
Abstract
Introduction
Dupilumab, a fully human anti-IL-4Rα monoclonal antibody, inhibits signaling of both interleukin (IL)-4 and IL-13, which are key drivers of type 2-mediated inflammation. Dupilumab is approved in the EU, USA, and other countries for the treatment of adults with inadequately controlled moderate-to-severe atopic dermatitis. Following positive phase 2 results in asthma, the phase 3 Liberty Asthma QUEST trial was initiated to provide further evidence for dupilumab efficacy and safety in patients with uncontrolled, moderate-to-severe asthma.
Methods
Liberty Asthma QUEST is a phase 3, multinational, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial (NCT02414854) in patients with persistent asthma who are receiving continuous treatment with inhaled corticosteroids (ICS) plus one or two other asthma controller medicines. A total of 1902 patients (aged ≥ 12 years) were randomized in a 2:2:1:1 ratio to receive 52 weeks of add-on therapy with subcutaneously administered dupilumab 200 or 300 mg every 2 weeks or matched placebo. The study consisted of a 4 ± 1-week screening period, 52-week randomized treatment period, and 12-week post-treatment follow-up period. All patients continued to receive their prescribed ICS plus up to two additional controller medications. The primary efficacy endpoints were annualized rate of severe exacerbation events during the 52-week treatment period and absolute change from baseline in pre-bronchodilator FEV1 at week 12.
Conclusion
Uncontrolled asthma patients with persistent symptoms represent a population of significant unmet need, for whom new treatments are required. Patients with severe asthma are at high risk of asthma exacerbations, and face an accelerated decline in lung function and impaired quality of life. QUEST examines the efficacy of dupilumab in this at-risk patient population; it is the largest placebo-controlled study in uncontrolled, moderate-to-severe asthma with a biologic agent to date, and the only phase 3 study of a biologic therapy of asthma that enrolled patients irrespective of baseline type 2 inflammatory biomarker levels.
Funding
Sanofi and Regeneron Pharmaceuticals, Inc.
Clinical Trials.gov Identifier
NCT02414854.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Asthma is a chronic inflammatory airway disease characterized by airway hyper-responsiveness, bronchoconstriction, airway edema, and mucus plugging. Approximately 5–10% of patients with asthma remain uncontrolled despite receiving the maximum recommended treatment. These patients are at increased risk of frequent and severe exacerbations, have impaired lung function and impaired quality of life, and account for a high healthcare cost burden [1]. As such, they represent a population with unmet needs, for whom new treatments are required.
Asthma phenotypes, including inflammatory, clinical, and trigger-related subtypes, have been identified and defined by interactions between genetic and environmental factors [2,3,4,5]. Type 2-high asthma is a common subtype, characterized by the release of the inflammatory prototypical cytokines interleukin (IL)-4, IL-5, and IL-13 from immune cells [6]. Several biomarkers are linked to type 2 airway inflammation, including fractional exhaled nitric oxide (FeNO), serum immunoglobulin E (IgE), periostin, and blood and sputum eosinophils [5]. Identification of inflammatory pathways involved in asthma pathophysiology has produced therapeutic approaches to severe asthma that target type 2 inflammatory mediators, including antibodies directed at IL-5, a key cytokine involved in eosinophil proliferation and recruitment to sites of inflammation.
Dupilumab is a fully human monoclonal antibody (mAb) directed against IL-4 receptor alpha (IL-4Rα). Dupilumab inhibits signaling of IL-4 and IL-13, cytokines that are key drivers of type 2 immune diseases (e.g. atopic/allergic diseases) such as atopic dermatitis (AD), asthma, allergic rhinitis, chronic rhinosinusitis (with or without nasal polyps), and food allergies that are often associated as comorbidities [7]. Dupilumab is approved in the EU, USA, and other countries for the treatment of adults with inadequately controlled moderate-to-severe AD.
In a phase 2a proof-of-concept study, dupilumab added to medium-to-high-dose inhaled corticosteroids (ICS) plus a long-acting β2-agonist (LABA) in patients with uncontrolled persistent eosinophilic asthma [8] significantly reduced the number of patients with asthma exacerbation by 87% and improved lung function compared to placebo. Given these promising findings, a phase 2b dose-ranging study assessed dupilumab as add-on therapy in a patient population including both eosinophilic and non-eosinophilic patients. Dupilumab was well tolerated, and doses given every 2 weeks improved lung function (least squares [LS] mean change in forced expiratory volume in 1 s [FEV1] from baseline to week 12 of 0.31 and 0.28 L for 200 and 300 mg of dupilumab, respectively, and 0.12 L for placebo), reduced severe exacerbations (70.0% to 70.5% risk reduction vs. placebo), improved asthma control (LS mean change in the 5-item asthma control questionnaire [ACQ-5] score from baseline to week 24 of − 1.49 and − 1.45 for 200 and 300 mg of dupilumab, respectively, and − 1.14 for placebo) [9], and reduced levels of type 2 inflammatory biomarkers including FeNO (LS mean change − 18.69 and − 20.86 ppb for 200 and 300 mg doses, respectively, and − 3.90 ppb for placebo), plasma eotaxin-3 (− 32.45 and − 30.63 pg/mL in dupilumab doses, − 1.84 pg/mL in placebo), and serum IgE (− 235.40 and − 207.95 IU/mL in dupilumab doses, 6.21 IU/mL increase in placebo) over a 24-week treatment period. While this study established that a 2-weekly dosing regimen was optimum, it did not establish the most appropriate dose [9].
Following positive phase 2 results, a phase 3 program was initiated with the objective of providing further evidence of the efficacy and safety of dupilumab in patients with uncontrolled, moderate-to-severe asthma.
Methods
Liberty Asthma QUEST Study Design
QUEST is a phase 3 multinational, multicenter, randomized, double-blind, placebo-controlled, parallel-group trial in patients with uncontrolled, moderate-to-severe asthma who are receiving continuous treatment with ICS plus one or two other asthma controller medicines (Clinical Trials.gov Identifier: NCT02414854). Database lock was planned to have data on the 52-week treatment period of at least 1638 randomized patients. Patients were randomized at 413 sites in 22 countries globally, making QUEST the largest placebo-controlled study ever performed to date in uncontrolled persistent asthma with a biologic agent. Subsequent to the findings of the phase 2b study that dupilumab was effective for patients with eosinophilic and non-eosinophilic asthma, the primary enrolled patient population in QUEST was based on clinical criteria alone, without any pre-specified biomarker requirement (an upper baseline blood eosinophil count limit of > 1500/µL was included). This enabled a full, unbiased analysis of the influence of baseline biomarker and clinical characteristics on treatment response. The key inclusion and exclusion criteria for QUEST are listed in Table 1.
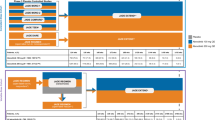
The study consisted of three periods: a 4 ± 1-week screening period, a 52-week randomized treatment period, and a 12-week post-treatment follow-up period (unless patients entered an open-label extension study) (Fig. 1). Patient eligibility and level of asthma control were established during the screening period. The study was sponsored by Sanofi and Regeneron Pharmaceuticals, Inc., and conducted in accordance with the principles established in the Declaration of Helsinki and the International Conference on Harmonization guidelines for good clinical practice. All study documents and procedures were approved by the appropriate institutional review board/ethics committees at each study site, and written informed consent was obtained from all patients before initiation into the study.

LIBERTY Asthma QUEST study design. IP investigational product, q2w every 2 weeks, sc subcutaneously
Treatment/Dosing
A total of 1902 patients (≥ 12 years) were randomized in a 2:2:1:1 ratio to receive 52 weeks of add-on therapy with subcutaneously administered dupilumab 200 mg (loading dose 400 mg) or 300 mg (loading dose 600 mg) every 2 weeks (q2w), or matched placebo corresponding to the respective volumes of each dupilumab pre-filled syringe (1.14 and 2 mL for the 200 and 300 mg doses, respectively). Hence, approximately 634 patients for each dupilumab dose group and 317 patients for each matching placebo group were randomized.
Patients were stratified at randomization by age (< 18, ≥ 18 years), blood eosinophil count at screening (< 300, ≥ 300 cells/μL), baseline ICS dose (medium versus high), and country. For the duration of the study, all patients continued to receive their prescribed ICS plus up to two additional asthma controller medications, without change. Throughout the study, patients were permitted to use a short-acting β2-adrenergic receptor agonist (either salbutamol or levosalbutamol) as relief medication for asthma symptoms as needed.
Objectives and Outcome Measures
A summary of the study outcome measures is provided in Table 2. The two primary efficacy endpoints were annualized rate of severe exacerbation events during the 52-week treatment period and absolute change from baseline in pre-bronchodilator FEV1 at week 12. A severe asthma exacerbation was defined as a deterioration of asthma requiring treatment for ≥ 3 days with systemic corticosteroids, or hospitalization or an emergency room visit because of asthma, requiring systemic corticosteroids. The key secondary endpoint was the percentage change from baseline to week 12 in pre-bronchodilator FEV1. Additional lung function assessments throughout the study were the percentage predicted FEV1, morning/evening peak expiratory flow (measured at home using an electronic peak flow meter), forced vital capacity, forced expiratory flow 25–75%, post-bronchodilator FEV1, and post-bronchodilator slope analysis on FEV1 to characterize the loss of lung function. Patient-reported outcome measures of asthma control (ACQ-5) and quality of life, including the asthma quality of life questionnaire (AQLQ), were assessed throughout the study period. Safety and tolerability were evaluated by assessing the incidence of adverse events (AEs) and serious AEs, and by examination of vital signs and physical assessment, clinical laboratory testing, and 12-lead electrocardiography (ECG). Blood samples for exploratory genetic analysis of DNA/RNA were also collected and stored for future investigation.
Statistical Considerations
Efficacy analyses were performed on the intention-to-treat (ITT) population (defined as all randomized patients according to the treatment allocated), regardless of whether treatment was received. The annualized rate of severe exacerbation events was analyzed using a negative binomial regression model, with the total number of events occurring during the 52-week treatment period as the response variable, and the four treatment groups, age, region (pooled country), baseline eosinophil strata, baseline ICS dose level, and number of severe exacerbation events within 1 year prior to the study as covariates. Log-transformed observation duration was the offset variable.
Patients who discontinued the study medication were encouraged to return to the clinic for all remaining study visits, and all severe exacerbation events that happened up to week 52 were included in the primary analysis, regardless of whether or not the patient was receiving treatment.
Change from baseline in continuous endpoints such as FEV1 and patient-reported outcomes were analyzed using a mixed-effects model with repeated measures (MMRM). The model included changes from baseline values as response variables, and treatment, age, baseline eosinophil strata, baseline ICS dose level, visit, treatment-by-visit interaction, baseline value, and baseline-by-visit interaction as covariates. In addition, sex and baseline height were included as covariates only in the models for spirometry parameters. For patients who discontinued the study treatment and stayed in the study, off-treatment measurements were included in the primary model. An unstructured correlation matrix was used to model within-patient errors, and parameters were estimated using the restricted maximum likelihood method with the Newton–Raphson algorithm.
For subgroups defined in Table 3, treatment-by-subgroup interactions were analyzed using a negative binomial model/MMRM similar to the primary model, with subgroup and treatment-by-subgroup interaction added as covariates in the negative binomial models and subgroup, subgroup-by-treatment interaction, and subgroup-by-treatment-by-visit interaction added as covariates in the MMRM models. Summary statistics were also provided within each subgroup. Planned subgroups for analysis are listed in Table 3, including subgroups defined by baseline blood eosinophil counts, and by baseline FeNO and baseline periostin levels.
Time-to-event endpoints were analyzed using a Cox regression model, with time to event as the dependent variable and treatment, age, number of asthma exacerbation events in the previous year, region (pooled country), baseline eosinophil strata, and baseline ICS dose level as covariates. The Kaplan–Meier method was used to estimate the probability of a patient experiencing an event.
Descriptive statistics were used for demographic and clinical characteristics, pharmacodynamic variables, and for safety variables including AEs, vital signs and findings on physical examination, clinical laboratory testing, and ECG.
Sample Size Estimates
Assuming an annualized exacerbation rate in the placebo group of 0.6, a sample size of approximately 1638 patients was estimated to provide 99% power (two-tailed α level of 0.05) to detect a 55% relative risk reduction (i.e., annualized rate of 0.27 for the dupilumab group) in the annualized rate of severe exacerbations. This sample size was also expected to provide 98% power to the second primary endpoint, capable of detecting a treatment difference of 0.15 L in change of FEV1 from baseline to week 12. In a population of this size, approximately 84 adolescent patients and 690 (42%) patients with a baseline blood eosinophil count of ≥ 300 cells/μL were expected to be randomized.
Discussion
Moderate-to-severe asthma patients with persistent symptoms despite ICS plus LABA use represent a population of significant unmet need and one for whom new treatments are required. These patients are at high risk of asthma exacerbations and a substantial proportion has a history of life-threatening asthma attacks. Patients with severe asthma face an accelerated decline in lung function, which often impairs quality of life and interferes with their work and/or their daily activities. QUEST examined the efficacy of dupilumab in this at-risk patient population, and is the largest placebo-controlled study ever performed in uncontrolled, moderate-to-severe asthma with a biologic agent to date.
Rationale for Endpoint Choice
Annualized rate of severe exacerbation events is one of the most clinically relevant endpoints with which to evaluate asthma controller medications; by capturing this measure over a 52-week treatment period, any impact caused by seasonal variations in exacerbation rates was minimized. Absolute change from baseline in pre-bronchodilator FEV1 at week 12 was selected as the second primary endpoint; this is a well-accepted parameter to determine the effect of a drug on lung function, and is a predictor of mortality in asthma.
Rationale for Dose and Regimen
In the phase 2b dose-ranging study [9], both the 300 and 200 mg q2w dupilumab dose regimens provided better efficacy compared with the equivalent dupilumab dose regimens administered every 4 weeks on most efficacy endpoints, irrespective of eosinophil count at baseline. Both doses were well tolerated and (with the exception of an increased number of injection site reactions) had tolerability profiles comparable to those observed with placebo. Both regimens were assessed in the current trial to identify and further characterize the optimal dupilumab regimen in this patient population. Matching placebo arms were included to ensure the most robust assessment of the efficacy and safety of the dupilumab regimens.
There are currently limited treatment options for patients with uncontrolled, moderate-to-severe asthma. Add-on long-acting muscarinic receptor antagonists (tiotropium) [10], and anti-IgE (omalizumab) [11] and anti-IL-5 (reslizumab, mepolizumab, and benralizumab) [12,13,14,15] mAbs are all approved, and are added on to standard ICS plus LABA therapy, but are effective only in specific subgroups of patients. Omalizumab is indicated only for persistent asthma patients with a positive skin test or in vitro reactivity to a perennial aeroallergen [16, 17], and anti-IL-5 treatments have shown efficacy only in patients with eosinophilic asthma and are thus indicated for an eosinophilic phenotype [18,19,20].
Dupilumab is the first biologic to target both IL-4 and IL-13 type 2 cytokines and, as such, targets type 2 inflammation effectively, reducing not only eosinophil levels but also FeNO and periostin, two other type 2-associated biomarkers [9]. The observation that dupilumab was efficacious in patients with baseline eosinophil levels of both < 300 and ≥ 300 cells/μL in the phase 2b study was of particular interest, as prior to this, other biologics (such as the anti-IL-5 agents reslizumab, mepolizumab, and benralizumab) have shown that efficacy is limited to patients with eosinophilic asthma [14, 15, 18,19,20] and have been more effective in patients with higher baseline eosinophil levels. Such patients are typically associated with more severe asthma [21]. A recently published phase 2 study of tezepelumab, a mAb specific for the epithelial-cell-derived cytokine thymic stromal lymphopoietin, in uncontrolled asthma has also shown efficacy irrespective of baseline eosinophil count [22]. However, QUEST evaluates a biologic therapy for asthma with a trial design that facilitates entry into the study for all patients regardless of baseline eosinophil count, and is the first to assess the primary endpoint in the overall population, rather than a pre-specified eosinophil group. This is important, as it allows the adoption of a “real-world” approach to the treatment of asthma in a large patient population, thus ensuring that data are more representative of clinical practice. In addition, allowing patients the use of a third controller medicine if needed allows for a more accurate reflection of how patients are currently treated in clinical settings.
In terms of limitations of the QUEST study design, some of the subgroup analyses were not adequately powered. As such, any conclusions that may be drawn as a result of these analyses should be taken with caution until replicated with additional clinical trials.
Conclusions
QUEST was the largest phase 3 clinical trial to date to assess adults and adolescents (> 12 years of age) with uncontrolled, moderate-to-severe asthma, despite treatment with ICS plus other controller medications, and to enroll patients irrespective of baseline eosinophil levels or any other biomarker requirement. The study aims to confirm earlier findings, and further demonstrate that dupilumab elicits responses in a broad range of patients with uncontrolled, moderate-to-severe asthma, and to provide these patients with improvements in lung function and asthma control, reductions in severe exacerbations, and an improved quality of life.
References
Godard P, Chanez P, Siraudin L, Nicoloyannis L, Duru G. Costs of asthma are correlated with severity: a 1 year prospective study. Eur Respir J. 2002;19:61–7.
Haldar P, Pavord ID, Shaw DE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med. 2008;178:218–24.
Wenzel SE. Asthma phenotypes: the evolution from clinical to molecular approaches. Nat Med. 2012;18:716–25.
Ray A, Oriss TB, Wenzel SE. Emerging molecular phenotypes of asthma. Am J Physiol Lung Cell Mol Physiol. 2015;308:L130–40.
Robinson D, Humbert M, Buhl R, et al. Revisiting type 2-high and type 2-low airway inflammation in asthma: current knowledge and therapeutic implications. Clin Exp Allergy. 2017;47:161–75.
Brusselle GG, Maes T, Bracke KR. Eosinophils in the spotlight: eosinophilic airway inflammation in nonallergic asthma. Nat Med. 2013;19:977–9.
Gandhi NA, Bennett BL, Graham NMH, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. 2016;15:35–50.
Wenzel SE, Ford L, Pearlman D, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med. 2013;368:2455–66.
Wenzel SE, Castro M, Corren J, et al. Dupilumab efficacy and safety in adults with uncontrolled persistent asthma despite use of medium-to-high-dose inhaled corticosteroids plus a long-acting β2 agonist: a randomised double-blind placebo-controlled pivotal phase 2b dose-ranging trial. Lancet. 2016;388:31–44.
Kerstjens HA, Disse B, Schröder-Babo W, et al. Tiotropium improves lung function in patients with severe uncontrolled asthma: a randomized controlled trial. J Allergy Clin Immunol. 2011;128:308–14.
Hanania NA, Alpan O, Hamilos DL, et al. Omalizumab in severe allergic asthma inadequately controlled with standard therapy: a randomized trial. Ann Intern Med. 2011;154:573–82.
Castro M, Zangrilli J, Wechsler M, et al. Reslizumab for inadequately controlled asthma with elevated blood eosinophil counts: results from two multicentre, parallel, double-blind, randomised, placebo-controlled, phase 3 trials. Lancet Respir Med. 2015;3:355–66.
Ortega HG, Liu MC, Pavord ID, et al. Mepolizumab treatment in patients with severe eosinophilic asthma. N Engl J Med. 2014;371:1198–207.
Bleecker ER, FitzGerald JM, Chanez P, et al. Efficacy and safety of benralizumab for patients with severe asthma uncontrolled with high-dosage inhaled corticosteroids and long-acting beta2-agonists (SIROCCO): a randomised, multicentre, placebo-controlled phase 3 trial. Lancet. 2016;388:2115–27.
FitzGerald JM, Bleecker ER, Nair P, et al. Benralizumab, an anti-interleukin-5 receptor alpha monoclonal antibody, as add-on treatment for patients with severe, uncontrolled, eosinophilic asthma (CALIMA): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2016;388:2128–41.
Xolair 150 mg Solution for Injection - Summary of Product Characteristics (SPC)—updated 21 Sep 2016, Novartis Pharmaceuticals UK Ltd. Available from: http://www.medicines.org.uk/emc/medicine/24912/SPC/Xolair+150mg+Solution+for+Injection.
Xolair (omalizumab) for injection, for subcutaneous use. The United States Package Insert (USPI) Revised Jun 2017.
Pavord ID, Korn S, Howarth P, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet. 2012;380:651–9.
Ortega HG, Yancey SW, Mayer B, et al. Severe eosinophilic asthma treated with mepolizumab stratified by baseline eosinophil thresholds: a secondary analysis of the DREAM and MENSA studies. Lancet Respir Med. 2016;4:549–56.
Corren J, Weinstein S, Janka L, Zangrilli J, Garin M. Phase 3 study of reslizumab in patients with poorly controlled asthma: effects across a broad range of eosinophil counts. Chest. 2016;150:799–810.
Katz LE, Gleich GJ, Hartley BF, Yancey SW, Ortega HG. Blood eosinophil count is a useful biomarker to identify patients with severe eosinophilic asthma. Ann Am Thorac Soc. 2014;11:531–6.
Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377:936–46.
Global Initiative for Asthma (GINA). 2017 Global Strategy for Asthma Management and Prevention. Available from: http://ginasthma.org/wp-content/uploads/2016/01/wms-GINA-2017-main-report-tracked-changes-for-archive.pdf. Accessed December 2017.
Acknowledgements
Funding
Research sponsored by Sanofi and Regeneron Pharmaceuticals, Inc. Clinical Trials.gov Identifier: NCT02414854. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. Costs for article processing and Open Access were also funded by the study sponsors.
Medical Writing, Editorial, and Other Assistance
Editorial assistance in the preparation of this manuscript was provided by Adam J. Beech, PhD, of Excerpta Medica. Support for this assistance was funded by Sanofi Genzyme and Regeneron Pharmaceuticals, Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship of this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
William W. Busse has been paid consultant fees by Regeneron Pharmaceuticals, Inc. and Sanofi. Jorge F. Maspero is a consultant for AstraZeneca, Sanofi, and Teva; has received speaker fees from GSK, Menarini, Novartis, and Uriach; and has received research grants from Novartis. Klaus F. Rabe has been a consultant for and received speaker fees from AstraZeneca, Boehringer Ingelheim, Novartis, Sanofi, and Teva. Alberto Papi has received grants, personal fees, non-financial support and other from Chiesi, AstraZeneca, GSK, Boehringer Ingelheim, Mundipharma, and TEVA; and personal fees and non-financial support from Menarini, Novartis, and Zambon. Sally E. Wenzel has received research support from Sanofi and is an unpaid consultant for Regeneron Pharmaceuticals, Inc. Linda B. Ford has received grant support through her institution from 3M, Aimmune, AstraZeneca, DBV, Genentech, GSK, Glenmark, Hoffmann La Roche, Novartis, Pearl, Sanofi, and Teva; and is a national consultant for Sanofi. Ian D. Pavord has received speakers’ honoraria from Aerocrine, Almirall, AstraZeneca, Boehringer Inglehiem, GSK, Novartis, and Teva; assistance with organization of educational events from AstraZeneca and Teva; sits on advisory boards for Almirall, AstraZeneca, Boehringer Ingelheim, Chiesi, Circassia, Dey, Genentech, GSK, Knopp, Merck, MSD, Napp, Novartis, Regeneron Pharmaceuticals, Inc., Respivert, Sanofi, Schering-Plough, and Teva; has received travel grants from AstraZeneca, Boehringer Ingelheim, Chiesi, GSK, Napp, and Teva; and has received clinical trial support from Chiesi. Bingzhi Zhang is an employee of and may hold stock and/or stock options in Sanofi. Heribert Staudinger is an employee of and may hold stock and/or stock options in Sanofi. Gianluca Pirozzi is an employee of and may hold stock and/or stock options in Sanofi. Laurent Eckert is an employee of and may hold stock and/or stock options in Sanofi. Ariel Teper is an employee of and may hold stock and/or stock options in Sanofi. Nikhil Amin is an employee of and shareholder in Regeneron Pharmaceuticals, Inc. Bolanle Akinlade is an employee of and shareholder in Regeneron Pharmaceuticals, Inc. Jingdong Chao is an employee of and shareholder in Regeneron Pharmaceuticals, Inc. Neil M. H. Graham is an employee of and shareholder in Regeneron Pharmaceuticals, Inc.
Compliance with Ethics Guidelines
The study was conducted in accordance with the principles established in the Declaration of Helsinki and the International Conference on Harmonization guidelines for good clinical practice. All study documents and procedures have been approved by the appropriate institutional review board/ethics committees at each study site, and written informed consent was obtained from all patients before initiation into the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital content
To view enhanced digital content for this article go to https://doi.org/10.6084/m9.figshare.6139400.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Busse, W.W., Maspero, J.F., Rabe, K.F. et al. Liberty Asthma QUEST: Phase 3 Randomized, Double-Blind, Placebo-Controlled, Parallel-Group Study to Evaluate Dupilumab Efficacy/Safety in Patients with Uncontrolled, Moderate-to-Severe Asthma. Adv Ther 35, 737–748 (2018). https://doi.org/10.1007/s12325-018-0702-4
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-018-0702-4