
Abstract
Introduction
Major depressive disorder (MDD) is associated with a significant burden of disease in China. Awareness and better access to treatments could help alleviate the burden associated with MDD. Because variations have been observed in the pharmacokinetics (PK) of antidepressants across different races and ethnicities, evaluation of the clinical pharmacology of vortioxetine in diverse populations remains important to assess the potential need for dose adjustments.
Methods
Data were pooled from two phase I open-label PK studies in healthy Chinese subjects, and one phase III double-blind noninferiority study in Chinese patients with MDD to describe the PK and safety data for vortioxetine. Doses in these studies ranged from 10 mg (single dose) to 10 and 20 mg (multiple daily doses). A population PK analysis of vortioxetine in the Chinese population was conducted using nonlinear mixed-effect modeling.
Results
In total, 186 individuals were included in the PK analysis: 79 healthy Chinese subjects and 107 Chinese patients with MDD. No clinically significant differences in the PK of vortioxetine were observed between the Chinese population and the previous data in non-Chinese populations. Because of a generally lower weight in the Chinese population compared with the non-Chinese population, exposures were 19% and 18% higher in the Chinese population than in the non-Chinese population (for maximum observed plasma concentration and area under the plasma concentration–time curve, respectively), which is not considered clinically relevant. A high prevalence of pruritus was observed in one phase I PK study (56% overall); however, this was not reflected in the phase III study in Chinese patients with MDD (0.8%).
Conclusions
The PK parameters of vortioxetine in Chinese subjects were comparable to previous data in non-Chinese subjects. Overall, no new safety concerns were raised among the Chinese population. On the basis of this analysis, the tolerability profile of vortioxetine in Chinese healthy subjects and in patients with MDD is expected to be comparable to that in the non-Chinese population.
Funding
H. Lundbeck A/S, Valby, Denmark.
Trial Registration
NCT01676571.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
According to the World Health Organization’s Global Burden of Diseases, Injuries, and Risk Factors Study 2016, major depressive disorder (MDD) was one of the five leading causes of disability and constituted a major public health issue worldwide [1]. On the basis of a meta-analysis, the 12-month prevalence rate of MDD in China (2.3%) was lower than that in developing countries (4.0–10.4%) and similar to that in Japan (2.2%) [2]. Lack of clinical recognition of MDD among Chinese healthcare professionals may contribute to the low diagnosis and treatment rates of MDD [2, 3]. However, similar to the findings of the Global Burden of Disease Study, patients with MDD in China were reported to have poorer quality of life and greater productivity loss and resource utilization than nondepressed control subjects [4]. Awareness of MDD and better access to treatments could help alleviate the burden associated with MDD in China [4].
Vortioxetine (Trintellix®/Brintellix®, Takeda Pharmaceuticals America, Inc./H. Lundbeck A/S) is a multimodal antidepressant currently approved globally for the treatment of MDD [5, 6]. It has two different mechanisms of action: direct action at specific serotonin (5-hydroxytryptamine, 5-HT) receptors and serotonin reuptake inhibition [7]. In vitro, vortioxetine acts as a 5-HT3 and 5-HT7 receptor antagonist, 5-HT1B receptor partial agonist, 5-HT1A receptor agonist, and serotonin transporter (SERT) inhibitor [7]. Clinical pharmacodynamic (PD) studies of vortioxetine using positron emission tomography demonstrated that SERT was not fully saturated in therapeutic doses; these findings support a multimodal mechanism of action and suggest that vortioxetine may mediate its effects through actions on 5-HT receptors in addition to SERT inhibition [8, 9]. Moreover, preclinical studies have demonstrated that the antidepressant activity of vortioxetine is exerted via modulation of multiple neurotransmitters, including the serotonin, norepinephrine, dopamine, acetylcholine, histamine, glutamate, and gamma-aminobutyric acid systems [8].
Clinical dose-dependent efficacy and the safety profile of vortioxetine as a treatment for MDD have been established in multiple clinical studies over the dose range of 5–20 mg/day [10, 11]. In a recent systematic review and network meta-analysis of 21 antidepressants, an assessment of head-to-head trials determined that vortioxetine was among the more effective and tolerable agents for the acute treatment of adults with MDD [11].
Vortioxetine has linear and dose-proportional pharmacokinetics (PK) after administration of a single dose (10–75 mg/day) or multiple doses (2.5–60 mg/day) in healthy subjects [12]. Vortioxetine undergoes metabolism primarily via oxidation and subsequent glucuronic acid conjugation [12, 13]. The clinical PK of vortioxetine, as determined by noncompartmental analysis, are characterized by prolonged absorption, medium clearance (47 L/h), and a large volume of distribution (3.5 × 103 L) [12]. The absolute bioavailability for oral administration was found to be 75% [12]. Vortioxetine is mainly metabolized through cytochrome P450 2D6 (CYP2D6) and to a lesser degree through CYP2C19 [13].
A range of clinical pharmacology studies have demonstrated that administration of vortioxetine is not associated with clinically relevant induction and inhibition of CYP enzymes. In addition, drug–drug interaction studies showed no impact of vortioxetine on the PK of warfarin, diazepam, caffeine, tolbutamide, ethinylestradiol, levonorgestrel, midazolam, aspirin, ethanol, or lithium [8, 14].
Genes and their variants related to the serotonin transporter gene promoter polymorphism region (5-HTTLPR) and cytochrome P450 superfamily have been widely studied in association with antidepressant response [15]. According to the inherited CYP2D6 alleles, four categories are used to describe the phenotype: extensive metabolizers (EMs), intermediate metabolizers, poor metabolizers (PMs), and ultrarapid metabolizers [16, 17]. PMs have higher plasma concentrations of antidepressants metabolized by CYP2D6 than EMs and are therefore more likely to experience dose-dependent adverse drug reactions [18]. PK and PD studies of antidepressants have also reported variations across different races due to genetic polymorphisms [15, 19]. Thus, it is important that the clinical pharmacology of antidepressants be evaluated in diverse populations to assess the need for dose adjustments [20]. To date, the PK and safety profile of vortioxetine in Chinese subjects have not been reported.
The objective of the current analysis was to assess the PK and safety of vortioxetine in a Chinese population by gathering data from Chinese patients with MDD and healthy Chinese subjects. A population PK analysis of three studies of vortioxetine in both healthy Chinese subjects and patients with MDD was conducted and the results were compared with previous data from global trials in which non-Chinese individuals constituted the majority of subjects.
Methods
Study Data
PK and safety data included in this post hoc analysis were pooled from two phase I open-label vortioxetine PK studies in healthy Chinese subjects and from one phase III double-blind, noninferiority study in Chinese patients with MDD who were treated with vortioxetine [21].
Studies Included
Study 1 (Single-Dose PK Study)
This was a single-center, open-label PK study with a primary objective to determine the PK of vortioxetine after a single dose of 10 mg administered orally under fasting conditions in healthy Chinese men and women (Trial registration: www.ClinicalTrials.gov identifier, NCT01676571).
Study 2 (Single- and Multiple-Dose PK Study)
This was a randomized open-label, single-center, single-dose, and multiple-dose PK study with a primary objective to determine the PK parameters after 10- and 20-mg single-dose and 10- and 20-mg multiple-dose administration of vortioxetine in healthy Chinese men and women (Trial registration: www.ClinicalTrials.gov identifier, NCT02386488). In the single-dose part, 16 male and 16 female subjects were randomized 1:1 to receive a single oral dose of 10 or 20 mg of vortioxetine on day 1. In the multiple-dose part, 16 male and 16 female subjects were randomized 1:1 to receive an oral dose of 10 or 20 mg of vortioxetine, respectively, each day for 14 consecutive days. Safety and tolerability were assessed throughout the study.
Study 3 (Active Comparator Noninferiority Study)
This was a multinational interventional, multicenter, randomized, double-blind, parallel-group, fixed-dose, active-comparator noninferiority study in patients with MDD (Trial registration: www.ClinicalTrials.gov identifier, NCT01571453). The study included 443 randomized patients recruited from 31 psychiatric inpatient and outpatient specialist settings in four countries (China, South Korea, Taiwan, and Thailand) from April 2012 to October 2013. Of note, only the PK and safety results from the 124 Chinese patients treated with vortioxetine were included in the current study’s pooled analyses. Efficacy results from the study have been published previously [21].
Study details for all three studies are summarized in Table 1.
Study Participants
For studies 1 and 2, participants were screened for enrollment from 21 to 2 days before treatment. Participants who met the inclusion criteria for each study were eligible for trial participation: men and women aged between 18 and 45 years; body weight at least 50 kg (study 1) or at least 45 kg (study 2); and body mass index between 19 and 24 kg/m2 (study 1) or between 18.5 and 24 kg/m2 (study 2). Major exclusion criteria in studies 1 and 2 included the following: disallowed medication taken less than 1 week before the first dose of study drug, or less than 5 half-lives before the screening visit for any medication taken; clinically significant comorbidities that might complicate treatment or potentially confound the study results; known hypersensitivity or allergies to medications; or clinically significant vital sign, laboratory test, or electrocardiogram abnormality at screening.
For study 3, participants were screened for enrollment from 7 days before treatment. Inclusion and exclusion criteria details for study 3 have been published previously [21]. Briefly, patients with a primary diagnosis of recurrent MDD according to Diagnostic and Statistical Manual of Mental Disorders-IV-Text Revision (DSM-IV-TR™) criteria, a Montgomery–Åsberg Depression Rating Scale (MADRS) total score of at least 26, and a Clinical Global Impression–Severity (CGI-S) score of at least 4 with a reported duration of the current major depressive episode of at least 3 months were eligible. Participants were ineligible if they had any current anxiety disorder (DSM-IV-TR™ criteria) as assessed using the Mini International Neuropsychiatric Interview (MINI) or a current diagnosis or history of manic or hypomanic episode, schizophrenia, or any other psychotic disorder. Only the PK and safety results from the Chinese patients treated with vortioxetine (n = 124) in this study were included in the pooled analysis.
PK Evaluation
The population PK of vortioxetine were determined using nonlinear mixed-effect modeling with NONMEM® (ICON Development Solutions) version 7. The first-order conditional error with interaction (FOCE INTER) minimization method was used.
No formal statistically based strategy or rules for handling outliers among the plasma concentrations or covariate data were used. Visual inspection of the data was used to detect potential outliers to exclude from the analysis. The structural model used was a two-compartment model based on that previously described in the pooled-population PK analysis in healthy subjects [22] with lag time and with first-order absorption and elimination. The model, schematically shown in Fig. 1, was parameterized in terms of ALAG (lag time), ka (absorption rate constant), CL/F (oral clearance), V2/F (volume of distribution of central compartment), Q/F (intercompartmental clearance), and V3/F (volume of distribution of peripheral compartment).

Schematic of PK model for vortioxetine. ALAG lag time, CL/F oral clearance, ka absorption rate constant, PK pharmacokinetics, Q/F intercompartmental clearance, V2/F volume of distribution of central compartment, V3/F volume of distribution of peripheral compartment
The continuous covariates of age, weight, height, and creatinine clearance, and the categorical covariates of sex and healthy/patient were tested in the population PK model. Standard goodness-of-fit plots were generated to evaluate the fit of the base model to the data. Details of the visual predictive check plot and goodness-of-fit plots are provided in supplementary material. The model was evaluated using the normalized prediction distribution error and visual predictive check plots. The condition number was used to evaluate the stability of the model. The standard errors of the final model parameter values were estimated using the nonparametric bootstrap approach. The elimination half-life (t1/2) was estimated for each subject on the basis of the individual parameter values.
Safety Evaluation
All treatment-emergent adverse events (TEAEs) either observed by the investigator or reported spontaneously by the patients and healthy subjects were recorded. Qualified personnel coded TEAEs using the lowest-level term according to the Medical Dictionary for Regulatory Activities (MedDRA), version 16.0. Clinical safety laboratory tests, vital signs, weight, body mass index, electrocardiograms, and physical examination findings were also evaluated.
Compliance with Ethics Guidelines
All study protocols were approved by the local ethics committees and were conducted in compliance with the International Conference for Harmonization, Harmonized Tripartite Guideline E6 for Good Clinical Practice. Enrolled participants for each study provided written informed consent before undergoing any study procedures.
Results
Quantifiable plasma concentrations of vortioxetine from a total of 186 individuals were included in the analysis: 16 healthy Chinese subjects from study 1, 63 healthy subjects from study 2 (1 subject was excluded because of lack of quantifiable plasma concentrations), and 107 Chinese patients with MDD from study 3. No apparent clinically relevant differences in age, height, body weight, or body mass index at baseline were noted among the three studies. In study 3, the mean age of the subjects was slightly higher than in studies 1 and 2 (Table 2).
PK Evaluation
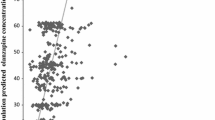
The base model described the data well after fixing lag time (ALAG) to the value of 0.781 found in the previous pooled phase I analysis [22]. Weight on central volume of distribution (V2/F) was the only significant covariate–parameter relationship, with an increase in V2/F with increasing weight. The PK model parameters were estimated with good precision, with a coefficient correlation of 0.60 between population predicted and observed plasma concentration. Observed plasma concentrations (dose-normalized to 10 mg) at steady state together with model-predicted median and 95% CI are shown in Fig. 2, whereas other goodness-of-fit plots, including the visual predictive check plot, are given in the supplementary material (Figs. S1–S9). On the basis of the final model parameters, area under the curve over 24 h (AUC0–24) and maximum concentration (Cmax) values at steady state for 10 mg were simulated for the analysis data set. Table 3 shows PK parameters, including Cmax and AUC0–24, for vortioxetine in the Chinese population, which were similar to the data from previous pooled analyses in which non-Chinese subjects constituted the majority [22, 23].

Observed plasma concentration (dose-normalized to 10 mg) at steady state together with model-predicted median and 95% CI. CI confidence interval
Safety Evaluation
The overall incidences (Table 4) and patterns of TEAEs with vortioxetine in Chinese patients and healthy subjects were comparable to those in the global clinical development program. In general, TEAEs with vortioxetine were rated as mild to moderate in intensity. Nausea is a common side effect of antidepressants, occurring at a rate of 21–34% in clinical trials of vortioxetine [5, 6]. Consistent with this result, nausea was observed across all three studies analyzed in this report [6% in study 1, 31.3% (10/32 subjects, multiple-dose group) in study 2, and 20.2% in study 3]. Dizziness was also seen across all three studies [6% in study 1, 3.1% (1/32 subjects, multiple-dose group) in study 2, and 9.7% in study 3]. Nausea and dizziness were also the two adverse events with the highest incidences in study 1 and study 3; however, in study 2, the adverse event with the highest incidence was pruritus (18/32 subjects; 56% overall). The incidence of pruritus was higher in the 20-mg vortioxetine group (13/16 subjects; 81.3%) than in the 10-mg vortioxetine group (5/16 subjects; 31.3%). Pruritus was not observed in the single-dose study (study 1) or in the single-dose group of study 2 and was reported in only 0.8% of Chinese patients (n = 124) in the vortioxetine group in study 3.
Discussion
Results of these analyses demonstrate that the PK and safety parameters of vortioxetine in Chinese subjects were similar to previous data in which non-Chinese individuals constituted the majority of subjects (Table 3) [22, 23]. Similar to our findings in this study, phase I trials conducted in healthy young Japanese adults and elderly Japanese subjects found no clinically significant differences in the PK of vortioxetine or its metabolites in Japanese adults compared with a non-Japanese population [20]. In the previously published pooled-population PK analysis (a meta-analysis of 26 phase I studies with data from 887 healthy subjects who received single doses of vortioxetine ranging from 2.5 to 75 mg or multiple doses of 2.5–60 mg), CYP2D6-predicted phenotype (ultrarapid, extensive, intermediate, or poor metabolizers), age on oral clearance, and height on central volume of distribution were identified as statistically significant covariate–parameter relationships [22]. Impact of CYP2D6 on oral clearance was the most significant; EMs were estimated to have approximately twice the oral clearance of PMs [22]. CYP2C19-predicted phenotype also had a significant impact on oral clearance, but to a lesser degree, with an EM to PM ratio of 1.4 [22]. Genotyping of CYP2D6/CYP2C19 was not included in the noninferiority study and hence could not be evaluated in the population PK analysis for Chinese subjects. The distribution of CYP2D6 alleles is different in the Chinese population than in a general non-Chinese population. The proportion of CYP2D6 PMs among European and US non-Chinese is 7%, and in other races it is 1–2% [24]. The frequency of CYP2D6 intermediate metabolizers is higher in Chinese subjects than in non-Chinese subjects. For CYP2C19, PMs are more frequent among Chinese subjects than among non-Chinese subjects [25]. Taken together, there is no reason to believe that the Chinese population in general should have a markedly different clearance of vortioxetine because of different distributions of CYP2D6 and CYP2C19 alleles, although it is anticipated that there will be fewer Chinese subjects at the lower end of the clearance range because there are fewer CYP2D6 PMs than among non-Chinese. For Chinese subjects evaluated in the current analysis compared with the healthy subjects in the previously performed pooled-population PK analysis, the values for observed oral clearance (31 L/h vs. 33 L/h) and mean elimination t1/2 (79.8 ± 26.8 L/h vs. 65.8 ± 26.8 L/h) also supported a lack of difference in vortioxetine PK between Chinese and non-Chinese populations.
In the present population PK analysis in Chinese subjects, the only significant covariate–parameter relationship was weight on V2/F. Because of a generally lower weight in the Chinese population (mean weight ± SD, 61.4 ± 10.8 kg for the population PK data set) than in the non-Chinese population (76 ± 19 kg and 73.6 ± 12.3 kg [22, 23]), exposure (in terms of Cmax and AUC) was 19% (Cmax) and 18% (AUC) higher in the Chinese population than in the non-Chinese population, which is not considered clinically relevant (relevant change is defined here as lower than 20% or higher than 25%, according to accepted bioequivalence criteria) [26].
Although Chinese healthy subjects and patients showed slightly greater exposure of vortioxetine compared with previous studies, the difference remained within bioequivalence criteria and thus clinical effects are expected to be similar to the non-Chinese population. Interestingly, results of a population PK/PD model suggest a nonlinear relationship between vortioxetine blood levels and response, with higher doses of vortioxetine on average predictive of larger improvements in depressive symptomatology. In addition, differences in the PK/efficacy model were also observed across geographic region, though the authors attribute this most likely to non-compliance observed across trials. Further, the results of PK/safety models suggest that nausea rates increase with increasing dose. Overall, these models support the appropriateness of a 5–20 mg therapeutic dose range that is adjustable on the basis of patient response and tolerability. These recommendations would apply for vortioxetine in any population, regardless of ethnicity [23].
Vortioxetine was generally well tolerated in these healthy young Chinese men and women and Chinese patients with MDD. A very high incidence of pruritus was noted in one phase I (single/multiple dose) PK study and appeared to be frequency and dose dependent; all reports of pruritus were of mild intensity. This adverse event may not be unique to the Chinese population, especially since it showed a large variation in frequency among the three studies. Also, because healthy subjects in phase I trials were monitored closely for detection of adverse events, differences in reporting of adverse events between the phase I and phase III studies might have contributed to this variability. Of note, the frequency of pruritus in the phase III study in Chinese patients with MDD was less than 1%, which is consistent with pruritus frequencies of less than 5% in patients in the global phase III study of vortioxetine in patients with MDD [27].
Findings from this study and those from the phase I trials conducted in healthy young Japanese adults and elderly Japanese subjects collectively indicate similar clinical pharmacology for vortioxetine irrespective of ethnicity. Thus, vortioxetine PK can be extrapolated to other Asian populations without the need for dose adjustments.
Study Limitations
The narrow inclusion and exclusion criteria used in clinical pharmacological studies could limit the generalizability of the study. One of the reasons age and creatinine clearance did not emerge as significant covariates could be the limited number of subjects in this population PK analysis for Chinese subjects compared with the larger pooled-population PK analyses. Because of the sparse PK sampling in study 3 in Chinese patients with MDD (maximum of two PK samples per patient), central volume of distribution (76%) and absorption rate constant (68%) could have been underestimated. This so-called ETA (individual parameter estimate) shrinkage for central volume of distribution affects the individual estimates of elimination t1/2 for the patients, especially the interindividual variability. For the phase I single-dose PK study, mean t1/2 was estimated (through noncompartmental analysis) to be 73 h, i.e., very close to the 66 h found in the pooled-population PK analysis in healthy subjects.
Conclusions
This pooled analysis of vortioxetine (10 mg [single dose] to 10 and 20 mg [multiple daily doses]) in a Chinese population showed PK parameters similar to previous data from studies in which non-Chinese people constituted the majority of subjects or patients. The slightly higher exposure (19% higher Cmax and 18% higher AUC) in the Chinese population compared with the non-Chinese population might be explained by a generally lower weight in the Chinese population. On the basis of this analysis, the tolerability profile of vortioxetine in Chinese healthy subjects and in patients with MDD is expected to be comparable to that in the non-Chinese population. No new safety concerns were raised among the Chinese population administered vortioxetine. These results support that there is no need to adjust the dosage of vortioxetine in Chinese patients with MDD.
References
GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211–59. https://doi.org/10.1016/s0140-6736(17)32154-2.
Gu L, Xie J, Long J, et al. Epidemiology of major depressive disorder in mainland China: a systematic review. PLoS One. 2013;8:e65356. https://doi.org/10.1371/journal.pone.0065356.
Hou Z, Jiang W, Yin Y, Zhang Z, Yuan Y. The current situation on major depressive disorder in China: research on mechanisms and clinical practice. Neurosci Bull. 2016;32:389–97. https://doi.org/10.1007/s12264-016-0037-6.
Gupta S, Goren A, Dong P, Liu D. Prevalence, awareness, and burden of major depressive disorder in urban China. Expert Rev Pharmacoecon Outcomes Res. 2016;16:393–407. https://doi.org/10.1586/14737167.2016.1102062.
TRINTELLIX [prescribing information]. Deerfield, IL: Takeda Pharmaceuticals America, Inc; 2018.
BRINTELLIX [summary of product characteristics]. Valby, Denmark: H. Lundbeck A/S; 2019.
Bang-Andersen B, Ruhland T, Jorgensen M, et al. Discovery of 1-[2-(2,4-dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): a novel multimodal compound for the treatment of major depressive disorder. J Med Chem. 2011;54:3206–21. https://doi.org/10.1021/jm101459g.
Chen G, Hojer AM, Areberg J, Nomikos G. Vortioxetine: clinical pharmacokinetics and drug interactions. Clin Pharmacokinet. 2018;57:673–86. https://doi.org/10.1007/s40262-017-0612-7.
Stenkrona P, Halldin C, Lundberg J. 5-HTT and 5-HT(1A) receptor occupancy of the novel substance vortioxetine (Lu AA21004). A PET study in control subjects. Eur Neuropsychopharmacol. 2013;23:1190–8. https://doi.org/10.1016/j.euroneuro.2013.01.002.
Thase ME, Mahableshwarkar AR, Dragheim M, Loft H, Vieta E. A meta-analysis of randomized, placebo-controlled trials of vortioxetine for the treatment of major depressive disorder in adults. Eur Neuropsychopharmacol. 2016;26:979–93. https://doi.org/10.1016/j.euroneuro.2016.03.007.
Cipriani A, Furukawa TA, Salanti G, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391:1357–66. https://doi.org/10.1016/S0140-6736(17)32802-7.
Areberg J, Sogaard B, Hojer AM. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012;111:198–205. https://doi.org/10.1111/j.1742-7843.2012.00886.x.
Hvenegaard MG, Bang-Andersen B, Pedersen H, Jorgensen M, Puschl A, Dalgaard L. Identification of the cytochrome P450 and other enzymes involved in the in vitro oxidative metabolism of a novel antidepressant, Lu AA21004. Drug Metab Dispos. 2012;40:1357–65. https://doi.org/10.1124/dmd.112.044610.
Chen G, Zhang W, Serenko M. Lack of effect of multiple doses of vortioxetine on the pharmacokinetics and pharmacodynamics of aspirin and warfarin. J Clin Pharmacol. 2015;55:671–9. https://doi.org/10.1002/jcph.456.
Crisafulli C, Fabbri C, Porcelli S, et al. Pharmacogenetics of antidepressants. Front Pharmacol. 2011;2:6. https://doi.org/10.3389/fphar.2011.00006.
Garcia-Barcelo M, Chow LY, Lam KL, Chiu HF, Wing YK, Waye MM. Occurrence of CYP2D6 gene duplication in Hong Kong Chinese. Clin Chem. 2000;46:1411–3.
Sachse C, Brockmoller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population: allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60:284–95.
Han KM, Chang HS, Choi IK, Ham BJ, Lee MS. CYP2D6 P34S polymorphism and outcomes of escitalopram treatment in Koreans with major depression. Psychiatry Investig. 2013;10:286–93. https://doi.org/10.4306/pi.2013.10.3.286.
Lee MS. Role of genetic polymorphisms related to neurotransmitters and cytochrome P-450 enzymes in response to antidepressants. Drugs Today (Barc). 2007;43:569–81. https://doi.org/10.1358/dot.2007.43.8.1130447.
Matsuno K, Nakamura K, Aritomi Y, Nishimura A. Pharmacokinetics, safety, and tolerability of vortioxetine following single- and multiple-dose administration in healthy Japanese adults. Clin Pharmacol Drug Dev. 2018;7:319–31. https://doi.org/10.1002/cpdd.381.
Wang G, Gislum M, Filippov G, Montgomery S. Comparison of vortioxetine versus venlafaxine XR in adults in Asia with major depressive disorder: a randomized, double-blind study. Curr Med Res Opin. 2015;31:785–94. https://doi.org/10.1185/03007995.2015.1014028.
Areberg J, Petersen KB, Chen G, Naik H. Population pharmacokinetic meta-analysis of vortioxetine in healthy individuals. Basic Clin Pharmacol Toxicol. 2014;115:552–9. https://doi.org/10.1111/bcpt.12256.
Naik H, Chan S, Vakilynejad M, et al. A population pharmacokinetic-pharmacodynamic meta-analysis of vortioxetine in patients with major depressive disorder. Basic Clin Pharmacol Toxicol. 2016;118:344–55. https://doi.org/10.1111/bcpt.12513.
de Leon J, Susce MT, Pan RM, Fairchild M, Koch WH, Wedlund PJ. The CYP2D6 poor metabolizer phenotype may be associated with risperidone adverse drug reactions and discontinuation. J Clin Psychiatry. 2005;66:15–27.
Yin SJ, Ni YB, Wang SM, Wang X, Lou YQ, Zhang GL. Differences in genotype and allele frequency distributions of polymorphic drug metabolizing enzymes CYP2C19 and CYP2D6 in mainland Chinese Mongolian, Hui and Han populations. J Clin Pharm Ther. 2012;37:364–9. https://doi.org/10.1111/j.1365-2710.2011.01298.x.
Davit B, Braddy AC, Conner DP, Yu LX. International guidelines for bioequivalence of systemically available orally administered generic drug products: a survey of similarities and differences. AAPS J. 2013;15:974–90. https://doi.org/10.1208/s12248-013-9499-x.
Baldwin DS, Chrones L, Florea I, et al. The safety and tolerability of vortioxetine: analysis of data from randomized placebo-controlled trials and open-label extension studies. J Psychopharmacol. 2016;30:242–52. https://doi.org/10.1177/0269881116628440.
Acknowledgements
The authors acknowledge Dr Qi Shen, working at the Phase I Unit of West China Hospital, for his work on the pharmacokinetics study in healthy volunteers.
Funding
This study and Rapid Service Fee were funded by H. Lundbeck A/S, Valby, Denmark. The sponsor was involved in study design and implementation, analysis, and interpretation of the data, writing of the report, and the decision to submit the article for publication. The authors retained full control of the content of the manuscript. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and/or Editorial Assistance
Medical writing support was provided by inVentiv Medical Communications, LLC, a Syneos Health™ group company. Support for this assistance was funded by H. Lundbeck A/S, Valby, Denmark. We thank the participants of the study.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Jia Miao is a professor working at the Phase I Unit of West China Hospital, Sichuan University, a nonprofit academic research center; these phase I trials are funded in part by unrestricted grants from pharmaceutical firms, as well as contract research organizations. Jia Miao reports no other potential conflicts of interest relevant to this article. Johan Areberg is a full-time employee of H. Lundbeck A/S. Anders Ettrup is a full-time employee of H. Lundbeck A/S. Astrid-Maria Højer is a full-time employee of H. Lundbeck A/S. Yan Zhao is a full-time employee of Lundbeck, Beijing, China. Gang Wang and Jie Hou have nothing to disclose.
Compliance with Ethics Guidelines
All study protocols were approved by the local ethics committees and were conducted in compliance with the International Conference for Harmonization, Harmonized Tripartite Guideline E6 for Good Clinical Practice. Enrolled participants for each study provided written informed consent before undergoing any study procedures.
Data Availability
The authors declare that the data supporting the findings of this study are available within the article. The authors may be contacted for further data sharing.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9771947.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Miao, J., Wang, G., Hou, J. et al. Pharmacokinetics and Safety of Vortioxetine in the Chinese Population. Adv Ther 36, 3134–3146 (2019). https://doi.org/10.1007/s12325-019-01092-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-019-01092-4