versus eGFR. Fig. 3a was adapted from Fig. 2. Forest plot of the ratios of zibotentan exposure in Tomkinson et al. [26]. Available at: https://doi.org/10.1186/1472-6904-11-3, licensed under CC BY 2.0 () and published under licence to BioMed Central Ltd. Glomerular filtration rate categories (mL/min/1.73 m2) were as follows: G1—normal or high (≥ 90), G2—mildly decreased (60–89), G3a—mildly to moderately decreased (45–59) and G3b—moderately to severely decreased (30–44). Predictions for the mean CL/F versus baseline eGFR, and associated 95% CIs, used model averaging on a dataset containing data from participants in the renal impairment and the hepatic impairment study (n = 80) (i.e., data available prior to conducting the combined renal and hepatic impairment study) as well as to a dataset containing data from participants in the renal, the hepatic and the combined impairment study (n = 103) (i.e., data available after conducting the combined renal and hepatic impairment study). AUC∞ area under the plasma concentration-time curve from zero to infinity, CI confidence interval, CL/F apparent total body clearance of drug from plasma after extravascular administration, Cmax maximum plasma drug concentration, eGFR estimated glomerular filtration rate, Gmean geometric mean
There was no clear difference in Cmax in participants with concurrent renal and hepatic impairment, those with only renal impairment and those with only hepatic impairment, compared with their respective healthy controls (Fig. 3b).
The CL/F in participants with concurrent renal and hepatic impairment was positively correlated with reduction of renal function as measured by eGFR at screening (Fig. 3c). Including the data from participants with concurrent hepatic and renal impairment resulted in a reduction in the width of the CIs for the predicted mean CL/F versus eGFR relationship in both subgroups, i.e., in participants with no hepatic impairment and, particularly, in participants with Child–Pugh class B. Although the regression lines of both subgroups were generally parallel to one another, the CI was wider in participants with Child–Pugh class B versus in those with no hepatic impairment. The CIs in both subgroups overlapped in the eGFR category G3b. Nonetheless, at similar eGFR levels, CL/F was at least 10 mL/h higher in participants with no hepatic impairment versus those with Child–Pugh class B.
3.2.2 Tolerability and Safety Endpoints
In total, 11 (47.8%) participants experienced TEAEs (participants with impairment, n = 7 [58.3%]; healthy controls, n = 4 [36.4%]). All TEAEs were mild to moderate in intensity and resolved before the end of the study. The most frequently reported TEAE in both cohorts was headache (participants with impairment, n = 6 [50.0%]; healthy controls, n = 4 [36.4%]), all events of which were considered by the investigators to be causally related to zibotentan. Other TEAEs were diarrhoea (participants with impairment, n = 1 [8.3%]) and asthenia, dizziness, circulatory collapse and hypotension (participant with impairment, n = 1 [8.3%]). The latter TEAEs were reported within the first hour after receiving zibotentan and resolved after 1 day, with the participant completing the study as per protocol. There were no serious TEAEs, TEAEs leading to discontinuation or deaths throughout the study. No worsening of ascites was observed, and there were no clinically significant changes in laboratory values, clinical chemistry, urinalysis results, vital signs, electrocardiogram measurements and physical examination, and none were reported as TEAEs.
4 Discussion
Previous PK studies showed increased zibotentan exposure in patients with hepatic impairment and in those with renal impairment [26] but were not able to inform on the effect of both impairments on the PK of zibotentan. Pharmacokinetics are not generally studied in populations with concurrent renal and hepatic impairment, as drugs that are metabolised via both renal and hepatic routes may not necessarily have target patient populations with both renal and hepatic impairment. Exclusion criteria for hepatic and/or renal impairment in clinical trials are also common [28, 29]. However, characterising the effect of impaired hepatic and/or renal function on PK as well as safety and efficacy is recommended to guide dosing and to ensure the applicability of the trial findings in such patient populations [28,29,30]. Our study demonstrates that, despite reduced clearance and higher exposure in participants with renal and hepatic impairment compared with healthy controls, a single dose of zibotentan 5 mg was generally well tolerated, in line with previous PK studies examining renal impairment and hepatic impairment as separate patient populations. The current study is the first to investigate zibotentan in a patient population with concurrent renal and hepatic impairment.
This study shows that concurrent renal and hepatic impairment leads to an approximate 2.10-fold increase in total exposure compared with healthy controls. This is slightly lower than the assumed 2.31-fold increase, which was based on the previously observed 1.45-fold AUC increase in moderate hepatic impairment and 1.59-fold AUC increase in moderate renal impairment [26]. Working out the expected increase by multiplying the effects of isolated impairment was a naïve approach chosen to illustrate intuitive expectations; more advanced approaches were not employed as the underlying pathophysiology is complex and renal and hepatic impairment are not independent conditions. Lack of information about the interplay of concurrent hepatic and renal impairment and the implications for the zibotentan exposure formed the motivation for the presented study. The similarity between the observed and the estimated exposure increases should, therefore, not be interpreted as confirmation of the simplistic approach.
The study results demonstrate that zibotentan AUC with concurrent moderate renal and hepatic impairment does not increase in a supra-additive way, even though both key metabolic pathways are impacted. There was no difference in Cmax between participants with impairment compared with healthy controls, as seen similarly in the previous studies of patients with renal impairment or with hepatic impairment [26], suggesting that any Cmax-based evaluation of tolerability and safety is independent of the presence of renal or hepatic impairment. Moreover, no new clinically significant tolerability or safety findings were observed compared with previous studies [26].
The repeated observation that zibotentan AUC, but not Cmax, is increased compared with the control group in patients with renal impairment alone, hepatic impairment alone or overlapping pathologies, is characteristic of drugs with low extraction by the liver. This suggests that any potential dose adjustments to reach a defined target exposure can be achieved by solely adjusting the maintenance dose, without the need for modifying the initial dose, thereby supporting adequate dosing strategies in future zibotentan trials in patients with liver cirrhosis [31].
The increased exposure to zibotentan observed in participants with concurrent renal and hepatic impairment compared with healthy controls appears to be primarily related to renal impairment and subsequent reduced renal clearance. Similarly, data from previous studies of patients with renal or hepatic impairment alone demonstrated that both moderate and severe renal impairment had a large impact on the PK profile of zibotentan in contrast to mild renal impairment, while only severe hepatic impairment, not mild or moderate hepatic impairment, had a large impact on the PK profile [26]. In the current study, while the geometric mean of CLNR/F (i.e., nonrenal clearance) was comparable between the cohorts, CL/F (i.e., total body clearance) in participants with impairment was approximately half of that in healthy controls, which was expected given total exposure was approximately doubled in participants with concurrent renal and hepatic impairment. The amount of unchanged drug, percentage of unchanged drug and renal clearance was 18.3, 18.3 and 8.8%, respectively, of that of healthy controls in participants with impairment, indicating impaired renal function mainly contributed to differences in clearance and exposure. In addition, the majority of participants with impairment had ascites, which may have contributed to a longer tmax and t½λz [32, 33]. These findings demonstrate how concurrent renal and hepatic impairment can impact PK and highlight the importance of investigating the effects of drugs in this patient population.
A strength of this study was the inclusion of patients based on an eGFR of 30–45 mL/min/1.73 m2 rather than 30–59 mL/min/1.73 m2, which ensured that participants would be representative of those with moderate renal impairment rather than milder renal impairment. This was, therefore, a conservative design decision, putting the study focus on the subpopulation where zibotentan exposure is expected to be high. Furthermore, given the influence of ascites on PK and volume of distribution, the inclusion of participants with ascites was a further strength providing additional information in this patient population. Patients with concurrent moderate hepatic and moderate renal impairment represent a previously unstudied patient population that may form part of study populations in future clinical development of zibotentan. Greater confidence in the predicted mean CL/F based on baseline eGFR in participants with and without hepatic impairment, after including data from patients with concurrent renal and hepatic impairment, highlights the value of studying PK in this population.
Limitations of the current study include the use of a single dose of zibotentan. Although accumulation of zibotentan is not expected in healthy participants, the effects of multiple dosing in participants with concurrent renal and hepatic impairment are less clear. Future studies should therefore evaluate the repeated-dose PK of zibotentan in patients with these impairments. Another limitation was that the study size was limited to 11 patients and 11 healthy controls. Related to this limited size, not the full spectrum of Child–Pugh B was included, all patients had either a score of 7 or 8, and no one had a score of 9. In addition, all participants were White, with the study conducted at a single centre in Bulgaria; hence, this study population may not be representative of wide, more diverse patient populations. Including cohorts with only hepatic impairment or only renal impairment would have improved comparability between the different patient populations, but this has been investigated previously [26].
The data from this study may be applied to mechanistic or physiology-based modelling to improve the understanding of zibotentan PK. These findings also provide unique insights into the PK of zibotentan in patients with renal and hepatic impairment both separately and combined, which may be informative for studies of other drugs intended for use in patients with renal and hepatic impairment or other ETA receptor antagonists in development.
5 Conclusions
The findings from this study will support the future clinical development of zibotentan by informing, together with to-be-generated efficacy and safety information, the dose strategy in the more vulnerable subpopulation of patients with concurrent renal and hepatic impairment.
References
GBD 2017 Cirrhosis Collaborators. The global, regional, and national burden of cirrhosis by cause in 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol Hepatol. 2020;5(3):245–66.
D’Amico G, Garcia-Tsao G, Pagliaro L. Natural history and prognostic indicators of survival in cirrhosis: a systematic review of 118 studies. J Hepatol. 2006;44(1):217–31.
Francoz C, Durand F, Kahn JA, Genyk YS, Nadim MK. Hepatorenal syndrome. Clin J Am Soc Nephrol. 2019;14(5):774–81.
Maiwall R, Pasupuleti SSR, Bihari C, Rastogi A, Singh PK, Naik V, et al. Incidence, risk factors, and outcomes of transition of acute kidney injury to chronic kidney disease in cirrhosis: a prospective cohort study. Hepatology. 2020;71(3):1009–22.
Chen CY, Lin CJ, Lin CS, Sun FJ, Pan CF, Chen HH, et al. The prevalence and association of chronic kidney disease and diabetes in liver cirrhosis using different estimated glomerular filtration rate equation. Oncotarget. 2018;9(2):2236–48.
Wong F, Reddy KR, O’Leary JG, Tandon P, Biggins SW, Garcia-Tsao G, et al. Impact of chronic kidney disease on outcomes in cirrhosis. Liver Transpl. 2019;25(6):870–80.
European Association for the Study of the Liver. EASL clinical practice guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69(2):406–60.
Møller S, Gülberg V, Henriksen JH, Gerbes AL. Endothelin-1 and endothelin-3 in cirrhosis: relations to systemic and splanchnic haemodynamics. J Hepatol. 1995;23(2):135–44.
Riezebos J, Watts IS, Vallance PJ. Endothelin receptors mediating functional responses in human small arteries and veins. Br J Pharmacol. 1994;111(2):609–15.
Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, et al. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature. 1988;332(6163):411–5.
Zipprich A, Gittinger F, Winkler M, Dollinger MM, Ripoll C. Effect of ET-A blockade on portal pressure and hepatic arterial perfusion in patients with cirrhosis: a proof of concept study. Liver Int. 2021;41(3):554–61.
Schelman WR, Liu G, Wilding G, Morris T, Phung D, Dreicer R. A phase I study of zibotentan (ZD4054) in patients with metastatic, castrate-resistant prostate cancer. Invest New Drugs. 2011;29(1):118–25.
Cognetti F, Bagnato A, Colombo N, Savarese A, Scambia G, Sehouli J, et al. A Phase II, randomized, double-blind study of zibotentan (ZD4054) in combination with carboplatin/paclitaxel versus placebo in combination with carboplatin/paclitaxel in patients with advanced ovarian cancer sensitive to platinum-based chemotherapy (AGO-OVAR 2.14). Gynecol Oncol. 2013;130(1):31–7.
Chouaid C, Nathan F, Pemberton K, Morris T. A phase II, randomized, multicenter study to assess the efficacy, safety, and tolerability of zibotentan (ZD4054) in combination with pemetrexed in patients with advanced non-small cell lung cancer. Cancer Chemother Pharmacol. 2011;67(5):1203–8.
AstraZeneca. Zibotentan and dapagliflozin for the treatment of CKD (ZENITH-CKD Trial) (ZENITH-CKD). 2021. Available from: https://clinicaltrials.gov/ct2/show/NCT04724837. Accessed: 8 Aug 2022.
Vercauteren M, Trensz F, Pasquali A, Cattaneo C, Strasser DS, Hess P, et al. Endothelin ETA receptor blockade, by activating ETB receptors, increases vascular permeability and induces exaggerated fluid retention. J Pharmacol Exp Ther. 2017;361(2):322–33.
Heerspink HJL, Parving HH, Andress DL, Bakris G, Correa-Rotter R, Hou FF, et al. Atrasentan and renal events in patients with type 2 diabetes and chronic kidney disease (SONAR): a double-blind, randomised, placebo-controlled trial. Lancet. 2019;393(10184):1937–47.
Stern EP, Host LV, Wanjiku I, Escott KJ, Gilmour PS, Ochiel R, et al. Zibotentan in systemic sclerosis-associated chronic kidney disease: a phase II randomised placebo-controlled trial. Arthritis Res Ther. 2022;24(1):130.
Ohara K, Masuda T, Murakami T, Imai T, Yoshizawa H, Nakagawa S, et al. Effects of the sodium-glucose cotransporter 2 inhibitor dapagliflozin on fluid distribution: a comparison study with furosemide and tolvaptan. Nephrology (Carlton). 2019;24(9):904–11.
Heerspink HJL, Kohan DE, de Zeeuw D. New insights from SONAR indicate adding sodium glucose co-transporter 2 inhibitors to an endothelin receptor antagonist mitigates fluid retention and enhances albuminuria reduction. Kidney Int. 2021;99(2):346–9.
Veenit V, Heerspink HJL, Ahlstrom C, Greasley PJ, Skritic S, van Zuydam N, et al. The sodium glucose co-transporter 2 inhibitor dapagliflozin ameliorates the fluid-retaining effect of the endothelin A receptor antagonist Zibotentan. Nephrol Dial Transplant. 2023. (accepted for publication).
Shimizu M, Suzuki K, Kato K, Jojima T, Iijima T, Murohisa T, et al. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes Metab. 2019;21(2):285–92.
Rockey DC, Chung JJ. Endothelin antagonism in experimental hepatic fibrosis. Implications for endothelin in the pathogenesis of wound healing. J Clin Invest. 1996;98(6):1381–8.
Clarkson-Jones JA, Kenyon AS, Kemp J, Lenz EM, Oliver SD, Swaisland H. Disposition and metabolism of the specific endothelin A receptor antagonist zibotentan (ZD4054) in healthy volunteers. Xenobiotica. 2012;42(4):363–71.
Swaisland HC, Oliver SD, Morris T, Jones HK, Bakhtyari A, Mackey A, et al. In vitro metabolism of the specific endothelin-A receptor antagonist ZD4054 and clinical drug interactions between ZD4054 and rifampicin or itraconazole in healthy male volunteers. Xenobiotica. 2009;39(6):444–56.
Tomkinson H, Kemp J, Oliver S, Swaisland H, Taboada M, Morris T. Pharmacokinetics and tolerability of zibotentan (ZD4054) in subjects with hepatic or renal impairment: two open-label comparative studies. BMC Clin Pharmacol. 2011;11:3.
Claeskens G, Hjort N. Model selection and model averaging. Cambridge: Cambridge University Press; 2008.
U.S. Food & Drug Administration. Pharmacokinetics in patients with impaired renal function—study design, data analysis, and impact on dosing and labeling. 2020. Available from: https://www.fda.gov/media/78573/download. Accessed: 25 Aug 2022.
European Medicines Agency. Guideline on the evaluation of the pharmacokinetics of medicinal products in patients with impaired hepatic function. 2005. Available from: https://www.ema.europa.eu/en/evaluation-pharmacokinetics-medicinal-products-patients-impaired-hepatic-function-scientific. Accessed: 28 Apr 2023.
U.S. Food & Drug Administration. Pharmacokinetics in patients with impaired hepatic function: study design, data analysis, and impact on dosing and labeling. 2003. Available from: https://www.fda.gov/media/71311/download. Accessed: 25 Aug 2022.
Delcò F, Tchambaz L, Schlienger R, Drewe J, Krähenbühl S. Dose adjustment in patients with liver disease. Drug Saf. 2005;28:529–45.
el Touny M, el Guinaidy M, Abdel Bary M, Osman L, Sabbour MS. Pharmacokinetics of cefodizime in patients with liver cirrhosis and ascites. Chemotherapy. 1992;38(4):201–5.
Shepherd AN, Bouchier IA. The influence of ascites on the pharmacokinetics of piretanide in cirrhotic patients. Eur J Clin Pharmacol. 1985;28(5):581–3.
Acknowledgements
This study was sponsored by AstraZeneca. Medical writing support, under the direction of the authors, was provided by Sonya Frazier, PhD, CMC Connect, a division of IPG Health Medical Communications, and funded by AstraZeneca, in accordance with Good Publication Practice (GPP 2022) guidelines (Ann Intern Med. 2022;175(9):1298–1304. https://doi.org/10.7326/M22-1460).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This study was sponsored by AstraZeneca. Authors who are employees of AstraZeneca contributed to the conduct of the study and analysis described in the manuscript and assisted with the preparation of the manuscript.
Conflict of interest
All authors are employees and shareholders of AstraZeneca.
Ethics approval
This study was conducted in accordance with the ethical principles of the Declaration of Helsinki, consistent with the International Conference on Harmonisation Guideline for Good Clinical Practice and applicable regulatory requirements. The study protocol was approved by the local Independent Ethics Committee.
Consent to participate
All participants provided written informed consent prior to enrolment. The informed consent documents were approved by the local Independent Ethics Committee.
Consent for publication
All authors agree to publication.
Availability of data and materials
Data underlying the findings described in this manuscript may be obtained in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Code availability
Not applicable.
Author contributions
MS, AKM and VERP contributed to the conception or design of the study. All authors contributed to the acquisition, analysis, or interpretation of data. All authors contributed to the edit and review of the manuscript and approved the final version.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Mercier, AK., Sunnåker, M., Ueckert, S. et al. Pharmacokinetics and Tolerability of Zibotentan in Patients with Concurrent Moderate Renal and Moderate Hepatic Impairment. Clin Pharmacokinet 62, 1713–1724 (2023). https://doi.org/10.1007/s40262-023-01306-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-023-01306-7