
Abstract
Background/Objectives
This study will aim to characterise disease behaviour during the peri-diagnostic period in patients with suspected interstitial lung disease (ILD), including idiopathic pulmonary fibrosis (IPF), using daily home spirometry and accelerometry. Additionally, this study will aim to increase collaboration between secondary and tertiary centres using a digital collaboration platform.
Methods
The STARLINER study (NCT03261037) will enrol approximately 180 symptomatic patients aged 50 years or more with radiological evidence of ILD/IPF from community and tertiary centres in Canada and Europe. Approximately two-thirds of sites will be community centres. Patients will be followed during pre-diagnosis (inclusion to diagnosis; up to a maximum of 12 months) and post-diagnosis (diagnosis to treatment initiation; up to a maximum of 6 months). The study will be facilitated by a digital ecosystem consisting of the devices used for home-based assessments and a digital collaboration platform enabling communication between community and tertiary centres, and between clinicians and patients.
Planned Outcomes
The primary endpoint will be time-adjusted semi-annual change in forced vital capacity (FVC; in millilitres) during the peri-diagnostic period. Physical functional capacity and patient-reported outcomes (PROs) will also be assessed. FVC and physical functional capacity will be measured using daily home spirometry and accelerometry, and at site visits using spirometry and the 6-min walk test. PROs will be assessed prior to, or during, site visits and will always be completed in the same order.
Conclusions
Findings from this study may help to facilitate the early and accurate diagnosis of ILDs by increasing knowledge about disease progression, enabling collaboration between community and tertiary centres and improving communication between clinicians and patients.
Trial Registration Number
NCT03261037.
Funding
F. Hoffmann-La Roche, Ltd., Basel, Switzerland.
Plain Language Summary
Plain language summary available for this article.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Plain Language Summary
Interstitial lung diseases (ILDs) are a group of disorders involving inflammation and/or scarring of the lungs. The cause of ILD can be unclear in some patients. Diagnosis of ILD can be complicated, and patients sometimes have to wait more than 12 months before being diagnosed.
Reasons why patients might wait a long time for a diagnosis include:
-
The symptoms of ILD can be confused with those of other conditions. Therefore, patients with ILD are sometimes told they have another disease first.
-
Patients with ILD are usually treated at specialist hospitals by doctors who are experts in ILD. Some patients have to wait a long time for an appointment at these specialist centres and have to travel long distances to their nearest centre.
If doctors could learn more about ILD from clinical studies and how symptoms change over time, they might be able to diagnose ILD faster.
This paper describes the STARLINER study, which will include patients whose doctors think they might have an ILD. An online system will be used to track measurements that patients take at home, and will allow communication between community and specialist centres and between doctors and patients. STARLINER will assess changes in how well patients’ lungs work, which patients will measure at home. Patients will also provide their daily step count and calorie use by wearing a smartwatch. It is hoped that the findings from this study will increase understanding of how ILD develops, leading to earlier diagnosis and improving communication between doctors and patients.
Introduction
The interstitial lung diseases (ILDs) are a diverse group of disorders characterised by inflammation and/or fibrosis of the lungs [1, 2]. Although some patients may have a known or suspected cause of ILD, such as an underlying connective tissue disease or certain environmental or occupational exposures, other patients may lack a clear cause [1, 2]. Idiopathic pulmonary fibrosis (IPF), which is a progressive and fatal disease of unclear aetiology, is among the most common of the ILDs [3, 4]. IPF is generally more prevalent in men and current or ex-smokers, and is primarily diagnosed in the sixth or seventh decade of life [4,5,6]. There are currently no available treatments for IPF that can reverse lung damage; however, two antifibrotics, pirfenidone and nintedanib, have been shown to significantly reduce disease progression in patients with IPF vs. placebo [7,8,9,10]. With the availability of therapies that can slow disease progression, earlier diagnosis and treatment of IPF may potentially lead to improved patient outcomes, particularly if patients can be diagnosed before substantial irreversible lung damage has occurred [11, 12].
The diagnosis of ILDs, including IPF, is a complex process, and many patients experience delays of over 12 months [13,14,15]. Misdiagnosis is a key cause of delays, with evidence suggesting that 55% of patients experience at least one misdiagnosis prior to a diagnosis of ILD [13]. Factors leading to misdiagnosis include the non-specific nature of initial symptoms in ILD [13] and geographical barriers to accessing specialist centres and multidisciplinary teams (MDTs) [16]. Delayed access to specialist centres is associated with an increased risk of mortality independent of pulmonary function [14] and may lead to the use of inappropriate or ineffective treatments. In addition, previous studies have shown that input from an MDT can lead to more accurate and confident diagnosis of ILD [17, 18]. Therefore, there is a need for increased collaboration between secondary and tertiary centres to increase access to MDTs and subsequently improve the diagnosis of ILDs, with virtual MDTs holding some promise, particularly for the diagnosis of IPF [19].
An additional barrier to the early diagnosis and treatment of ILD is the current lack of evidence characterising ILD behaviour and progression during the early stages of disease [20]. In general, clinical trials investigating patients with ILD have collected data after diagnosis and initiation of treatment, with clinical measurements performed approximately every 3–6 months [8, 9, 21]. As such, there is a clear need for study designs utilising more frequent data collection, with a focus on defining disease trajectories during the pre- and post-diagnostic periods (peri-diagnostic period) in patients with ILD. Observational studies utilising daily home spirometry and accelerometry have shown that these measurements by patients with IPF are feasible to perform [22,23,24,25,26], and can be used to predict mortality [23, 26]. In combination with the benefit of allowing daily measurements, these observations suggest that home-based assessments could be utilised for the characterisation of early disease behaviour in ILDs, potentially guiding diagnostic decisions [27].
This paper describes the design of a study aiming to characterise disease behaviour in patients with ILD, including IPF, during the peri-diagnostic period using daily home-based measurements. Communication between community and ILD tertiary centres will be facilitated by a digital collaboration platform.
Methods
Study Design
The STARLINER study (ClinicalTrials.gov NCT03261037) is a multicentre, ongoing study that plans to enrol approximately 180 patients across approximately 40 community and tertiary clinical centres in Canada, France, Ireland, Italy, the Netherlands and Russia (for further details, visit https://clinicaltrials.gov/ct2/show/study/NCT03261037). To reflect clinical practice, where many patients may not be treated at ILD tertiary centres (i.e. specialist centres), this study will aim for approximately two-thirds of sites to be community centres.
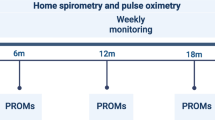
This study will assess disease behaviour in patients with suspected ILD, including IPF, during the peri-diagnostic period, which will consist of the pre-diagnostic period (from inclusion to diagnosis; up to a maximum of 12 months) and the post-diagnostic period (from diagnosis to treatment initiation; or up to 6 months after diagnosis) (Fig. 1). Disease behaviour will be assessed in terms of pulmonary function and physical functional capacity, which will be measured at home using daily spirometry and accelerometry, respectively, and at site visits using site spirometry and the 6-min walk test (6MWT), respectively.

Study procedures. ILD interstitial lung disease, IPF idiopathic pulmonary fibrosis. aPatients will leave the study if there is no diagnosis within 12 months of inclusion or a non-ILD diagnosis
Patients will leave the study if they do not receive a diagnosis within 12 months of inclusion or if they receive a non-ILD diagnosis (Fig. 1). Patients diagnosed with ILD within 12 months of inclusion will remain in the study for a maximum of 6 months after the patient receives a diagnosis from the investigator, or until the date treatment commences. Therefore, the total length of the study will vary for each patient (up to a maximum of 18 months) depending on their individual patient journey and local clinical practice.
The study design will be facilitated by a digital ecosystem consisting of two pillars (Fig. 2). Pillar 1 will comprise the spirometers and accelerometers that will be used for home-based assessments, which will have a real-time connection to a tablet computer, allowing patients to view their results in real time. Pillar 2 will include the digital collaboration platform, which will enable virtual MDT discussions between community and tertiary sites (Fig. 2). Additionally, the tablet computer will act as a bridge between the two pillars, linking the patient data to the collaboration platform, where investigators will be able to access their patients’ data in real time (Fig. 2).

Digital ecosystem. FVC forced vital capacity, ILD interstitial lung disease, IPF idiopathic pulmonary fibrosis, MDT multidisciplinary team
Sample Selection
Symptomatic patients (unexplained dyspnoea on exertion and/or cough) with suspected ILD/IPF aged 50 years or more with radiological evidence (on chest radiograph or computed tomography scan) of ILD/IPF will be eligible for inclusion. In accordance with the investigators’ judgement, patients must be able to comply with the study protocol and must provide signed informed consent to the investigators. Patients will be excluded if they participate in any investigational study within 28 days prior to inclusion, have a history of clinically significant cardiac disease that could explain their symptomatology in the opinion of the investigators, or have a known history of any connective tissue disease, including, but not limited to, rheumatoid arthritis, scleroderma, systemic lupus erythematosus or mixed connective tissue disease.
Measurements
No investigational medicinal product will be assessed in this study, and patients will be managed according to the standard of care at the discretion of the treating clinician. All medications taken for comorbid conditions will be continued as necessary and will be recorded throughout the study. Three mandatory site visits will be conducted at baseline, diagnosis and end of study for each individual patient, and additional site visits will be scheduled at the discretion of the investigator. This study will be conducted under real-life conditions, and therefore the timing of all mandatory and additional site visits will not be pre-determined, although additional site visits should include at least one visit during each study period (pre- and post-diagnosis). Mandatory assessments will consist of pulmonary function, physical functional capacity and patient-reported outcome (PRO) measures (see Table S1 in the Electronic Supplementary Material). Any other assessments will be performed at the discretion of investigators, according to local clinical practice.
For home-based assessments, daily measurement of forced vital capacity (FVC; in millilitres) will be performed using a portable handheld spirometer (Spirobank® Smart; Medical International Research, Rome, Italy) (Table 1). Patients will receive training on how to perform spirometry at enrolment and will perform “test blows” until they can demonstrate a reliable manoeuvre. Patients will also be provided with a manual and will have access to a digital avatar to help them complete the spirometry assessments successfully. Physical functional capacity will be assessed using continuous daily measurement of steps per day and calorie expenditure with an accelerometer resembling a watch (Steel HR; Withings, Issy-les-Moulineaux, France) (Table 1), which will be worn around the wrist. Patients will receive training on how to use the accelerometer at enrolment and will be provided with an instruction manual for the device.
At site visits, pulmonary function and physical functional capacity will be assessed, according to the instructions provided to investigators in the study procedure manual, using site spirometry and the 6MWT, respectively (Table 1; see also Table S1 in the Electronic Supplementary Material). The 6MWT will only be performed at sites where a formalised process is available and the test can be performed under safe conditions. Patients will complete the PRO assessments at baseline and at the request of the investigator just prior to, or during, site visits, and they will always be completed in the same order. PROs will include the King’s Brief Interstitial Lung Disease questionnaire, the modified Medical Research Council Dyspnoea Scale, the 5-Level EuroQol 5-Dimension questionnaire, the Fatigue Assessment Scale, a Cough-Visual Analogue Scale (VAS), an Urge-to-Cough VAS and a Fatigue-VAS (Table 1).
Digital Collaboration Platform
At study enrolment, each patient will receive a handheld spirometer, an accelerometer, a tablet computer and user manuals. Patients will be able to view their spirometry and accelerometry data on their tablet computers, which will also act as a conduit for real-time data transmission to the collaboration platform, where investigators will be able to access their patients’ data (Fig. 2). In the event that patients do not wish to have access to their clinical data, or become distressed as a result of viewing it, investigators will have the option to block the patient’s data access on the tablet. Investigators will be notified via email and text message if a patient has not measured their pulmonary function for 3–4 consecutive days or if more than 10% relative decline from baseline in FVC (in millilitres) is recorded for three consecutive days. Patients will also be able to request for their site investigators to contact them via a “call back” feature on their tablet computer. Investigators from community sites will be able to share data (such as high-resolution computed tomography images and home assessment results) with other centres via the collaboration platform, allowing virtual MDT discussions to take place between community and tertiary sites (Fig. 2). Discussions between centres can take place at any time during the study, with all diagnoses at the discretion of the investigators. Participation in the virtual MDT is optional, and the clinical experts included will depend on the requirements of the individual sites. The study sponsor will not have access to the digital collaboration platform. Patient data from the tablet computers will be anonymised and transferred to the study database for analysis.
Planned Outcomes
The primary endpoint of this study is the time-adjusted semi-annual change in FVC (in millilitres) in patients with IPF, measured using daily home spirometry, during the peri-diagnostic period. A full list of study endpoints is presented in Table S2 in the Electronic Supplementary Material.
Secondary endpoints include changes in FVC in patients with ILD, excluding patients with IPF, measured using daily home spirometry; changes in FVC in patients with ILD and IPF, measured using site spirometry; changes in physical functional capacity in patients with ILD and IPF, measured using daily home accelerometry and site 6MWT; and the correlation between home and site measurements for pulmonary function and physical functional capacity. Changes in health status and quality of life, measured using PROs, and the incidence of non-elective hospitalisations, investigator-reported acute exacerbations and mortality will also be recorded (see Table S2 in the Electronic Supplementary Material).
A number of exploratory endpoints will also be assessed, including an analysis of baseline characteristics and changes in FVC by ILD subgroup. At the end of the study, patients and investigators will complete structured surveys that will collect data on the usefulness and practicality of the collaboration platform and the digital ecosystem; for example, their general experiences with the technology and whether investigators collaborated with a tertiary site. Any non-compliance to home assessments (for seven or more consecutive days) will be counted objectively throughout the study period.
This study has no investigational medicinal product and patients will leave the study prior to initiating pharmacological treatment for ILD; therefore, there will be no safety objectives. Any events thought to be related to a medicinal product used during the course of a patient’s standard medical treatment will be reported to the market authorisation holder or local health authority, according to local regulatory requirements.
Data Analysis
The planned sample size of 180 patients is based on an assumption that approximately 40% of enrolled patients will be diagnosed with IPF, 50% will be diagnosed with non-IPF ILD and 10% will receive a non-ILD diagnosis or will remain undiagnosed 12 months post-enrolment.
The primary endpoint of time-adjusted semi-annual change in FVC (in millilitres) in patients with IPF will be presented descriptively, with two-sided 95% confidence intervals. FVC change for individual patients will be estimated by applying a linear regression model to all data points. The estimated FVC change for each individual patient will then be used to calculate the mean FVC change in the overall cohort of patients with IPF. A repeated-measures mixed model will be applied to estimate the FVC change over 6 months as a sensitivity analysis.
The secondary endpoint of time-adjusted semi-annual change in FVC (in millilitres) in patients with ILD, excluding patients with IPF, measured using home spirometry, will be analysed using the same method as for the primary endpoint. Changes in physical functional capacity and PROs will be summarised descriptively over time. The correlation between home and site measurements of FVC and physical functional capacity will be analysed using Pearson’s correlation coefficient, taking the individual mean FVC changes and accelerometry values for each patient into account.
Data Monitoring Committee and Interim Analyses
The study will be conducted under the leadership of a steering committee. In the absence of the use of any investigational medicinal product, an independent review committee or a data safety monitoring board/data monitoring committee will not be needed. Interim analyses will be performed at least once per year. The first interim analysis is planned for when 20 enrolled patients are diagnosed with IPF and can be fully evaluated with regard to the pre-diagnostic period.
Ethics and Dissemination
This study will be conducted in full conformance with the International Conference on Harmonisation (ICH) E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki, or the laws and regulations of the country where the research is conducted, whichever affords the greater protection to the individual. The study will comply with the requirements of the ICH E2A guideline (Clinical Safety Data Management: Definitions and Standards for Expedited Reporting).
Discussion
To our knowledge, this will be the first study to assess disease behaviour during the peri-diagnostic period in patients with ILD, including IPF. Whereas the majority of clinical trials have collected data from patients after they have received a confirmed diagnosis of ILD and initiated pharmacological treatment, this study has been designed to collect meaningful clinical data on the natural history of ILDs at the start of the diagnostic process. Furthermore, ILD clinical trials have typically investigated longitudinal outcomes, collecting data every 3–6 months. To investigate and identify disease progression during the immediate pre- and post-diagnostic periods, there is a clear need for study designs that allow more frequent data collection. As such, in addition to the site assessments, this study will utilise daily home spirometry and accelerometry to assess short-term changes in pulmonary function and physical functional capacity during the peri-diagnostic period in patients with ILD.
Previous observational studies utilising home spirometry have shown that home measurements have excellent correlation with hospital-obtained readings [26] and are feasible to perform for the majority of patients [24,25,26, 28]. Furthermore, the rate of FVC decline measured using handheld spirometry has been associated with mortality. For example, in a cohort of 50 patients with IPF, the rate of change in FVC assessed at 3 months, 6 months and 12 months was predictive of subsequent mortality [26]. Few studies have also assessed physical activity using accelerometry in patients with ILD [22, 23] with the available evidence suggesting that physical activity predicts mortality in patients with IPF [23].
The daily pulmonary function and physical activity data collected in this study will help to address a number of key unmet needs that have been identified by patients with ILD and clinicians involved in their care. First, there is a lack of data characterising the early course of ILDs, including IPF. It has been suggested that detailed information on ILD disease behaviour is required to establish a working diagnosis and decide on further patient management, with a recent Fleischner Society White Paper recommending that disease behaviour should be considered during multidisciplinary diagnosis [27]. The data collected in this study will increase our knowledge of disease behaviour during the peri-diagnostic period in patients with ILD, and it is hoped that this knowledge will help clinicians to form working diagnoses and reassess patients when disease behaviour is discordant to the previously established diagnosis [27].
Second, many patients with ILD report a need for improved access to diagnosis and treatment [15, 16, 29]. For example, in a qualitative survey of patients with IPF, the majority of respondents reported waiting over 12 months for a diagnosis [15]. Similarly, other surveys of patients with ILD and their caregivers reported delays of at least 12 months between symptom onset and a final diagnosis [13, 30]. In particular, delays can be introduced in the time period between symptom onset and referral to a tertiary centre [14, 16]. The reasons for delayed referral to an ILD specialist identified in previous studies included initial misdiagnosis with another condition and geographical barriers to accessing an MDT [13, 16, 31]. In this study, the digital collaboration platform will allow community and tertiary sites to communicate with each other via virtual MDT discussions, with community centres able to share clinical data with tertiary centres for a second opinion on diagnosis. This design feature has the potential to empower community centres to deliver improved care for patients with ILD, thereby reducing misdiagnoses and unnecessary delays associated with restricted access to tertiary centres. While the lack of a control group in our study will preclude any definite conclusions regarding the benefit of the digital collaboration platform on diagnostic delays, the existing literature base describing diagnostic delays in ILD will provide a context within which we will be able to critically evaluate the diagnostic processes observed in our study [13,14,15, 30]. Furthermore, this platform will reduce the need for patients to travel to tertiary centres to access specialist diagnostic services and input. In geographical areas where access to specialist centres is limited, our study has the potential to significantly reduce the inconvenience experienced by patients with ILD and increase patient satisfaction with their access to specialist care without having to travel.
A third key unmet need, which has been highlighted in the European IPF Patient Charter and the Irish National Patient Charter for IPF, is the need to provide patients with more information regarding their disease, the available treatments and support services [16, 32]. The digital collaboration platform used in this study will facilitate two-way communication between patients and clinicians. For example, clinicians will be alerted automatically to deterioration in their patients’ FVC and will be able to contact the patient, while the “call back” feature on patients’ tablet computers will also enable patients to request contact from their clinician. This “call back” feature may also help patients to feel more comfortable asking questions and could help them to become more familiar with their clinical data. Empowering patients through access to their real-time clinical data could provide patients with further insights into their disease and may also help with adherence to the study. It is also possible that the availability of real-time daily clinical data may also have future applications; for example, similar to technology used in chronic obstructive pulmonary disease [33, 34], the platform could become a useful tool for predicting acute exacerbations, prompting patients to seek a consultation on the basis of patterns in their clinical data.
This study has some novel design aspects, including minimal inclusion and exclusion criteria, the assessment of disease behaviour pre-diagnosis, the use of home-based assessments, few mandatory assessments and site visits, and the virtual MDT. An additional novel aspect of this study will include the assessment of multiple, simple PRO measures in patients with ILD. Of particular interest, this study will assess fatigue, which is among the most common and limiting symptoms for patients with ILD [35,36,37], but to our knowledge is yet to be assessed in a clinical trial. The subjective experiences of patients with ILD are important because the individual patient’s perception of how they feel and function, and how the disease impacts their life, is not only central to the patient’s well-being but can also influence treatment success [38]. Furthermore, PRO measures can help to facilitate communication between patients and clinicians, detect unrecognised problems, guide treatment choices and monitor treatment response [38]. However, despite the importance of these data, there is very little knowledge in this area from clinical trials. The PRO measures that will be assessed in this study are short, easy to complete and cover a number of the important aspects of living with ILD, including dyspnoea, psychological symptoms, mobility, pain, discomfort, cough and fatigue. The combination of clinical data and PROs assessed will ensure that this study provides meaningful information characterising the multidimensional aspects of ILD during the peri-diagnostic period.
Conclusions
To our knowledge, this study will be the first to characterise disease behaviour in patients with ILD, including IPF, during the peri-diagnostic period using real-time, home-based assessments. This study aims to address a number of key unmet needs experienced by patients with ILD and the healthcare professionals involved in their care. In particular, it is hoped that this study will help to facilitate the early and accurate diagnosis of ILDs by increasing knowledge about disease behaviour, increasing communication between community and tertiary centres and improving communication between clinicians and patients. In the future, the virtual MDT platform used in this study could be used in clinical practice to facilitate collaboration between community and tertiary centres, while reducing inconvenience and delays in diagnoses for patients.
References
Ryerson CJ, Collard HR. Update on the diagnosis and classification of ILD. Curr Opin Pulm Med. 2013;19(5):453–9.
Skolnik K, Ryerson CJ. Unclassifiable interstitial lung disease: a review. Respirology. 2016;21(1):51–6.
European Respiratory Society. Interstitial lung diseases. In: Gibson GJ, Loddenkemper R, Sibille Y, Lundbäck B, Fletcher M, editors. European lung white book. Wakefield: Charlesworth; 2013. http://www.erswhitebook.org. Accessed 26 Oct 2018.
Meltzer EB, Noble PW. Idiopathic pulmonary fibrosis. Orphanet J Rare Dis. 2008;3:8.
Margaritopoulos GA, Harari S, Caminati A, Antoniou KM. Smoking-related idiopathic interstitial pneumonia: a review. Respirology. 2016;21(1):57–64.
Prasad R, Gupta N, Singh A, Gupta P. Diagnosis of idiopathic pulmonary fibrosis: current issues. Intractable Rare Dis Res. 2015;4(2):65–9.
Caminati A, Cassandro R, Torre O, Harari S. Severe idiopathic pulmonary fibrosis: what can be done? Eur Respir Rev. 2017;26(145):170047.
King TE Jr, Bradford WZ, Castro-Bernardini S, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2083–92.
Noble PW, Albera C, Bradford WZ, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet. 2011;377(9779):1760–9.
Richeldi L, du Bois RM, Raghu G, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med. 2014;370(22):2071–82.
Cottin V, Richeldi L. Neglected evidence in idiopathic pulmonary fibrosis and the importance of early diagnosis and treatment. Eur Respir Rev. 2014;23(131):106–10.
Thickett DR, Kendall C, Spencer LG, et al. Improving care for patients with idiopathic pulmonary fibrosis (IPF) in the UK: a round table discussion. Thorax. 2014;69(12):1136–40.
Cosgrove GP, Bianchi P, Danese S, Lederer DJ. Barriers to timely diagnosis of interstitial lung disease in the real world: the INTENSITY survey. BMC Pulm Med. 2018;18(1):9.
Lamas DJ, Kawut SM, Bagiella E, Philip N, Arcasoy SM, Lederer DJ. Delayed access and survival in idiopathic pulmonary fibrosis: a cohort study. Am J Respir Crit Care Med. 2011;184(7):842–7.
Russell AM, Ripamonti E, Vancheri C. Qualitative European survey of patients with idiopathic pulmonary fibrosis: patients’ perspectives of the disease and treatment. BMC Pulm Med. 2016;16:10.
Bonella F, Wijsenbeek M, Molina-Molina M, et al. European IPF patient charter: unmet needs and a call to action for healthcare policymakers. Eur Respir J. 2016;47(2):597–606.
De Sadeleer LJ, Meert C, Yserbyt J, et al. Diagnostic ability of a dynamic multidisciplinary discussion in interstitial lung diseases: a retrospective observational study of 938 cases. Chest. 2018;153(6):1416–23.
Walsh SLF, Wells AU, Desai SR, et al. Multicentre evaluation of multidisciplinary team meeting agreement on diagnosis in diffuse parenchymal lung disease: a case-cohort study. Lancet Respir Med. 2016;4(7):557–65.
De Boer K, Aravena C, Sood R, et al. Virtual multidisciplinary discussion: feasibility and diagnostic concordance with face-to-face multidisciplinary discussion. QJM Int J Med. 2016;109(1):S43–4.
Wells AU. Managing diagnostic procedures in idiopathic pulmonary fibrosis. Eur Respir Rev. 2013;22(128):158–62.
Tashkin DP, Roth MD, Clements PJ, et al. Mycophenolate mofetil versus oral cyclophosphamide in scleroderma-related interstitial lung disease (SLS II): a randomised controlled, double-blind, parallel group trial. Lancet Respir Med. 2016;4(9):708–19.
Bahmer T, Kirsten AM, Waschki B, et al. Clinical correlates of reduced physical activity in idiopathic pulmonary fibrosis. Respiration. 2016;91(6):497–502.
Bahmer T, Kirsten AM, Waschki B, et al. Prognosis and longitudinal changes of physical activity in idiopathic pulmonary fibrosis. BMC Pulm Med. 2017;17(1):104.
Moor CC, van Manen MJG, Tak NC, van Noort E, Wijsenbeek MS. Development and feasibility of an eHealth tool for idiopathic pulmonary fibrosis. Eur Respir J. 2018;51(3):1702508.
Moor CC, Wapenaar M, Miedema JR, Geelhoed JJM, Chandoesing PP, Wijsenbeek MS. A home monitoring program including real-time wireless home spirometry in idiopathic pulmonary fibrosis: a pilot study on experiences and barriers. Respir Res. 2018;19(1):105.
Russell AM, Adamali H, Molyneaux PL, et al. Daily home spirometry: an effective tool for detecting progression in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2016;194(8):989–97.
Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society White Paper. Lancet Respir Med. 2018;6(2):138–53.
Johannson KA, Vittinghoff E, Morisset J, Lee JS, Balmes JR, Collard HR. Home monitoring improves endpoint efficiency in idiopathic pulmonary fibrosis. Eur Respir J. 2017;50(1):1602406.
Maher TM, Molina-Molina M, Russell AM, et al. Unmet needs in the treatment of idiopathic pulmonary fibrosis-insights from patient chart review in five European countries. BMC Pulm Med. 2017;17(1):124.
Collard HR, Tino G, Noble PW, et al. Patient experiences with pulmonary fibrosis. Respir Med. 2007;101(6):1350–4.
Wuyts WA, Cavazza A, Rossi G, Bonella F, Sverzellati N, Spagnolo P. Differential diagnosis of usual interstitial pneumonia: when is it truly idiopathic? Eur Respir Rev. 2014;23(133):308–19.
Irish Lung Fibrosis Association. National patient charter for idiopathic pulmonary fibrosis; 2015. http://www.ilfa.ie/docs/ILFA_CharterBooklet_lores.pdf. Accessed 26 Oct 2018.
van der Heijden M, Lucas PJ, Lijnse B, Heijdra YF, Schermer TR. An autonomous mobile system for the management of COPD. J Biomed Inform. 2013;46(3):458–69.
Mohktar MS, Redmond SJ, Antoniades NC, et al. Predicting the risk of exacerbation in patients with chronic obstructive pulmonary disease using home telehealth measurement data. Artif Intell Med. 2015;63(1):51–9.
Carvajalino S, Reigada C, Johnson M, Dzingina M, Bajwah S. Symptom prevalence of patients with fibrotic interstitial lung disease: a systematic literature review. BMC Pulm Med. 2018;18(1):78.
De Kleijn W, De Vries J, Lower E, Elfferich M, Baughman R, Drent M. Fatigue in sarcoidosis: a systematic review. Curr Opin Pulm Med. 2009;15(5):499–506.
van Manen MJ, Geelhoed JJ, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2017;11(3):157–69.
Wijsenbeek M, van Manen M, Bonella F. New insights on patient-reported outcome measures in idiopathic pulmonary fibrosis: only PROMises? Curr Opin Pulm Med. 2016;22(5):434–41.
Acknowledgements
The authors would like to thank all the patients and colleagues involved in the preparation and conduct of this study.
Funding
This study is sponsored by F. Hoffmann-La Roche, Ltd., Basel, Switzerland, which will provide clinical operations management, data management and medical monitoring, and will be involved in the interpretation of the data. F. Hoffmann-La Roche, Ltd., Basel, Switzerland also funded the journal’s article processing charges and Open Access fee.
Authorship
All authors were involved in the design of this study. All authors contributed to the manuscript from the outset, and read and approved the final draft. All authors vouch for the accuracy of the content included in the final manuscript. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing, Editorial and Other Assistance
Medical writing support was provided by Gráinne Faherty of CMC AFFINITY, a division of Complete Medical Communications, Ltd., Glasgow, UK, funded by F. Hoffmann-La Roche, Ltd.
Disclosures
Marlies Wijsenbeek has received unrestricted grants and speaker fees from Boehringer Ingelheim and F. Hoffmann-La Roche, Ltd., and advisory board fees from Galapagos. All grants and fees were paid to the Erasmus University Medical Center in Rotterdam, the Netherlands. Elisabeth Bendstrup has received speaker fees and/or advisory board fees from Boehringer Ingelheim and F. Hoffmann-La Roche, Ltd., and has received a PI fee from Sanofi. Claudia Valenzuela has received speaker fees from Boehringer Ingelheim and F. Hoffmann-La Roche, Ltd. Michael T. Henry has received speaker fees and/or advisory board fees from Boehringer Ingelheim, F. Hoffmann-La Roche, Ltd., GlaxoSmithKline and Menarini. Catharina Moor has no conflicts of interest to disclose. Carlo Vancheri has received grants, speaker and personal fees from Boehringer Ingelheim and F. Hoffmann-La Roche, Ltd. Klaus-Uwe Kirchgaessler is an employee of Roche-Genentech and holds shares. Monica Bengus is an employee of Roche-Genentech and holds shares. Andras Perkesi is an employee of Roche-Genentech. Frank Gilberg is an employee of Roche-Genentech.
Steering Committee
Marlies Wijsenbeek (the Netherlands), Elisabeth Bendstrup (Denmark), Claudia Valenzuela (Spain), Michael T. Henry (Ireland) and Carlo Vancheri (Italy).
Compliance with Ethics Guidelines
This study will be conducted in full conformance with the International Conference on Harmonisation (ICH) E6 guideline for Good Clinical Practice and the principles of the Declaration of Helsinki, or the laws and regulations of the country where the research is conducted, whichever affords the greater protection to the individual. The study will comply with the requirements of the ICH E2A guideline (Clinical Safety Data Management: Definitions and Standards for Expedited Reporting). All patients must provide signed informed consent to the investigator before any study procedures are performed.
Data Availability
Qualified researchers may request access to individual patient-level data through the clinical study data request platform (http://www.clinicalstudydatarequest.com). Further details on Roche’s criteria for eligible studies are available at https://clinicalstudydatarequest.com/Study-Sponsors/Study-Sponsors-Roche.aspx. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, visit https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
Additional Protocol Information
See Appendices A and C in the Electronic Supplementary Material.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7358393.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Wijsenbeek, M., Bendstrup, E., Valenzuela, C. et al. Design of a Study Assessing Disease Behaviour During the Peri-Diagnostic Period in Patients with Interstitial Lung Disease: The STARLINER Study. Adv Ther 36, 232–243 (2019). https://doi.org/10.1007/s12325-018-0845-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0845-3