
Abstract
Introduction
Obeticholic acid (OCA), a potent and selective farnesoid X receptor agonist, is indicated for the treatment of primary biliary cholangitis (PBC). We investigated the potential drug–drug interaction effect of OCA on metabolic CYP450 enzymes and drug transporters.
Methods
Five phase 1 single-center, open-label, fixed-sequence, inpatient studies were conducted in healthy adult subjects to evaluate the effect of oral daily doses of 10 or 25 mg OCA on single-dose plasma pharmacokinetics of specific probe substrates for enzymes CYP1A2 (caffeine, R-warfarin), CYP3A (midazolam, R-warfarin), CYP2C9 (S-warfarin), CYP2D6 (dextromethorphan), CYP2C19 (omeprazole), and drug transporters, BCRP/OATP1B1/OATP1B3 (rosuvastatin), and P-gp (digoxin).
Results
OCA showed no substantial suppression/inhibition of S-warfarin, digoxin, and dextromethorphan and weak interactions with caffeine, omeprazole, rosuvastatin, and midazolam. The maximal pharmacodynamic responses (E max) to warfarin-based INR, PT, and aPTT were reduced by 11%, 11%, and 1%, respectively, for the 10-mg dose group and by 7%, 7% and 0%, respectively, for the 25-mg dose group. Overall, drugs dosed in combination with OCA were well tolerated, and most adverse events were mild in severity. No clinically important trends were noted in laboratory evaluations, vital signs, or 12-lead ECGs.
Conclusion
In these studies, OCA showed weak to no suppression/inhibition of metabolic enzymes and drug transporters at the highest recommended therapeutic dose in patients with PBC. On the basis on these analyses, monitoring and maintenance of target INR range are required during coadministration of OCA with drugs that are metabolized by CYP1A2 (R-warfarin).
Funding
Intercept Pharmaceuticals, Inc.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Obeticholic acid (OCA) is a modified bile acid that has shown promise for treatment of progressive non-viral liver diseases. OCA has been most thoroughly evaluated for the treatment of patients with primary biliary cholangitis (PBC). PBC is a rare autoimmune liver disease characterized by inflammation and progressive impairment of bile flow, resulting in cholestasis and the eventual development of cirrhosis, end-stage liver disease, and death [1]. PBC disproportionately affects women versus men (approximately 10:1) and is typically diagnosed in patients between 40 and 60 years of age [2]. Ursodeoxycholic acid (UDCA), a bile acid constituent, was until recently the only approved treatment for PBC. However, up to 50% of UDCA-treated patients either fail to respond or have a suboptimal response, and consequently a higher risk of progressing to hepatocellular carcinoma [3], demonstrating an unmet medical need. OCA recently received FDA approval for the treatment of patients with PBC in combination with UDCA for those with an inadequate response to UDCA, or as monotherapy in those intolerant to UDCA. The suggested initial dose for patients with PBC, who do not have moderate or severe hepatic impairment, is 5 mg OCA once daily, with dose titration to 10 mg according to tolerability at 6 months [4].
Another progressive liver disease, nonalcoholic steatohepatitis (NASH), is much more prevalent and is a leading cause of liver transplantation [5]. There are currently no therapies approved for the treatment of NASH. OCA is being investigated as a new potential therapy for NASH. The phase 2 Farnesoid X Receptor Ligand Obeticholic Acid in NASH Treatment (FLINT) trial demonstrated promising results [6] and the phase 3 Randomized Global Phase 3 Study to Evaluate the Impact on NASH with Fibrosis of Obeticholic Acid Treatment (REGENERATE) trial is ongoing (ClinicalTrials.gov Identifier: NCT02548351).
OCA is a potent agonist for the farnesoid X receptor (FXR), a nuclear receptor expressed at high levels in the liver and intestine, and has demonstrated greater selectivity for FXR compared with other receptors. OCA is a semi-synthetic analog of the primary bile acid chenodeoxycholic acid (CDCA), the endogenous FXR ligand, but OCA has 100-fold greater potency than CDCA at activating FXR [7]. FXR (NR1H4) controls bile acid synthesis, influx and efflux in the liver–gut axis, and has been recognized as a master regulator of bile acid homeostasis. FXR agonists directly induce small heterodimer partner/protein (SHP) (NR0B2) and fibroblast growth factor (FGF-19) expression in hepatocytes and enterocytes, respectively. Both SHP and FGF-19 suppress cholesterol 7-alpha-hydroxylase (CYP7A1), the enzyme for the rate-limiting conversion of cholesterol to bile acids in the liver [8]. In addition, activated FXR promotes bile acid-dependent bile flow (choleretic effect) by upregulating bile salt excretory pump (BSEP), the major bile acid efflux transporter on the canalicular membrane of hepatocytes [9]. FXR-responsive elements are also located in the promoter regions of both organic solute transporter α/β (OST α/β) genes [10]. The heterodimers of OSTα/OSTβ transport bile acids into blood, thereby reducing hepatic intracellular levels of bile acids and reducing the potential for bile acid induced cytotoxicity [10]. After absorption from the gastrointestinal tract, OCA is rapidly taken up by the liver, where it is conjugated with glycine or taurine and secreted into bile. OCA conjugates, which are the primary circulating forms of OCA, are pharmacologically active and are reabsorbed from the small intestine, leading to enterohepatic circulation. Excretion is primarily through feces.
Drug-metabolizing cytochrome P450 (CYP) enzymes and drug transporters are regulated by nuclear receptors. Drug inhibition or induction/suppression of CYP enzymes and/or drug transporters can potentially lead to drug–drug interaction (DDI) in clinical settings, resulting in adverse drug events. Therefore, to investigate the potential for pharmacokinetic (PK) or pharmacodynamic (PD) interactions between OCA and other drugs, five clinical studies using sensitive CYP (CYPs 1A2, 3A, 2C9, 2C19, and 2D6) and transporter (multidrug resistance protein 1 [MDR1]/ATP-binding cassette subfamily B member 1 [ABCB1]/P-glycoprotein [P-gp], breast cancer resistance protein [BCRP], organic anion transporting polypeptide [OATP] 1B1, and OATP1B3) substrates were conducted. In addition, the effects of OCA on the pharmacodynamics of warfarin were assessed.
Methods
Subjects
Healthy male and female subjects were screened up to 21 days before check-in (day 0). Subjects were eligible for study participation if all inclusion/exclusion criteria were met. Subjects were 18–55 years of age with a body mass index (BMI) of 18–30 kg/m2. Subjects were required to be in good general health as determined by a standard physical exam, 12-lead electrocardiogram (ECG), and clinical laboratory testing. Subjects were excluded for any known exposure to OCA and any of its components, probe substrates, or any comparable or similar product. Additional exclusion criteria were specific to study.
Study Design
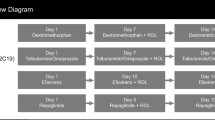
These five studies were conducted as single-center, open-label, fixed-sequence, parallel-design, inpatient studies in healthy volunteers. Each study was designed to evaluate the single-dose PK of the substrate probe alone and in combination with repeat doses (at least 14 days) of 10 and 25 mg OCA, the two highest OCA doses intended for commercialization across indications (Table 1). To reduce the total number of studies required to assess seven probe substrates, we combined the assessment of midazolam and caffeine into one study and dextromethorphan and omeprazole in another study, evaluating each probe substrate separately on different days. OCA and probe substrates were administered in the fasted state.
Eligible subjects were randomized in a 1:1 ratio to one of two OCA treatment arms (10 or 25 mg) receiving OCA once daily. All subjects remained at the clinic from day 0 to the final day of dosing which varied depending on the time needed to properly evaluate the PK of each probe substrate, which was dependent on the plasma half-life of the probe substrate. Safety analyses were conducted on all subjects who received at least one dose of study drug.
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. All subjects provided informed consent before any study procedures or assessments were performed. The studies were not registered as this is not a requirement for phase 1 studies.
Study I: Specific Substrates for CYP1A2 (Caffeine) and for CYP3A (Midazolam)
Subjects were required to avoid meals and fluids containing caffeine, theobromine, and xanthine for at least 72 h before day 0 and during the inpatient portion of the study. Subjects received a single oral dose of 2 mg midazolam in syrup form on day 1 and day 19 and a single oral dose of 200 mg caffeine in caplet form on day 3 and day 21 under fasting conditions (10-h overnight fast). On day 5 through day 23, oral doses of 10 or 25 mg OCA were administered daily under fasting conditions. Blood samples were collected at predose and at 0.25, 0.5, 1, 1.25, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h after dosing of midazolam on day 1 and day 19 for the measurement of plasma concentrations of midazolam and its metabolite (1-hydroxymidazolam). Similarly, blood samples were collected at predose and at 0.25, 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, 24, 36, and 48 h after dosing of caffeine on day 3 and day 21 for the measurement of plasma concentrations of caffeine and its metabolite (paraxanthine).
Study II: Specific Substrates for CYP2C9 (S-warfarin) and for CYP1A2 and CYP3A (R-warfarin)
Subjects were excluded if they were a poor metabolizer of warfarin as determined by CYP2C9 genotype testing, if they were highly sensitive to warfarin as determined by genetic testing for vitamin K epoxide reductase complex, subunit 1 (VKORC1) or had a prothrombin time (PT), activated partial thromboplastin time (aPTT), international normalized ratio (INR), protein C, or protein S value(s) outside the normal ranges at screening for which the results were clinically significant.
Several published DDI studies used a racemic warfarin single dose of 25 mg to produce well-characterized PK and PD responses [11,12,13]. In this study, a 25-mg dose of racemic warfarin was expected to provide sufficient plasma concentrations of both enantiomers to characterize their plasma PK, and also produce measurable PD effects with no undue risk to the subjects. Subjects were administered a single oral dose of 25 mg racemic warfarin (R- and S-warfarin) in tablet form on day 1 and day 21 under fasting conditions. Daily oral administration of OCA at doses of 10 or 25 mg was initiated on day 8 and continuing through day 27. PK blood samples for measuring plasma concentrations of R- and S-warfarin were collected at predose and at 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, 24, 36, 48, 60, 72, 96, 120, 144, and 168 h (i.e., over a 7-day period) after dosing of racemic warfarin on day 1 and day 21. The PD of racemic warfarin using the serial blood samples (predose and 12, 24, 36, 48, 60, 72, 96, 120, 144, and 168 h after dosing on day 1 and day 21) was assessed by measuring coagulation parameters PT, aPTT, and INR.
Study III: Specific Substrates for CYP2D6 (Dextromethorphan) and for CYP2C19 (Omeprazole)
CYP2D6 is a genetically controlled polymorphic enzyme such that approximately 5–10% of people of white ethnicity express no functional enzyme and are characterized as poor metabolizers. Subjects were excluded if they were a poor metabolizer of dextromethorphan as determined by CYP2D6 genotyping or omeprazole as determined by CYP2C19 genotyping or screened positive for Helicobacter pylori (H. pylori).
Subjects received a single oral 30-mg dose of dextromethorphan in capsule form on day 1 and day 18. On the basis of the terminal half-life (t 1/2) of dextromethorphan (2–4 h), blood samples for the measurement of plasma concentrations of dextromethorphan and its metabolite (dextrorphan) were collected at predose and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 12, 24, 48, and 72 h after dosing of dextromethorphan on days 1 and 18.
A single 20-mg dose of omeprazole in capsule form was administered orally on day 4, and once-daily dosing was initiated on days 21 through 28 as a second test treatment. Blood samples for the measurement of plasma concentrations of omeprazole and its metabolite (5-hydroxyomeprazole) were collected at predose and at 0.25, 0.5, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, and 12 h after dosing of omeprazole on days 4 and 21, and at 3 h postdose on day 28. OCA was administered orally once daily, beginning on day 5 and continued through day 28.
Study IV: BCRP, OATP1B1, and OATP1B3 Substrate (Rosuvastatin)
Rosuvastatin is a recommended substrate for assessment of inhibition of transporters responsible for its distribution and elimination by BCRP, OATP1B1, and OATP1B3 [14]. Several reported drug interaction studies have used rosuvastatin single doses ranging from 10 to 80 mg [15,16,17,18,19]. On the basis of the terminal half-life of rosuvastatin (20 h) and the bioanalytical plasma assay sensitivity, a single dose of 20 mg rosuvastatin was selected to provide concentrations of rosuvastatin sufficient to characterize the PK while minimizing risk to the subjects.
Subjects were administered a single oral dose of 20 mg rosuvastatin in tablet form on day 1 and day 19 under fasting conditions. Daily administration of oral OCA (10 or 25 mg) was initiated on day 6 and continued for 18 days. Blood samples for the measurement of plasma concentrations of rosuvastatin and its metabolite (N-desmethyl rosuvastatin) were collected at predose and at 0.5, 1, 2, 3, 3.5, 4, 4.5, 5, 5.5, 6, 7, 8, 10, 12, 24, 36, 48, 72, 96, and 120 h after dosing of rosuvastatin on day 1 and day 19.
Study V: P-gp Substrate (Digoxin)
The subjects who were sensitive to digoxin or other digitalis preparations were excluded. Digoxin (immediate release formulation) is a sensitive and recommended substrate for the assessment of P-gp, and is eliminated as unchanged drug primarily via the kidney [20]. Subjects were administered a single oral dose of 0.25 mg digoxin in tablet form on day 1 and day 19 under fasting conditions. Daily administration of OCA was initiated on day 6 and continued for 18 days. On the basis of the half-life of digoxin (1.5–2 days), blood samples for the measurement of plasma concentrations of digoxin were collected at predose and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 24, 36, 48, 60, 72, 96, and 120 h after dosing on day 1 and day 19.
In each study, blood samples for the measurement of plasma concentrations of OCA and its metabolites (glyco-OCA and tauro-OCA) were collected predose on the first day of OCA administration (reference treatment), periodically throughout the dosing period, and on days when OCA and probe drugs were administered concomitantly (test treatment).
Analytical Methods
The plasma concentrations of each probe substrate and of OCA and its metabolites were determined by high-performance liquid chromatography (HPLC) with tandem mass spectrometric detection (LC–MS-MS) using validated analytical methods.
Pharmacokinetic and Pharmacodynamic Parameters
For all studies, the reported PK parameters included the area under the plasma concentration–time curve (AUC) from time 0 to the last sampling time with measurable analyte concentration (AUC0−t ), AUC from time 0 to infinity (AUC∞), and maximum observed plasma concentration (C max). All PK parameters were measured using Phoenix® WinNonlin® (Version 6.2 or later).
The PD parameters for PT, aPTT, and INR were calculated in study II (racemic warfarin), including the area under the effect curve from time zero to the last sampling collection time point (0–168 h) (AUEC), maximum observed effect (E max), and time to reach E max (TEmax).
Statistical Analysis
Statistical analyses were performed using SAS® Version 9.2. The primary PK endpoints (AUC0−t , AUC∞, and C max) were analyzed using a linear mixed effect model on the natural log (ln) transformed PK parameters of the different probe substrates. Geometric least square mean (GLSM) and ratios for each of the substrate probes were obtained from the ln-transformed AUC0−t , AUC∞, and C max. No apparent drug–drug interaction was concluded if the GLSM ratio was contained within the bounds (0.80–1.25) of the 90% confidence interval (CI) [14].
PD parameters AUEC and E max for PT, aPTT, and INR were analyzed using the same statistical analyses performed on the PK parameters.
Results
Subjects
The demographic and baseline characteristics of subjects participating in each study are summarized in Table 2. Subjects participating in the studies were predominantly non-hispanic, white males ranging from approximately 33 to 38 years of age. The discontinuation incidence by study ranged from 0% to 6% with one discontinuation due to a treatment-emergent adverse event (TEAE) (Table 2 and Supplemental Table S7).
Pharmacokinetic Evaluation
Statistical comparisons of the primary PK parameters (AUC∞ and C max) following a single dose of probe substrates administered alone and with 10 or 25 mg OCA are presented in Table 3.
PK data for all substrates are presented in Supplemental Tables S1–S5.
CYP1A2 (Caffeine)
Mean plasma caffeine concentrations over time were higher in the presence of 10 and 25 mg OCA (Fig. 1). In the presence of 10 mg OCA, the GLSM ratios ([caffeine + OCA]/[caffeine alone]) demonstrated a 42% increase in AUC∞ and a 6% increase for C max (Table 3). In the 25 mg OCA group, the GLSM ratios for caffeine demonstrated increases of 65% for AUC∞ and 10% for C max. Together, the PK exposure data indicated weak suppression/inhibition of CYP1A2 when administering caffeine in the presence of OCA (Table 3).

Pharmacokinetic profile of caffeine. Data are mean (+SD) plasma caffeine concentrations versus time following a single 200-mg dose of caffeine administered alone (solid line, triangles) and with 10 mg (a) or 25 mg (b) OCA (dotted line, squares). Main figures display caffeine plasma concentration on a linear scale, and insets display log scale. Includes patients from caffeine PK population (N = 45)
CYP3A (Midazolam)
Mean plasma midazolam concentrations over time in the presence of 10 mg OCA were similar to midazolam alone, and slightly higher in the presence of 25 mg OCA (Fig. 2). The GLSM ratios for the primary midazolam PK parameters in the OCA 10 mg group were generally comparable to midazolam alone. In the 25 mg OCA group, the GLSM ratios demonstrated a 26% increase in AUC∞ and 17% increase for C max compared with midazolam alone, suggesting weak suppression/inhibition of CYP3A at the higher dose of OCA (Table 3). Although the 26% increase in exposure is incrementally higher than the 25% cutoff, the effect was not considered clinically significant (Table 3).

Pharmacokinetic profile of midazolam. Data are mean (+SD) plasma midazolam concentrations versus time following a single 2-mg dose of midazolam administered alone (solid line, circles) and with 10 mg (a) or 25 mg (b) OCA (dotted line, x). Main figures display midazolam plasma concentration on a linear scale, and insets display log scale. Includes patients from midazolam PK population (N = 47)
CYP2C9 (S-warfarin)
Mean plasma S-warfarin concentrations over time were generally similar for warfarin alone and warfarin in the presence of 10 or 25 mg OCA (Fig. 3). In the presence of 10 mg OCA, the GLSM ratios for S-warfarin demonstrated weak increases in exposure: 13% for AUC∞ and 12% for C max (Table 3). In the 25 mg OCA group, the GLSM ratios for S-warfarin demonstrated increases of 18% for AUC∞ and 6% for C max. All 90% CIs were within the 80% to 125% bounds, suggesting that OCA does not have a clinically meaningful effect on CYP2C9 (Table 3).

Pharmacokinetic profile of warfarin. Data are mean (+SD) plasma S-warfarin (a, b) and R-warfarin (c, d) concentrations versus time following a single 25-mg dose of warfarin administered alone (solid line, circles) and with 10 mg (a, c) or 25 mg (b, d) OCA (dotted line, x). Main figures display warfarin plasma concentration on a linear scale, and insets display log scale. Includes patients from PK/PD population (N = 43)
CYP1A2 and CYP3A (R-warfarin)
Mean plasma R-warfarin concentrations over time were modestly higher in the presence of OCA than warfarin alone (Fig. 3). The GLSM ratios for R-warfarin after repeated administration of 10 mg OCA demonstrated a modest increase in exposure of 21% for AUC∞ and 11% for C max (Table 3). The GLSM ratios in the 25 mg OCA dose demonstrated an increase of 32% for AUC∞ and 5% for C max. On the basis of these modest increases in exposure, and the 90% CIs exceeding the 80% to 125% bounds in some instances, these data demonstrate that OCA has a weak effect on the enzymes primarily responsible for R-warfarin metabolism (CYP1A2 and/or CYP3A) (Table 3) and is consistent with the weak inhibition of CYP1A2 observed in study I for caffeine.
CYP2D6 (Dextromethorphan)
Mean plasma dextromethorphan concentrations over time were generally similar for dextromethorphan alone and dextromethorphan in the presence of 10 and 25 mg OCA (Fig. 4). The GLSM ratios for dextromethorphan demonstrated an 11% and 12% decrease in AUC∞ and C max, respectively, in the presence of 10 mg OCA compared with dextromethorphan alone. In the 25 mg OCA group, the GLSM ratios for dextromethorphan demonstrated an 11% and 17% decrease in AUC∞ and C max, respectively (Table 3). The decreases were not dose-related, suggesting no clinically relevant effect of OCA on CYP2D6.

Pharmacokinetic profile of dextromethorphan. Data are mean (+SD) plasma dextromethorphan concentrations versus time following a single 30-mg dose of dextromethorphan administered alone (solid line, circles) and with 10 mg (a) or 25 mg (b) OCA (dotted line, x). Main figures display dextromethorphan plasma concentration on a linear scale, and insets display log scale. Includes patients from dextromethorphan PK population (N = 47)
CYP2C19 (Omeprazole)
Mean plasma omeprazole concentrations over time were higher in the presence of 10 and 25 mg OCA (Fig. 5). The GLSM ratios demonstrated a 32% and 33% increase in AUC∞ and C max, respectively, in the 10 mg OCA group, and a 37% and 15% increase in AUC∞ and C max, respectively, in the OCA 25 mg group, compared to omeprazole alone. The increases indicate a mild but not dose-related suppression/inhibition of CYP2C19 (Table 3).

Pharmacokinetic profile of omeprazole. Data are mean (+SD) plasma omeprazole concentrations versus time following a single 20-mg dose of omeprazole administered alone (solid line, triangles) and with 10 mg (a) or 25 mg (b) OCA (dotted line, squares). Main figures display omeprazole plasma concentration on a linear scale, and insets display log scale. Includes patients from omeprazole PK population (N = 47)
BCRP, OATP1B1, and OATP1B3 (Rosuvastatin)
Mean plasma rosuvastatin concentrations over time were modestly higher in the presence of 10 and 25 mg OCA (Fig. 6). The GLSM ratios for rosuvastatin after repeated administration of 10 mg OCA demonstrated a modest increase in exposure of 22% for AUC∞ and 27% for C max (Table 3). The GLSM ratios in 25 mg OCA demonstrated an increase of 30% for AUC∞ and 26% for C max (Table 3). The increases, which were not dose-related, were deemed to have no clinically relevant effect of OCA on these transporters.

Pharmacokinetic profile of rosuvastatin. Data are mean (+SD) plasma rosuvastatin concentrations versus time following a single 20-mg dose of rosuvastatin administered alone (solid line, circles) and with 10 mg (a) or 25 mg (b) OCA (dotted line, x). Main figures display rosuvastatin plasma concentration on a linear scale, and insets display log scale. Includes patients from rosuvastatin PK population (N = 48)
P-gp Substrate (Digoxin)
Mean plasma digoxin concentrations over time in the presence of 10 mg OCA were similar to digoxin alone, and slightly higher in the presence of 25 mg OCA (Fig. 7). The GLSM ratios for the primary digoxin PK parameters in the 10 mg OCA group indicate comparable exposure for digoxin with OCA versus digoxin alone. In the 25 mg OCA group, the GLSM ratios demonstrated a 7% increase in AUC∞ and a 24% increase for C max compared to digoxin alone. On the basis of these weak increases in exposure of less than 25%, these data demonstrate that OCA has no effect on digoxin exposure at 10 mg and only a slight effect at 25 mg (Table 3).

Pharmacokinetic profile of digoxin. Data are mean (+SD) plasma digoxin concentrations versus time following a single 0.25-mg dose of digoxin administered alone (solid line, circles) and with 10 mg (a) or 25 mg (b) OCA (dotted line, x). Main figures display digoxin plasma concentration on a linear scale, and insets display log scale. Includes patients from digoxin evaluable PK population (N = 48)
Overall, PK parameters for metabolites of probe drugs (1-hydroxymidazolam for midazolam, paraxanthine for caffeine, dextrorphan for dextromethorphan, 5-hydroxyomeprazole for omeprazole, and N-desmethyl rosuvastatin for rosuvastatin) concurred with the PK data for the corresponding primary drug (Supplemental Table S6).
Mean total OCA plasma concentrations over time in midazolam, caffeine, warfarin, dextromethorphan, omeprazole, rosuvastatin, and digoxin populations are shown in Supplemental Fig. S2. A visual representation of overall effects of OCA on various substrates is shown as a forest plot in Supplemental Fig. S3.
Pharmacodynamic Evaluation
The warfarin PD coagulation parameters PT, aPTT, and INR were evaluated in the presence of 25 mg warfarin administered alone and with 10 or 25 mg OCA over 7 days. As expected, mean PT, aPTT, and INR values generally increased after administration of warfarin alone but returned to baseline values, indicating that a 25-mg dose was sufficient for determining the effect of OCA on the pharmacological response of warfarin (Supplemental Fig. S1).
Modest decreases were observed in PT E max with OCA at 10 mg (11%) and 25 mg (7%), and in INR E max with OCA at 10 mg (11%) and 25 mg (7%) (Table 4). There were no notable or significant changes in aPPT.
Safety
Administration of OCA with the different probe substrates was generally well tolerated. No clinically meaningful changes in clinical laboratory parameters, vital signs, 12-lead ECGs, or physical examination were observed in any of the five drug–drug interaction studies. The overall frequency of adverse events (AEs) was low in all studies and most AEs were mild in severity. The incidence of AEs, serious adverse events, and AEs leading to treatment or study discontinuation are provided in Supplemental Table S7. No dose-dependent changes in the incidence of AEs were reported in any of the studies.
In study I, infrequent bowel movements (13% of subjects), nausea (6%), and headache (6%) were the most frequently reported TEAEs. In study II, infrequent bowel movements (9%), headache (5%), and fatigue (5%) were the most frequently reported TEAEs. In study III, pruritus (4%) and headache (4%) were the most frequently reported TEAEs. In study IV, the most common TEAE was infrequent bowel movements (4%). In study V, headache (10%), nausea (8%), dizziness (6%), and abdominal discomfort (6%) were the most frequently reported TEAEs.
There were no deaths, serious AEs, or TEAEs that led to discontinuation in studies I, III, IV, or V. In study II, one subject experienced an AE of abdominal pain that resulted in study discontinuation and two serious AEs of acute cholecystitis and cholelithiasis.
Discussion
Five phase 1 single-center, open-label, fixed-sequence, inpatient studies were conducted to evaluate the potential for drug–drug interactions between OCA and specific probe substrates for an array of CYP enzymes and transporters. Two weeks of OCA administration were considered sufficient for OCA to reach steady state exposure and for any induction or suppression of enzymes to occur, based on the pharmacokinetics of OCA and the half-lives of CYP enzymes. At steady state, OCA showed no substantial suppression/inhibition of the PK exposure parameters of S-warfarin (CYP2C9), digoxin (P-gp), and dextromethorphan (CYP2D6) at both the 10 and 25 mg OCA doses; all changes were considered not clinically relevant.
Very weak interactions were seen for caffeine. This weak interaction is most likely due to a modest suppression of CYP1A2 that was observed in in vitro hepatocyte studies (data not shown). A potential mechanism that would relate this weak interaction with FXR is the potential of SHP to interfere with the transcriptional regulation of CYP1A2 via the aryl hydrocarbon receptor (AhR) in conjunction with the aryl hydrocarbon receptor nuclear translocator (ARNT) [21, 22].
A modest effect of OCA on the PK of rosuvastatin was observed, with inhibition occurring only at the 25-mg dose, and not at the 10-mg dose. OCA 10 and 25 mg produced weak suppression/inhibition of CYP2C19, as demonstrated by modest increases in the mean plasma concentrations of omeprazole. The clinical significance of these modest increases is unknown. OCA treatment did not suppress/inhibit CYP3A at 10 mg, but an increase in midazolam exposure was observed at 25 mg. This effect is unlikely to produce clinically relevant increases in exposure of midazolam or other CYP3A substrates when co-administered with OCA.
Racemic warfarin is an anticoagulant comprising an S-enantiomer and R-enantiomer in a 1:1 mixture. The pharmacological effects of warfarin are elicited mainly by the S-enantiomer, which exhibits 2–5 times more anticoagulant activity than the R-enantiomer. The R- and S-enantiomers of warfarin are metabolized differently by human P450 enzymes with CYP2C9 primarily metabolizing S-warfarin and CYP1A2 and CYP3A primarily metabolizing R-warfarin. OCA demonstrated no meaningful increases in S-warfarin exposure, which is a CYP2C9 substrate. These data indicate that OCA is not an inhibitor or suppressor of CYP2C9, and is not expected to inhibit the metabolism of other CYP2C9 substrates consistent with in vitro results (data not shown). Repeat administration of OCA at daily doses of 10 and 25 mg resulted in modest dose-related increases in R-warfarin exposure which is consistent with results in study I showing a weak interaction of OCA on CYP1A2.
Repeat administration of OCA at daily doses of 10 and 25 mg resulted in weak changes in plasma PD coagulation parameters INR and PT and no changes in aPTT. Oral doses of 10 and 25 mg OCA administered alone and in combination with warfarin appeared to be generally safe and well tolerated; given the observed clinical changes, INR and PT should be monitored with coadministration of OCA and warfarin.
A limitation for consideration is that these studies were completed predominantly in healthy male subjects approximately 33 to 38 years of age. PBC is typically diagnosed in women between 40 and 60 years of age [2].
Conclusion
Overall, the results demonstrate that OCA shows weak to no suppression/inhibition of most major metabolic enzymes and transporters. The results suggest that, with the exception of narrow therapeutic index CYP1A2 substrates, OCA can be administered with medications that are substrates of most major metabolic enzymes and transporters without producing clinically significant drug–drug interactions in healthy subjects.
References
Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353(12):1261–73.
Mistry P, Seymour CA. Primary biliary cirrhosis—from Thomas Addison to the 1990s. Q J Med. 1992;82(299):185–96.
Kuiper EM, Hansen BE, Adang RP, et al. Relatively high risk for hepatocellular carcinoma in patients with primary biliary cirrhosis not responding to ursodeoxycholic acid. Eur J Gastroenterol Hepatol. 2010;22(12):1495–502.
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67(1):145–172.
Charlton MR, Burns JM, Pedersen RA, Watt KD, Heimbach JK, Dierkhising RA. Frequency and outcomes of liver transplantation for nonalcoholic steatohepatitis in the United States. Gastroenterology. 2011;141(4):1249–53.
Neuschwander-Tetri BA, Loomba R, Sanyal AJ, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial [published corrections appear in Lancet. 2015;385:946 and Lancet. 2016;387;1618]. Lancet. 2015;385(9972):956–65.
Pellicciari R, Fiorucci S, Camaioni E, et al. 6alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45(17):3569–72.
Modica S, Petruzzelli M, Bellafante E, et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology. 2012;142(2):355–65.
Ananthanarayanan M, Balasubramanian N, Makishima M, Mangelsdorf DJ, Suchy FJ. Human bile salt export pump promoter is transactivated by the farnesoid X receptor/bile acid receptor. J Biol Chem. 2001;276(31):28857–65.
Baghdasaryan A, Chiba P, Trauner M. Clinical application of transcriptional activators of bile salt 5 transporters. J Mol Aspects Med. 2013;37:57–76.
Yates RA, Wong J, Seiberling M, Merz M, Marz W, Nauck M. The effect of anastrozole on the single-dose pharmacokinetics and anticoagulant activity of warfarin in healthy volunteers. Br J Clin Pharmacol. 2001;51(5):429–35.
Walker G, Mandagere A, Dufton C, Venitz J. The pharmacokinetics and pharmacodynamics of warfarin in combination with ambrisentan in healthy volunteers. Br J Clin Pharmacol. 2009;67(5):527–34.
Malhotra B, Alvey C, Gong J, Li X, Duczynski G, Gandelman K. Effects of fesoterodine on the pharmacokinetics and pharmacodynamics of warfarin in healthy volunteers. Br J Clin Pharmacol. 2011;72(2):257–62.
FDA. Guidance for Industry: drug interaction studies—study design, data analysis, implications for dosing, and labeling recommendations. 2012.
Cooper KJ, Martin PD, Dane AL, Warwick MJ, Raza A, Schneck DW. Lack of effect of ketoconazole on the pharmacokinetics of rosuvastatin in healthy subjects. Br J Clin Pharmacol. 2003;55(1):94–9.
Schneck DW, Birmingham BK, Zalikowski JA, et al. The effect of gemfibrozil on the pharmacokinetics of rosuvastatin. Clin Pharmacol Ther. 2004;75(5):455–63.
Polli JW, Hussey E, Bush M, et al. Evaluation of drug interactions of GSK1292263 (a GPR119 agonist) with statins: from in vitro data to clinical study design. Xenobiotica. 2013;43(6):498–508.
Haberer LJ, McSherry I, Cargill A, McCarthy L. Effects of vercirnon on the activity of CYP3A4, CYP2C19 and CYP2C8 enzymes and BCRP and OATP1B1 transporters using probe substrates. Eur J Clin Pharmacol. 2014;70:37–45.
Zhu T, Awni WM, Hosmane B, et al. ABT-335, the choline salt of fenofibric acid, does not have a clinically significant pharmacokinetic interaction with rosuvastatin in humans. J Clin Pharmacol. 2009;49(1):63–71.
Fenner KS, Troutman MD, Kempshall S, et al. Drug–drug interactions mediated through P-glycoprotein: clinical relevance and in vitro–in vivo correlation using digoxin as a probe drug. Clin Pharmacol Ther. 2009;85(2):173–81.
Beischlag TV, Morales JL, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr. 2008;18(3):207–50.
Klinge CM, Jernigan SC, Risinger KE, et al. Short heterodimer partner (SHP) orphan nuclear receptor inhibits the transcriptional activity of aryl hydrocarbon receptor (AHR)/AHR nuclear translocator (ARNT). Arch Biochem Biophys. 2001;390(1):64–70.
Acknowledgements
Sponsorship for this study, article processing charges, and open access fee were funded by Intercept Pharmaceuticals, Inc. All authors had full access to all of the data in this study and take complete responsibility for the integrity of the data and accuracy of the data analysis. Editorial assistance in the preparation of this manuscript was provided by Kjersti Swearingen (Intercept Pharmaceuticals, Inc.). All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. The authors would like to thank the healthy subjects for their participation in these studies.
Disclosures
Jeffrey E. Edwards is a full time employee of and stockholder in Intercept Pharmaceuticals, Inc. Sharon Karan is a full time employee of and stockholder in Intercept Pharmaceuticals, Inc. Leigh MacConell is a full time employee of and stockholder in Intercept Pharmaceuticals, Inc. Lise Eliot has received travel support, fees for review activities, and was a consultant for Intercept Pharmaceuticals, Inc. Andrew Parkinson is a consultant for Intercept Pharmaceuticals, Inc.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1964, as revised in 2013. All subjects provided informed consent before any study procedures or assessments were performed.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/F37AF060185C9D9C.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Edwards, J.E., Eliot, L., Parkinson, A. et al. Assessment of Pharmacokinetic Interactions Between Obeticholic Acid and Caffeine, Midazolam, Warfarin, Dextromethorphan, Omeprazole, Rosuvastatin, and Digoxin in Phase 1 Studies in Healthy Subjects. Adv Ther 34, 2120–2138 (2017). https://doi.org/10.1007/s12325-017-0601-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12325-017-0601-0