
Abstract
Introduction
Nicotine replacement therapy (NRT) benefits smokers who wish to quit; nicotine gum represents one NRT. New formulations of nicotine gum have been developed to consider consumer preferences and needs. A new mint-flavored nicotine gum with a different texture was developed that may provide a more appealing taste and chewing experience. This study evaluated this new nicotine gum (2 and 4 mg strengths) for bioequivalence versus the original flavor sugar-free nicotine gum at corresponding dosages.
Methods
All subjects randomized in this crossover study received a single dose of all treatments, i.e., 2 and 4 mg doses of test and reference gums, separated by 2–7 days of washout between treatments. Subjects’ maximal plasma nicotine concentration (Cmax) and extent of nicotine absorption (AUC0–t) following the administration of each treatment were calculated from plasma nicotine concentrations. Ratios of test/reference for Cmax and AUC0–t were calculated to evaluate bioequivalence between the two products.
Results
Both 2 and 4 mg doses of the new mint-flavored nicotine gum were bioequivalent to the dose-matched reference product as determined by the ratio of the geometric means and their 90% confidence intervals for Cmax and AUC0–t as well as secondary pharmacokinetic parameters. The safety profiles of the test and reference gums were similar; all treatments were well tolerated.
Conclusions
A new mint-flavored nicotine gum with modified taste and texture is bioequivalent to the original flavor sugar-free nicotine gum at both the 2 and 4 mg dosage strengths and has a similar safety profile.
Funding
GlaxoSmithKline.
Trial Registration
ClinicalTrials.gov identifier NCT01847443.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Despite the well-known risks, smoking continues to be the single largest preventable cause of early morbidity and mortality in the USA, killing more than 480,000 Americans annually [1, 2]. Smoking adversely affects nearly all organs and organ systems of the body [1]. Unfortunately, the rapid pharmacologic effects of nicotine are addictive, making the act of quitting very difficult for many people [3]. Despite the challenges, many people who currently smoke would like to stop [4].
Nicotine replacement therapy (NRT) is recommended by evidence-based clinical guidelines as a first-line treatment for smoking cessation [5] and is available in multiple different forms, including transdermal patch, nasal spray, lozenge, and gum. These different formulations have different pharmacokinetic (PK) and aesthetic characteristics, enabling smokers to select one or more options on the basis of their personal needs and preferences [6]. Nicotine replacement gum has been shown to increase the success rate for quitting and may also reduce harm among smokers who are not ready to quit abruptly [7,8,9]. A new mint-flavored NRT gum has been developed that provides consumers with another NRT quit option that may be more appealing, also potentially leading to better adherence and higher quit rates.
Herein we report details of a bioequivalence analysis of this new mint-flavored nicotine gum (2 and 4 mg doses) relative to the original flavor sugar-free nicotine gum to determine if the two products are bioequivalent in terms of the maximal observed plasma nicotine concentration (Cmax) and the area under the concentration–time curve from time 0 to the time of the last observed/measured non-zero concentration (AUC0–t).
Methods
Study Design and Subjects
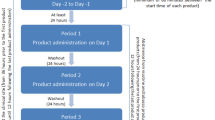
This was a single-center, open-label, randomized, four-treatment, four-period crossover study conducted in May and June of 2013. An independent ethics committee operating in accordance with current local regulations reviewed and approved the protocol and consent procedures before the initiation of the study. The trial was registered on ClinicalTrials.gov (NCT01847443) and conducted in full compliance with all relevant laws and regulations of the country in which the research was conducted and with the requirements specified in the Declaration of Helsinki, and according to International Council for Harmonisation Guideline for Good Clinical Practice. This study was also approved by the Medicines and Healthcare Products Regulatory Agency (MHRA), European Union Drug Regulating Authorities Clinical Trials (EudraCT) number 2013-000207-16. Voluntary written informed consent was provided by all subjects prior to any study procedures being performed.
All enrolled subjects were current heavy smokers who smoked daily for at least 1 year, smoked their first cigarette within 30 min of awakening, and who were in otherwise good general health in the investigator’s opinion. Heavy smokers were recruited in the study to eliminate unnecessary exposure of healthy non-smokers to nicotine. Subjects were between 18 and 55 years of age, inclusive, with a body mass index of 19–30 kg/m2. Enrolled female subjects were either practicing a reliable method of contraception or were otherwise unable to become pregnant during the study.
Any woman known to be pregnant, intending to become pregnant, having a positive serum pregnancy test at screening, or who was breastfeeding was ineligible for study entry. Any subjects with known or suspected allergies or intolerances to the study compounds or any medical condition with the potential to interfere with absorption, distribution, metabolism, or excretion of nicotine were also excluded. Subjects who attempted smoking cessation within the last 12 months, were currently trying to quit, or planned to quit smoking within the next 3 months were excluded. Subjects were ineligible if they were taking hepatic enzyme-altering agents within 30 days, prescription medications (except hormonal contraceptives or hormone replacement therapy, which were permitted) within 14 days, or over-the-counter medications within 24 h of each study treatment visit; use of paracetamol was permitted during the confinement period at the investigator’s discretion. Subjects who donated blood or experienced significant blood loss within 90 days of the screening visit, had a current diagnosis of anemia, or had positive serologies for hepatitis B virus, hepatitis C virus, or human immunodeficiency virus were not eligible. Finally, abuse of drugs or alcohol within the previous year or a positive urine drug screen for illicit substances or alcohol was also exclusionary.
Subject Restrictions
All study subjects were required to fast for 8 h leading up to the initial screening visit, which included clinical laboratory testing. Fasting was also required for 8 h before and 2 h after receiving the study treatments; no liquids could be consumed in the period from 20 min before dosing until 60 min after drug administration. Standardized food and water was provided 2 h after the drug administration and at set times through the day. Consumption of caffeine or xanthine-containing beverages during treatment periods or alcoholic beverages within 24 h of the beginning of the treatment periods was prohibited. During each study session, all nicotine use was prohibited for 36 h before treatment administration and continuing until the last blood sample was obtained. Smoking abstinence was verified by measuring exhaled carbon monoxide (CO) levels both before and after dosing with a Smokerlyzer® apparatus (Bedfont Scientific Ltd., Maidstone, UK). Expired CO levels could not be greater than or equal to 10 parts per million (ppm) prior to study drug administration at each treatment period or rise above 10 ppm during any of three random tests conducted during each treatment period after dosing, or the subject would be withdrawn from the study. Finally, all study subjects had to agree to refrain from strenuous physical activity.
Study Treatments
The test treatments in this study consisted of 2 and 4 mg mint-flavored nicotine gums [GSK Consumer Healthcare (GSKCH), Weybridge, UK, manufactured by Fertin Pharma, Vejle, Denmark]. Nicorette® 2 and 4 mg original flavor sugar-free nicotine gums (GSKCH, Brentford, UK) served as reference products. On day −1 of the first treatment period all study participants underwent a chew familiarization session with a confectionary gum (Wrigley’s Extra® peppermint sugar-free gum; Wm. Wrigley Jr. Company, Chicago, Illinois, USA) to maximize the conformity of gum chewing and reduce variability in the pharmacokinetic results obtained using the test and reference products. The standards for gum chewing included a steady pace of 15 chews/min (about one chew every 4 s) for 30 min; timers with audible signals were used to mark off intervals of 4 s. Participants chewed on alternate sides of their mouths every 30 s and were permitted to swallow saliva, if they wished, every 30 s. After completing the 8-h fasting period, participants were randomized to one of the four treatment sequences according to a computer-generated randomization schedule provided by the GSKCH Biostatistics Department. An additional 12 h of smoking abstinence took place after dosing during the confinement period. The treatment phase consisted of four treatment periods. Given the terminal elimination half-life of approximately 2 h for nicotine, the four treatment periods were separated by a 2- to 7-day washout period beginning at the time of the last dose in the preceding treatment period. Following this washout period, the subject returned to the study site for the next treatment in their respective sequence until all four treatments were administered.
Study Assessments
Screening Visit
Subjects were recruited by the study site (Celerion, Belfast, UK) from the site’s volunteer database using a standard telephone screening interview, and informed consent was obtained from those potential subjects who were eligible and interested in participating. During the initial visit, each subject underwent screening procedures. After each subject’s demographic information, medical history, and smoking history were obtained, the level of nicotine dependence was determined using the Fagerström Test for Nicotine Dependence (FTND) [10]. Screening laboratories included assessment of hematology, biochemistries, viral serology testing, serum FSH, serum pregnancy testing, urinalysis, and urine testing for illicit drugs and alcohol. A physical examination including vital signs and a 12-lead electrocardiogram rounded out the screening assessments.
Treatment Visits
Study subjects checked in at the study site 2 days before each treatment session (day −2) to ensure a minimum of 36 h abstinent from cigarettes before administration of the study treatment. After check-in, all subjects underwent urine testing for drug and alcohol use and pregnancy, if appropriate, and each participant’s compliance with eligibility criteria and lifestyle restrictions was verified. Exhaled CO levels were checked immediately before administration of the study drug and at three random times after dosing using a Smokerlyzer device. Subjects who had expired CO levels greater than or equal to 10 ppm were withdrawn from the study. Vital signs were checked on day 1 of each treatment session.
Sample Collection
Approximately 312 mL of blood was collected from each subject for PK analysis, another 20 mL for screening tests, and 5 mL of blood collected from each woman at each treatment visit check-in for serum pregnancy testing, as needed. Pharmacokinetic sampling involved collection of 13 blood samples obtained during treatment sessions from a forearm vein. Samples were collected 15 min before dosing and at 5, 15, 30, and 45 min, and 1, 1.5, 2, 3, 4, 6, 8, and 12 h after dosing. Blood was collected in tubes containing K2EDTA and centrifuged at 3000 revolutions per minute for 15 min at 4 °C to collect the plasma for analysis.
Plasma samples were split into two aliquots and frozen within 120 min of being collected. The plasma was then stored at −20 ± 10 °C for transport to the laboratory analytic facility (Celerion, Lincoln, Nebraska, USA). Plasma nicotine concentrations were determined by a certified bioanalytical laboratory using a validated method of solid-phase extraction and liquid chromatography with tandem mass spectroscopy (LC–MS/MS) detection, and the analytical range of the assay was 0.200–10.0 ng/mL.
Safety Evaluation
The safety evaluation included documentation of all adverse events (AEs) during the study, defined as any untoward medical occurrence in a subject following administration of an investigational product, which did not necessarily have a causal relationship with this treatment. Reported AEs were further graded by intensity (mild, moderate, or severe) and the likelihood that the AE was related to the study treatment ranging from “not related” to “highly probable.” AEs were categorized as serious (SAEs) if they were life-threatening, resulted in death, required hospitalization or prolongation of existing hospitalization, resulted in disability or incapacity, or were congenital anomalies or birth defects. Vital sign abnormalities were described.
Data Analysis
Sample Size
At least 80 subjects were planned to be enrolled and randomly assigned to the four treatment sequences so that 72 subjects would complete each of the four treatment periods. A previously conducted bioequivalence study (study S1640123; data on file, GlaxoSmithKline Healthcare) with a similar four-way crossover design with four treatments and a similar variablility of 0.060 for log Cmax and 0.045 for log AUC0–t was used to inform sample size. Calculated coefficient of variation (CV) was 24.9% and 21.5% for Cmax and AUC0–t, respectively. The sample size was calculated using estimates of geometric means (log scale) with two one-sided test (TOST). With a 25% CV, the expected ratios of test/reference were between 0.90 and 1.10 and provided a power of at least 88% to reject bioequivalence for Cmax and AUC0–t. Because of the PK sampling profile for this study, no more than 30 inpatients were studied at one time, and therefore three cohorts were used in the study. Eligible subjects were randomized to one of the four treatment sequences in each cohort.
Analysis Populations
The bioequivalence-evaluable population consisted of subjects who were randomized, received at least one dose and had primary PK parameter data (Cmax and AUC0–t) for both the study and reference treatments at the 2 mg and/or 4 mg dose, had no vomiting during the first 4 h following treatment administration, were in compliance with all lifestyle restrictions, had no expired CO level greater than or equal to 10 ppm prior to study drug administration, and did not have a pre-dose nicotine concentration greater than 5% of Cmax. The safety population included any subject who was randomized and received at least one study treatment in a given treatment period.
Statistical Analysis
The primary PK parameters calculated in this study, Cmax and AUC0–t, were derived using unadjusted plasma nicotine concentrations. PK variables were computed using Phoenix Win-Nonlin® Version 6.3. Bioequivalence was determined using the standard bioequivalence limits of 0.80 and 1.25 for the 90% confidence interval (CI) of the ratio of geometric means of the PK parameters. Analysis of variance (ANOVA) testing was conducted using a linear fixed effects model for a four-way crossover design and conducted separately for each primary PK variable (log-transformed Cmax and AUC0–t). The terms sequence, subjects nested within sequence, period, and product were included as fixed effects in the model.
The differences of the least square means (LSMs) of log Cmax and log AUC0–t for the 2 and 4 mg products and their 90% CIs were exponentiated to calculate the ratio of geometric means and their 90% CIs for the primary PK variables for the 2 and 4 mg products. The test and reference treatments were determined to be bioequivalent if the 90% CI of the ratio of geometric means for Cmax and AUC0–t fell within the limits of 0.80 and 1.25.
The secondary PK parameters of apparent terminal elimination half-life of the drug (t1/2), time to maximal plasma concentration (Tmax), and area under the concentration–time curve from time 0 extrapolated to infinity (AUC0–∞) were calculated using unadjusted plasma nicotine concentrations. The parameters t1/2 and Tmax were analyzed using the non-parametric Wilcoxon signed rank test based on the within-subject difference to compare test and reference treatments at the 5% significance level. AUC0–∞ was analyzed in the same way as the primary PK parameters. The apparent terminal elimination rate constant (Kel) was also calculated on the basis of unadjusted nicotine concentrations and analyzed by the Wilcoxon signed rank test based on the within-subject difference comparing test and reference treatments at the 5% significance level.
A sensitivity analysis was performed to compare results of bioequivalence stemming from PK parameters based on unadjusted nicotine concentration (primary PK endpoints) to those based on baseline-adjusted nicotine concentrations. A separate analysis was planned to study the effect of potential outliers of PK parameters (i.e., including those with baseline nicotine concentrations greater than 5% of Cmax).
Safety data, including AEs and serious AEs, were summarized by treatment group.
Results
Subject Disposition
A total of 160 potential study subjects were screened, of whom 84 were randomized and 77 (91.7%) completed all four study treatments. The bioequivalence-evaluable subject population consisted of 62 individuals for the 2 mg treatment products and 73 individuals for the 4 mg products. The primary reasons why subjects were excluded from the bioequivalence-evaluable population were for having a baseline plasma nicotine concentration greater than 5% of Cmax for a given treatment or not receiving both the test and reference treatment at the 2 or 4 mg dose. The safety population for this study consisted of 82 subjects for the 2 mg test treatment, 77 subjects for the 2 mg reference treatment, 79 subjects for the 4 mg test treatment, and 81 subjects for the 4 mg reference treatment.
The demographic and baseline characteristics of the study population are summarized in Table 1. Study subjects included both men and women aged 19–51 years; all but one subject were white (98.8%). The subjects enrolled in this study had no clinically significant medical or surgical findings at the screening process, as determined by the investigator.
PK Results
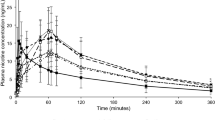
The unadjusted arithmetic mean plasma nicotine concentrations over time for the test and reference treatments are shown in Fig. 1 (2 mg treatments) and Fig. 2 (4 mg treatments). Descriptive statistics for the primary PK parameters, Cmax and AUC0–t, are summarized in Table 2 for the 2 mg treatments and in Table 3 for the 4 mg treatments.

Mean over time of nicotine concentration for the 2 mg dose products, bioequivalence-evaluable subject population

Mean over time of nicotine concentration for the 4 mg dose products, bioequivalence-evaluable subject population
PK analysis based on the unadjusted plasma nicotine concentrations of test to reference 2 mg treatment comparison showed that the two treatments were bioequivalent on the basis of the ratio of geometric means and 90% CIs for Cmax and AUC0–t (Table 2) falling within the standard bioequivalence acceptance range of 0.80–1.25. Likewise, the 90% CIs of the ratio of geometric means of Cmax and AUC0–t for the 4 mg test gum relative to the 4 mg reference gum were also within the range of 0.80–1.25, demonstrating bioequivalence between these two 4 mg products (Table 3).
The secondary PK parameter AUC0–∞ was also calculated using unadjusted nicotine concentrations and analyzed in the same manner as AUC0–t (Tables 2, 3). The 90% CIs of these geometric mean ratios for the 2 and 4 mg test products vs the 2 and 4 mg reference products, respectively, also fell within the bioequivalence limits, supporting the finding of bioequivalence based on the primary PK parameters.
The secondary PK parameters of Tmax, t1/2, and Kel are summarized in Tables 4 and 5 for the respective 2 and 4 mg dosages. There were no significant differences between test and reference products for the secondary PK parameters at either dose strength.
The planned sensitivity analysis, for which outliers were to be included, was not conducted because no Cmax outliers were found in this study. Given that finding, a post hoc sensitivity analysis based on unadjusted nicotine concentrations and including only subjects who had completed all four treatment periods was conducted. The results of this sensitivity analysis were very similar to the results of the primary analysis of bioequivalence (see Tables 2 and 3). Baseline-adjusted comparisons found geometric mean ratios (test/reference) and 90% CIs of 1.047 (1.003–1.092) for Cmax and 1.111 (1.074–1.149) for AUC0–t for the 2 mg products and 1.070 (1.027–1.115) for Cmax and 1.097 (1.061–1.135) for AUC0–t for the 4 mg products. The post hoc sensitivity analysis found geometric mean ratios (test/reference) and 90% CIs of 1.056 (1.008–1.105) for Cmax and 1.116 (1.076–1.156) for AUC0–t for the 2 mg products and 1.079 (1.030–1.130) for Cmax and 1.107 (1.069–1.148) for AUC0–t for the 4 mg products. Thus, results from both sensitivity analyses (predefined and post hoc) were very similar to those from the primary bioequivalence analysis, reinforcing the robustness of the primary bioequivalence results calculated using unadjusted nicotine data.
A sensitivity analysis of the secondary variables Tmax, t1/2, AUC0–∞, and Kel for both the 2 and 4 mg products produced very similar results to the primary analysis of the secondary parameters. The 90% CI of the ratio of geometric means for AUC0–∞ was within the interval of 0.80–1.25, and the differences between products in Tmax, t1/2, and Kel were not statistically different (P > 0.05) for the 2 mg product comparisons and 4 mg product comparisons (data not shown).
Safety Results
A total of 75 treatment-emergent AEs (TEAEs) were reported in 31 subjects, including five AEs reported by five subjects that were considered by the investigator to be treatment related. Treatment-emergent AEs that occurred in more than one subject in any treatment group are listed in Table 6. Of the 75 TEAEs, 59 were mild in intensity and 16 were of moderate intensity. AEs resulted in study withdrawal for two subjects: abdominal pain during the 4 mg nicotine gum reference treatment period in one subject and rash during the 2 mg nicotine gum test treatment period in one subject. The profile of the most common AEs was similar to that observed in other clinical trials of NRT including gastrointestinal disorders, nervous system disorders, general disorders and administration site conditions, and respiratory, thoracic, and mediastinal disorders. There were no deaths, no SAEs, or any other significant effects in this study.
Discussion
Nicotine replacement therapies are effective agents for smokers who wish to achieve abstinence [5]. Preferences for smoking cessation aids differ among smokers; different NRT options with varying delivery methods, sizes, and flavors offer smokers a variety of options that may increase the likelihood of success.
In India, where approximately 30% of adults use tobacco [11], three prototype 4 mg nicotine lozenges with different dissolution and nicotine release profiles were recently compared in a bioequivalence study versus a marketed product [12]. Investigators found that each prototype nicotine lozenge was bioequivalent to the marketed product in terms of Cmax and AUC0–t, but one of the prototypes demonstrated significantly faster Tmax compared with the marketed product [12]. Faster nicotine delivery may provide an advantage in terms of acute nicotine craving relief. Similarly, a single-dose bioequivalence study found cherry-flavored 2 and 4 mg nicotine mini lozenges to be bioequivalent to marketed mint-flavored nicotine mini lozenges according to standard criteria, thus providing another potential NRT treatment to achieve smoking cessation [13].
This study demonstrated that a new mint-flavored nicotine gum in 2 and 4 mg dose strengths is bioequivalent to the original flavor sugar-free nicotine gum at those respective doses. The secondary PK parameter of AUC0–∞ supported the bioequivalence of the test and reference products observed in the primary analysis, as did test to reference comparisons of Tmax, t1/2, and Kel, where no statistically significant differences for either the 2 or 4 mg dose were noted.
Sensitivity analyses showed that the bioequivalence results were not altered significantly by using either baseline-adjusted nicotine concentrations or when analyzing the population of subjects who completed all of the four treatments. The AEs reported during this study were mild in most cases and typical of those associated with NRT in general. Both the 2 and 4 mg test gums were shown to have a safety profile similar to their respective reference products and were well tolerated by the study subjects; no new safety signals were noted.
Limitations of the study include its open-label nature and crossover design. The 2- to 7-day washout period between each of the four treatment sequences was provided to minimize carryover effects from one treatment session to another. Results from this study in otherwise healthy smokers may not be generalizable to smokers with comorbid conditions. Lastly, there was no measure of treatment efficacy specified for this study. By virtue of the bioequivalence results, however, the new mint-flavored nicotine gum would be expected to be as effective as the marketed original flavor sugar-free nicotine gum, and hopefully will provide another option for consumers who wish to stop smoking.
Conclusions
The results of this clinical study demonstrate that the new mint-flavored formulation of nicotine gum available in 2 and 4 mg doses is bioequivalent to an original flavor sugar-free nicotine gum. The new mint-flavored formulation is well tolerated at both doses and does not present any new safety concerns beyond those already known to be associated with NRT. This new nicotine gum formulation may provide smokers a new and appealing option when choosing an NRT to aid in smoking cessation.
References
U.S. Department of Health and Human Services. The health consequences of smoking: a report of the Surgeon General. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 2004.
Burden of Tobacco Use in the U.S. Atlanta, GA: Office on Smoking and Health, National Center for Chronic Disease Prevention and Health Promotion, Centers for Disease Control and Prevention; 2017. https://www.cdc.gov/tobacco/campaign/tips/resources/data/cigarette-smoking-in-united-states.html. Accessed 19 June 2018.
Benowitz NL, Hukkanen J, Jacob P 3rd. Nicotine chemistry, metabolism, kinetics and biomarkers. Handb Exp Pharmacol. 2009;192:29–60.
Tobacco and smoking: Gallup; 2016. http://www.gallup.com/poll/1717/tobacco-smoking.aspx. Accessed 19 June 2018.
Fiore MC, Jaév XP, Baker TB, et al. Treating tobacco use and dependence: 2008 update. Clinical Practice Guideline Rockville, MD: US Department of Health and Human Services; May 2008. http://bphc.hrsa.gov/buckets/treatingtobacco.pdf. Accessed 19 June 2018.
Du D, Nides M, Borders J, Selmani A, Waverczak W. Comparison of nicotine oral soluble film and nicotine lozenge on efficacy in relief of smoking cue-provoked acute craving after a single dose of treatment in low dependence smokers. Psychopharmacology. 2014;231(22):4383–91.
Shiffman S, Ferguson SG, Strahs KR. Quitting by gradual smoking reduction using nicotine gum: a randomized controlled trial. Am J Prev Med. 2009;36(2):96–104.e1.
Batra A, Klingler K, Landfeldt B, Friederich HM, Westin A, Danielsson T. Smoking reduction treatment with 4-mg nicotine gum: a double-blind, randomized, placebo-controlled study. Clin Pharmacol Ther. 2005;78(6):689–96.
Kralikova E, Kozak JT, Rasmussen T, Gustavsson G, Le Houezec J. Smoking cessation or reduction with nicotine replacement therapy: a placebo-controlled double blind trial with nicotine gum and inhaler. BMC Public Health. 2009;9:433.
Fagerstrom KO. Towards better diagnoses and more individual treatment of tobacco dependence. Br J Addict. 1991;86(5):543–7.
Rani M, Bonu S, Jha P, Nguyen SN, Jamjoum L. Tobacco use in India: prevalence and predictors of smoking and chewing in a national cross sectional household survey. Tob Control. 2003;12(4):e4.
Sukhija M, Srivastava R, Kaushik A. Pharmacokinetic characterization of three novel 4-mg nicotine lozenges. Int J Clin Pharmacol Ther. 2018;56(3):113–9.
Rasmussen SC, Becker WD, Shanga GM. Single-dose bioequivalence of two mini nicotine lozenge formulations. Clin Pharmacol Drug Dev. 2018;7(5):498–505.
Acknowledgements
The author wishes to thank the participants of this study.
Funding
This study was sponsored by GlaxoSmithKline Consumer Healthcare. GlaxoSmithKline Consumer Healthcare paid for journal processing charges and open access fees and provided a full review of the article. The author had full access to all of the data in this study and takes complete responsibility for the integrity of the data and accuracy of the data analysis.
Medical Writing and/or Editorial Assistance
Medical writing assistance was provided by Erik MacLaren, PhD, of Peloton Advantage and was funded by GlaxoSmithKline Consumer Healthcare.
Authorship
The author meets the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, takes responsibility for the integrity of the work as a whole, and gave his approval for this version to be published.
Disclosures
Daniel Du is a former employee of GlaxoSmithKline Consumer Healthcare and was employed during the time this study was conducted. Daniel Du’s current affiliation is Hua Medicine (Shanghai) Ltd, 275 AiDiSheng Rd, Pudong, Shanghai, PR China.
Compliance with Ethics Guidelines
The trial was conducted in full compliance with all relevant laws and regulations of the country in which the research was conducted and with the requirements specified in the Declaration of Helsinki, and according to International Council for Harmonisation Guideline for Good Clinical Practice. This study was also approved by the Medicines and Healthcare Products Regulatory Agency (MHRA), European Union Drug Regulating Authorities Clinical Trials (EudraCT) number 2013-000207-16. Informed consent was obtained from those potential subjects who were eligible and interested in participating in the study.
Data Availability
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.6731291.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Du, D. A Single-Dose, Crossover-Design Bioequivalence Study Comparing Two Nicotine Gum Formulations in Healthy Subjects. Adv Ther 35, 1169–1180 (2018). https://doi.org/10.1007/s12325-018-0752-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0752-7