
Abstract
Immune-mediated cerebellar ataxias (IMCAs) have diverse etiologies. Patients with IMCAs develop cerebellar symptoms, characterized mainly by gait ataxia, showing an acute or subacute clinical course. We present a novel concept of latent autoimmune cerebellar ataxia (LACA), analogous to latent autoimmune diabetes in adults (LADA). LADA is a slowly progressive form of autoimmune diabetes where patients are often initially diagnosed with type 2 diabetes. The sole biomarker (serum anti-GAD antibody) is not always present or can fluctuate. However, the disease progresses to pancreatic beta-cell failure and insulin dependency within about 5 years. Due to the unclear autoimmune profile, clinicians often struggle to reach an early diagnosis during the period when insulin production is not severely compromised. LACA is also characterized by a slowly progressive course, lack of obvious autoimmune background, and difficulties in reaching a diagnosis in the absence of clear markers for IMCAs. The authors discuss two aspects of LACA: (1) the not manifestly evident autoimmunity and (2) the prodromal stage of IMCA’s characterized by a period of partial neuronal dysfunction where non-specific symptoms may occur. In order to achieve an early intervention and prevent cell death in the cerebellum, identification of the time-window before irreversible neuronal loss is critical. LACA occurs during this time-window when possible preservation of neural plasticity exists. Efforts should be devoted to the early identification of biological, neurophysiological, neuropsychological, morphological (brain morphometry), and multimodal biomarkers allowing early diagnosis and therapeutic intervention and to avoid irreversible neuronal loss.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Immune-mediated cerebellar ataxias (IMCAs) have diverse etiologies [1,2,3,4,5,6] (Table 1). IMCAs are divided into two groups: (1) those in which the trigger of autoimmunity leading to cerebellar damage are known, such as infection (e.g., post-infectious cerebellar syndrome, PiCS, post-infectious cerebellitis), neoplasm (e.g., paraneoplastic cerebellar degeneration, PCD), and gluten sensitivity (gluten ataxia, GA); and (2) those with no clear triggers but with serological markers strongly suggestive of IMCAs (e.g., anti-GAD ataxia). When immune-mediated mechanisms leading to cerebellar damage are strongly suspected, but a serological profile does not match any of the known etiologies, the patients are categorized as having primary autoimmune cerebellar ataxia (PACA) [7].
Patients with IMCAs generally develop a cerebellar motor syndrome, characterized mainly by gait ataxia, with an acute or subacute clinical course. Identification of well-characterized antibodies (Abs) is essential in the diagnosis of some IMCAs: for example, onconeural Abs in PCD, anti-gliadin and anti-TG6 Abs in GA, and high-titer of anti-GAD antibodies (anti-GAD Abs) in anti-GAD ataxia [1,2,3,4,5,6]. In contrast, some patients exhibit ataxia with a slowly progressive time-course, without obvious autoimmune background [8,9,10]. Neurological symptoms other than ataxia sometimes precede the development of ataxia, suggestive of the existence of a prodromal stage in IMCAs [9,10,11].
The condition is analogous to latent autoimmune diabetes in adults (LADA), which is characterized by an atypical presentation of autoimmune type 1 diabetes mellitus (DM), slow and progressive course, and sometimes fluctuating association of anti-GAD Abs, the sole autoimmune biomarker [12,13,14,15]. Patients are initially diagnosed with type 2 DM and gradual decompensation due to islet autoimmunity and insulin deficiency leading to insulin dependency [16]. In analogy with LADA, we propose a novel clinical concept of latent autoimmune cerebellar ataxia (LACA) to underline two conditions: “not manifestly evident autoimmune pathologies” and “prodromal stage of IMCAs.”
This novel clinical concept, LACA, will provide a framework for early intervention during a period when cerebellar reserve, capacities for compensation and restoration, is preserved [17]. The current consensus paper aims to discuss the validity of the LACA hypothesis. From this point of view, we revisit clinical profiles of CA associated with low-titer of anti-GAD Abs CA, GA, and PCD.
Latent Autoimmune Cerebellar Ataxia (Mario Mano, Marios Hadjivassiliou, Hiroshi Mitoma)
Slowly Progressive Cerebellar Ataxia and “Not Manifestly Evident” Autoimmunity
There are reports of patients showing slowly progressive ataxia without definite autoimmune triggers or well-characterized Abs responding positively to immunotherapies [8,9,10]. Some researchers questioned if these patients have IMCAs. They argue that it is uncertain whether their autoimmune tendency is directly or indirectly responsible for the insult to the cerebellum based on the following two problems [18].
The first problem is the significance of the presence of autoantibodies. If such antibodies are not pathogenic, then autoantibodies and ataxia coexist, and the ataxia is caused by metabolic/toxic or degenerative mechanisms other than autoimmunity. It should also be noted that some autoantibodies such as anti-thyroid Abs and low-titer anti-GAD Abs can be found in healthy subjects, albeit at low frequencies [18]. In addition, some antibodies, e.g., anti-NH2-terminal of α-enolase (NAE) Abs, co-exist in patients with multiple system atrophy, suggesting degenerative changes might induce secondarily autoimmune processes [19].
The second problem is the lack of autoimmune features. For example, in diagnosis criteria of PACA, clinical features that suggest autoimmune etiology were proposed, including acute or subacute time course, midline cerebellar atrophy, CSF pleocytosis and/or positive CSF restricted IgG oligoclonal bands, history of other autoimmune disorders, or family history of autoimmune disorders [7]. However, some of these patients may not exhibit many of the autoimmunity-pointing features defined by PACA criteria.
Thus, the reported cases were diagnosed as IMCAs for the first time as a result of response to immunotherapies or intrathecal Ab synthesis [8,9,10] and the exclusion of other etiologies. Faced with such cases, clinicians may have difficulty making a diagnosis of IMCAs.
“Prodromal Stage” in Immune-Mediated Cerebellar Ataxias
Degenerative diseases, including Alzheimer’s, idiopathic Parkinson’s disease, progressive supranuclear palsy, usually develop gradually, over the course of months to years. It is well known that some non-neurological and neurological symptoms (e.g., anosmia in Parkinson’s disease) can be present at the prodromal stage [20, 21]. Consistently, a pre-symptomatic or prodromal stage was observed in an animal model of degenerative ataxia [22]. In patients with degenerative ataxia, pre-ataxic symptoms have recently been reported in gait stability [23] and ocular movements [24]. Time-course of development of ataxia in spinocerebellar ataxia (SCA) types 1, 2, 3, and 6 mutation carriers was traced longitudinally and revealed marginal progression in the prodromal period, followed by increasing progression once ataxia is established [25]. On the other hand, it should be acknowledged that particular neurological symptoms such as brainstem attacks preceded the manifestation of ataxia in some patients with IMCAs [9,10,11]. Some organ-specific autoimmune disorders can also be present as prodromal [9].
The Analogy with Latent Autoimmune Diabetes in Adults/Slowly Progressive Insulin-Dependent Diabetes Mellitus
Anti-GAD Abs are specifically associated with type 1 DM, characterized by acute onset with an immediate requirement for insulin therapy and immune-mediated destruction of pancreatic beta-cells. Interestingly, anti-GAD Abs are also detected in ~10% of patients with adult-onset DM initially diagnosed as type 2 DM [13,14,15,16]. Thyroid and gastric auto-immunity are also frequently associated. These patients, who initially do not require insulin treatment, gradually become insulin-dependent. This atypical subset of autoimmune DM is referred to as slowly progressive insulin-dependent DM (SPIDDM) [26] or latent autoimmune diabetes in adults (LADA) [12, 27]. The presence of anti-GAD Abs reflects the autoimmune-mediated inflammation in the islet.
LADA shows a lack of manifestly evident autoimmunity. In addition to atypical clinical courses (slow progression to insulin dependency), anti-GAD Abs are not repeatedly detected. It was reported that after 3 years of follow-up, 37% of LADA patients remained anti-GAD Ab positive, 20% fluctuated between positivity and negativity, and the remaining 43% became anti-GAD Ab negative [15].
The time course of LADA is now classified into three stages [16]. At the first stage of the disease, genetically triggered autoimmunity slowly destroys the beta-cell and reduces insulin secretion. At a second stage, exposure to an unhealthy lifestyle causes insulin resistance and overload of beta-cells. Eventually, any compensations by beta-cells fail to meet the increasing insulin need, resulting in hyperglycemia and, finally, an insulin-dependent status. In other words, the slowly progressive immune-mediated inflammation ultimately disrupts beta-cells’ reserve capacities [13]. Thus, clinicians often struggle to reach an early diagnosis. Careful estimations on insulin deficiency (e.g., C-peptide test) are recommended in LADA/SPIDDM [13, 14]. Recent studies using ELISA methods show that the titer of anti-GAD Abs has no relevance to insulin-requiring diabetes [14]. The progression to a stage of beta-cell destruction occurs in patients with high-titer and low-titer. Furthermore, an early intervention targeting anti-GAD Ab-positive individuals without manifest diabetes is proposed [16].
In conclusion, a clinical notion of LADA/SPIDDM argues for the importance of early intervention by stressing two critical characteristics: (1) autoimmune etiology is present but easily overlooked, (2) beta-cell deficiency is potential but not active /symptomatic in the early stage (a prodromal stage).
Definition of Latent Autoimmune Cerebellar Ataxia (LACA)
We propose the concept of latent autoimmune cerebellar ataxia (LACA) analogous to LADA. The term LACA should be used in the following situations (see also Fig. 1):
-
(1)
An autoimmune etiology is present but not easily detectable since it is not associated with personal history of autoimmune diseases or well-characterized autoantibodies.
-
(2)
The ataxia is subclinical or so mild that is difficult to detect on clinical examination, and non-specific symptoms or other non-cerebellar neurological manifestations may precede the manifestation of ataxia. This stage can be retrospectively identified as prodromal.
-
LACA by definition is likely to follow a course of slow progression. Ultimately, the autoimmune mechanisms will affect the cerebellum, resulting in clinical ataxia and eventually marked cerebellar atrophy.
-
The notion of LACA is introduced to encourage clinicians to carefully examine the possibility of slow-evolving IMCA, as well as to stress the importance of the early intervention of immunotherapies during a period when there is cerebellar reserve.
-

A definition of LACA. Prodromal IMCAs and IMCAs are classified based on the manifestation of two factors: cerebellar ataxias and autoimmunity. LACA can be utilized to describe situations in which cerebellar ataxias are latent or the autoimmunity is latent. IMCAs, immune-mediated cerebellar ataxias; LACA, latent autoimmune cerebellar ataxia; MFS, Miller Fisher syndrome; PCD, paraneoplastic cerebellar degeneration; OMS, opsoclonus myoclonus syndrome; PACA, primary autoimmune cerebellar ataxia
A clinical course is shown in Fig. 2 and the comparisons between LADA and LACA are shown in Table 2.

A clinical course from presymptomatic stage and prodromal stage to ataxic stage. LACA can be used to describe the prodromal stage in IMCAs. During a period of the presymptomatic and prodromal stages, the internal model is preserved, leading to normal predictive operations, whereas, in ataxic stage, the internal model is impaired, resulting in a development of cerebellar ataxias. As the disease progresses, cerebellar reserve is lost. IMCAs, immune-mediated cerebellar ataxias; LACA, latent autoimmune cerebellar ataxia; MFS, Miller Fisher syndrome; PCD, paraneoplastic cerebellar degeneration; OMS, opsoclonus myoclonus syndrome; PACA, primary autoimmune cerebellar ataxia
“Not Manifestly Evident” Autoimmunity Observed in LACA
This section highlights “not manifestly evident” autoimmunity in LACA through examples of CA associated with anti-GAD Abs and paraneoplastic cerebellar degeneration (PCD). In the “Slowly Progressive Cerebellar Ataxia and “Not Manifestly Evident” Autoimmunity” section, we proposed two factors of “not manifestly evident” autoimmunity: (1) the presence of poorly characterized autoantibodies and (2) the lack of autoimmune features. Here we will discuss these two features in CA associated with anti-GAD Abs and PCD, respectively. The significance of poorly characterized Abs associated with cerebellar ataxia needs to be carefully followed in conjunction with the manifestation of other autoimmunity features.
Anti-GAD Autoantibodies: Titer and Epitope Specificity
High-Titer and Low-Titer Anti-GAD Ab and CA (Hiroshi Mitoma, Christiane S Hampe, Marios Hadjivassiliou)
High-titer anti-GAD65 Ab in serum (often also found in the CSF) leads to the diagnosis of anti-GAD ataxia [2, 4]. The titer is usually above10,000 U/mL (or 10- to 100-fold higher compared to those of patients with type 1 DM) [2, 4]. In patients with ataxia and serum anti-GAD Abs exceeding 2000 U/mL, one can safely consider anti-GAD ataxia [6]. In contrast, the significance of low-titer anti-GAD Ab is unclear. However, it should be acknowledged that a portion of patients with low-titer anti-GAD Ab and CA must have immune-mediated etiologies since immunotherapies improved are effective. We will use the terms “high-titer” and “low-titer” as defined in the respective studies rather than reporting specific values due to the use of different anti-GAD Ab assays.
The practical problem is that the autoimmune nature may not be easily detectable in CA associated with low-titer anti-GAD Ab [8,9,10, 28,29,30,31,32,33,34,35]. Only half of such patients have a history of autoimmune diseases, and very few showed intrathecal production of anti-GAD Abs [8,9,10, 28,29,30,31,32,33,34,35]. Moreover, all patients showed pan-cerebellar ataxia with insidious clinical courses [8,9,10, 28,29,30,31,32,33,34,35]. Due to these clinical presentations atypical for IMCAs, the presence of low-titer anti-GAD Ab does not provide obvious evidence that immune-mediated mechanisms insult the cerebellum. The autoimmune significance of low-titer anti-GAD Ab was determined only after confirming the responsiveness to immunotherapies that relied on these few clues [8,9,10, 28,29,30,31,32,33,34,35].
Thus, it is likely that the majority of patients with high-titer of anti-GAD Ab, and not, as yet, any clinical evidence of ataxia, may well have LACA, and that a smaller proportion of patients with low-titer anti-GAD Ab may also have LACA.
Notably, the epitope specificities of anti-GAD Abs in type 1 DM differ significantly from those in neurological diseases associated with anti-GAD Ab. In addition, it was suggested that high-titer anti-GAD Ab decreased GABA release, while low-titer anti-GAD Ab had no such pathogenic actions [32]. These characteristics of low-titer anti-GAD Ab and CA show the following principle: “The presence of auto-Abs alone may not support the diagnosis of IMCAs due to multiple epitopes, while low-titer auto-Abs are not necessarily correlated with only low-level insults on the cerebellum.” This principle should be considered when diagnosing “not manifestly evident” autoimmunity. Since epitope specificities have not been considered so far in the diagnosis of neuroimmune diseases, we will discuss methodological problems in defining epitopes in the next section.
Importance of Epitope Mapping of Anti-GAD Abs (Christiane S Hampe)
Epitope mapping of disease-specific anti-GAD Abs can support the correct diagnosis and prediction of disease, understanding of the underlying autoimmune response, identification of antibodies with pathologic potential, and development of therapeutics.
Disease-specific anti-GAD Ab epitopes in type 1 DM and neurological disorders, such as Stiff Person Syndrome (SPS), were evident already in the early 90s, when Baekkeskov et al. found that anti-GAD Abs in patients with SPS recognized both linear and conformational GAD epitopes, while those in patients with type 1 DM were dependent on the conformation integrity of the molecule [36]. Subsequent analyses utilized fusion proteins substituting regions of GAD65 with those of the slightly larger isoform GAD67. GAD65/67 fusion proteins [37] helped to identify two major regions in the middle and the C-terminus that contained conformational epitopes relevant to type 1 DM, while anti-GAD Ab in patients with SPS recognized epitopes located also at the N-terminus [38] (Fig. 3). However, there were considerable shortcomings associated with this epitope mapping approach. The use of GAD67 as a “scaffold” for GAD65 epitope regions depends on the relative lack of antigenicity of GAD67. Indeed, antibodies in type 1 DM only occasionally recognize GAD67 [39]. However, anti-GAD Ab in SPS and other neurological disorders frequently react with GAD67, and GAD65/67 fusion proteins are therefore of limited use for epitope analyses, especially in neurological disorders [31].

Anti-GAD Ab epitope domains recognized by antibodies in different diseases. Linear anti-GAD Ab epitopes recognized by patients with SPS (rectangular boxes) have been identified using both peptide mapping and ES-RBA. These epitopes are dispersed across the entire GAD molecule. Conformational anti-GAD Ab epitopes recognized by patients with SPS (yellow ovals) or patients with type 1 DM (blue circles) have been identified by the use of GAD65/67 fusion proteins and ES-RBA
An epitope mapping assay (epitope-specific radioligand binding assay (ES-RBA)) allows the detection of conformational and linear GAD65-specific antibody epitopes and was able to distinguish between anti-GAD Abs in sera of patients with type 1 DM, SPS, limbic encephalitis, CA, and anti-GAD Ab-positive epilepsy [40, 41] (Fig. 3). This epitope recognition was independent of anti-GAD Ab titer, a crucial aspect as anti-GAD Ab titers often vary considerably. Importantly, other studies using GAD65 fragments for epitope mapping in patients with the same neurological autoimmune diseases could not identify these disease-specific epitopes [42, 43], underlining the relevance of this approach for the identification of disease-specific anti-GAD Ab epitopes.
Therefore, the presence of anti-GAD Abs in itself is not sufficient for a precise diagnosis. Furthermore, low anti-GAD Ab titers do not always suggest a more benign pathogenesis. Other factors, such as anti-GAD Ab epitopes (linear and conformational), location of anti-GAD Ab production (systemic and intrathecal), and IgG subclasses, need to be part of a careful evaluation of the underlying autoimmune response. Future studies to identify anti-GAD Ab epitopes in LACA are needed to establish a disease-specific pattern, which may aide clinicians in the diagnosis and targeted therapy.
“Not Manifestly Evident” Autoimmunity in PCD (Sergio Muñiz-Castrillo, Alberto Vogrig, Bastien Joubert, Jérôme Honnorat)
Paraneoplastic cerebellar degeneration (PCD), often manifesting as rapidly progressive cerebellar syndrome, is defined as a severe subacute pancerebellar syndrome triggered by cancer [44, 45]. PCD has been reported as one of the most common paraneoplastic neurological syndromes (PNS), accounting for nearly 20–30% of those fulfilling the criteria for definite PNS [46,47,48,49].
Well and Poorly Characterized Onconeural Antibodies
A characteristic of PCD, as in other PNS, is the association with autoantibodies that target a great diversity of neural antigens (Table 3). Most of these antigens are located at the intracellular level; hence, the antibodies are thought not to be directly involved in the pathogenesis of the disease that is instead mostly mediated by cytotoxic T cells [50]. Nevertheless, most of these antibodies are reliable biomarkers of an underlying cancer and therefore useful to guide the tumor screening [44], as nearly 65% of these patients are not known to have cancer at the time of diagnosis [51, 52]. In two PCD series, the most common anti-neural Abs identified were, in decreasing order of frequency, Yo, Hu, CV2/CRMP5, Tr/DNER (delta/notch-like epidermal growth factor-related receptor), Ri, and Ma2 [51, 52]. Furthermore, the oncological accompaniments considerably differ between these Abs [53,54,55,56,57,58] (Table 3). Recently, anti-Kelch-like protein 11 (KLHL11) Abs have also been described in association with testicular tumors and brainstem-cerebellar involvement [59].
Besides, several additional antibodies have been identified in only a few PCD patients (Table 4), such as Abs directed against TRIM9/67 and metabotropic glutamate receptor 2 [60, 61]; their characterization therefore warrants further investigation. Finally, seronegative cases may account for approximately 20% of PCD, being associated with gynecological cancers, lymphomas (non-Hodgkin and Hodgkin), and lung cancer in women, and with lung, Hodgkin lymphoma, and genitourinary cancers in men [52]. Conversely, cancer is not found in about 10% of Ab-positive suspected PCD [51, 52].
Main Clinical Features and Temporal Patterns
PCD commonly presents as a truncal and appendicular ataxia developed over a matter of weeks [51]. Additionally, horizontal nystagmus is an almost constant feature, often with some vertical and torsional component, whereas down-beat nystagmus in particular has been proposed as a hallmark of PCD [55, 62]. Hyperacute presentations in less than 24 h have been described in patients with anti-Yo and anti-Hu Abs, though they only account for approximately 5% of all PCDs [56, 62,63,64]. Less commonly, the cerebellar presentation can be slowly progressive over months, especially in patients with anti-Ri and anti-Ma2 Abs [57, 65].
LACA: the Prodromal Stage
There has been cumulating evidence suggesting that some neurological symptoms precede the manifestation of ataxia in degenerative/genetic ataxias. Alteration of gait and postural sway control was detected in subjects with prodromal SCA2 using a wearable sensor-based system [23]. Pre-ataxic changes in SCA3 patients were reported in vestibulo-ocular reflex gain, main sequence of vertical volitional saccades, and slow-phase velocity of central and gaze-evoked (SPV-GE) nystagmus in SCA3 [24]. We argue the existence of a “prodromal stage” that precedes the manifestation of CAs also in IMCAs. We retrospectively review prodromal symptoms in low-titer anti-GAD ataxia (section “Prodromal Stage in Patients with Anti-GAD Antibodies (José Fidel Baizabal-Carvallo)”), PCD (section “Prodromal Stage in PCD (Sergio Muñiz-Castrillo, Alberto Vogrig, Bastien Joubert, Jérôme Honnorat)”), gluten ataxia (GA) (section “Prodromal Stage in Gluten Ataxia (Marios Hadjivassiliou)”), and post-infectious cerebellar syndrome (section “Prodromal Stage in Post-infectious Cerebellar Syndrome (PiCS) (José Fidel Baizabal-Carvallo)”).
Prodromal Stage in Patients with Anti-GAD Antibodies (José Fidel Baizabal-Carvallo)
The clinical course of LACA is largely unknown. Emerging evidence suggests that patients with LACA may present with systemic or organ-specific autoimmune disorders before the onset of progressive ataxia, including neurological and systemic manifestations (Table 5). Moreover, a prodromal stage characterized by nonspecific symptoms including malaise, fatigue, or even cognitive complaints may be seen before the onset of neurological symptoms.
Oculomotor manifestations may precede the development of overt ataxia. Multidirectional, horizontal, upbeat, and downbeat nystagmus may present in patients with low-titer anti-GAD Ab [28, 66, 67]. These patients should be evaluated carefully as they may initially show relatively minor signs of ataxia, such as difficulties with tandem gait or mild dysmetria. Such relatively subtle cerebellar manifestations may progress with time, leading to overt CA [68].
A clinical picture with more diffuse brainstem manifestations may also be a presentation of LACA [11]. Emerging evidence suggests that about 25% and 35% of patients with ataxia and high anti-GAD Ab develop episodes of transient neurological dysfunction involving brainstem nuclei and cerebellar connections [11]. These so-called brainstem attacks manifest with horizontal and/or vertical diplopia, nystagmus, vertigo, nausea, vomiting, dysarthria, paralysis of the posterior pharyngeal wall, gait, and limb ataxia [11, 31, 34]. These prodromal symptoms may last from several minutes to days but even weeks or months; their frequency may also be extremely variable, and the episodes may be isolated or with a paroxysmal character. Although these episodes usually resolve spontaneously and patients initially do not present with evident CA, an insidious progressive cerebellar syndrome usually appears within the following 3 months, but longer latencies of up to 2 years have also been reported [11, 31]. The reversibility of such symptoms suggests a transient autoimmune impairment apart from conspicuous neuronal loss that will eventually lead to progressive cerebellar damage.
Besides brainstem manifestations, axial or appendicular muscle rigidity, sometimes with superimposed muscle spasms may antedate the onset of a progressive CA [31, 69]. Such manifestations are consistent with the “classic” or “focal/segmental” subtypes of SPS antedating the cerebellar syndrome, sometimes underlying, by years [33, 70].
The coexistence of systemic or organ-specific autoimmune disorders [11, 35] with LACA suggests the presence of an active multi-organ autoimmune response with different degrees of active antibody production. Low and high-titer anti-GAD Abs may be identified in patients with gluten ataxia, which is a form of sporadic ataxia associated with anti-gliadin and anti-transglutaminase 6 Abs [71]. Forty percent of patients with gluten ataxia were anti-GAD Ab positive with a mean titer of 25 U/mL [71]. Whether such low-titers of anti-GAD Abs contributed to the cerebellar damage is difficult to define; however, the titer of anti-GAD Abs decreased in a third of these patients following a gluten-free diet, a phenomenon related to clinical improvement [71].
Similarly, 70% of patients with anti-GAD ataxia were found to have positive serology for gluten sensitivity, some of which responded well to a gluten-free diet without requiring immunosupression. This suggests a significant overlap between gluten ataxia and anti-GAD ataxia [72]. Current literature does not associate LACA with underlying neoplasia; however, few patients with CA and high-titer of anti-GAD Abs have been found with occult neoplasia and may exceptionally exists with low anti-GAD Ab titer [31, 33, 66].
The morphological appearance of the cerebellum assessed by neuroimaging seems a poor predictor of anti-GAD Ab titer, as up to 43% of patients with high Ab titer do not have cerebellar atrophy on MRI [73].
In patients with high titer anti-GAD antibodies, improvement of oculomotor abnormalities may follow treatment with IVIg, corticosteroids, or plasma exchange; however, the response may be incomplete or selective to specific form of ocular motor deficits. Cyclophosphamide may provide benefit in those who are refractory to treatment with IVIg, plasma exchange, or steroids [66]. However, it is unclear what proportion of these patients will eventually evolve to overt CA with high-titer anti-GAD Ab. Miller Fisher syndrome with negative anti-GQ1b and relatively low anti-GAD Ab (<2000 IU/mL) has been reported to improve clinically with IVIg alongside with a decrease of anti-GAD Ab titer [74].
Despite the low-titer of anti-GAD Abs, improvement with monthly courses of IVIg has been registered in selected patients coupled with improvement in cerebellar perfusion [75, 76]. Intravenous methylprednisolone 1000 mg/day for 5 days followed by oral prednisone for 2 months or oral corticosteroids alone may also provide clinical benefit on different ataxia scales [28, 29].
In summary, the brainstem manifestations either as transient deficits, i.e., “brainstem attacks” or as oculomotor dysfunction are among the most notable preceding symptoms in patients with ataxia associated with high-titer and low-titer anti-GAD Abs. SPS-like, systemic, or organ-specific autoimmune disease may also be present. Immunotherapy usually relates with good outcomes and clinical improvement associates with decrease in anti-GAD Ab titer, even if they are low at baseline. Whether this is explained by publication bias through successfully treated patients should be clarified in further studies.
Prodromal Stage in PCD (Sergio Muñiz-Castrillo, Alberto Vogrig, Bastien Joubert, Jérôme Honnorat)
PCD may be accompanied by extracerebellar involvement, the frequency, and severity of which largely depends on the associated antibody. In general, patients with anti-Yo Abs and those with anti-Tr/DNER Abs tend to manifest PCD as isolated or predominant neurological manifestation [51]. Nevertheless, a thorough neurological examination in patients with anti-Yo Abs can disclose mild signs or symptoms suggesting involvement of corticospinal tract (30–58%) [62, 77], brainstem disorders or cranial neuropathies (13%) [77], peripheral disorders ranging from hyporeflexia and mild sensory disturbances (54%) [62] to a clear diagnosis of peripheral neuropathy (10%) [77], or even gastrointestinal dysmotility in rare circumstances [77]. Sometimes, the cerebellar syndrome is so severe that it prevents the optimal mental status evaluation. Robust dysarthria and motor dysfunction interfere with the evaluation of the cognitive impairment. Despite these difficulties in the examination, approximately one-fifth of patients with anti-Yo Abs has clinical evidence of cognitive dysfunction, typically in the form of emotional lability and memory deficits [62]. It remains unclear if these deficits relate to the cognitive functions of the cerebellum (Schmahmann syndrome) or reflect the involvement of extra-cerebellar structures [78]. In a series of 28 patients with anti-Tr/DNER Abs, all but one patient had cerebellar involvement, which was isolated except for 2 cases (7%) who showed encephalopathy and sensory neuropathy [53].
PCD patients with other anti-neural Abs show distinctive associations with both central and peripheral neurological disorders (Table 3). Importantly, the extracerebellar involvement can be a clue for the associated antibody. For example, sensory neuronopathy with or without encephalomyelitis is typically associated with anti-Hu Abs [55], hearing loss and/or tinnitus is frequently associated with anti-KLHL11 Abs [59], opsoclonus-myoclonus in adults typically associate with anti-Ri antibodies [56, 57], while narcolepsy-cataplexy and hypopituitarism are common in patients with anti-Ma2 Abs [58].
Notably, these extra-cerebellar symptoms are sometimes subtle, and precede the manifestations of CA.
PCD is sometimes preceded by prodromal clinical symptoms such as nausea, vomiting, dizziness, and vertigo [79, 80]. In addition, some patients with anti-Ri Abs may present with an isolated action tremor that evolves into an overt PCD late in the disease [57]. Similarly, hearing loss or tinnitus antedate the development of cerebellar/brainstem dysfunction in nearly 25% of the patients with anti-KLHL11 Abs [59]. We observed that up to 37% of patients with ataxia and anti-KLHL11 Abs experienced transient, paroxysmal, episodes of vertigo, unbalance, and vomiting, which lasted from months to years prior to the development of a permanent cerebellar dysfunction [81].
Furthermore, the genetic abnormalities observed in ovary cancer of patients with anti-Yo PCD and leading to the immune breakdown responsible of PCD immune activation are present in some women many years before the development of the cerebellar symptoms [82]. These data suggest that in some patients the cerebellar immune reaction can be present many months and years before the clinical symptoms.
In conclusion, even in patients with PCD, many arguments suggest that the immune reaction is present in the cerebellum many weeks or months before the development of clinical symptoms suggesting that the concept of LACA can be extended to patients with PCD.
Prodromal Stage in Gluten Ataxia (Marios Hadjivassiliou)
GA refers to an IMCA triggered by the ingestion of gluten in gluten-sensitive individuals [83, 84]. By definition, such patients will have CA in the presence of serological markers of gluten sensitivity (one or more of antigliadin, TG2, and TG6 antibodies) [85]. The presence of enteropathy defines coeliac disease (CD) but it is not a prerequisite for the diagnosis of GA.
It has been shown that up to 47% of patients with newly diagnosed CD, presenting to the gastroenterologists have abnormal MR spectroscopy of the cerebellum [86]. Clinical evaluation showed that 29% of the patients had evidence of mild gait ataxia and 11% had nystagmus. None of these patients, however, had been referred to or seen by a neurologist even if on direct questioning 24% reported some gait instability. It could be argued that patients with LACA for whom the diagnosis of CD is based primarily on gastrointestinal symptoms, early treatment and good outcome can be expected given the retained cerebellar reserve.
In our experience at the Sheffield Ataxia Centre, we often get referrals of patients who have had brain imaging for various reasons (e.g., headache) who are then noted to have evidence of cerebellar atrophy. We have a cohort of such patients with positive serology for gluten sensitivity who on clinical examination have no evidence of any detectable ataxia. Often these patients have abnormal spectroscopy of the cerebellum that improves with the introduction of gluten-free diet. These examples represent LACA where the cerebellar reserve is sufficient to compensate for the cerebellar atrophy and the clinical intervention with gluten-free diet can result in complete recovery.
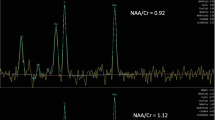
As such, the use of gluten sensitivity-related antibodies and in particular antigliadin and TG6 Abs may be a useful biomarker of some cases of LACA. Equally the use of MR spectroscopy of the cerebellum may identify patients with reduced NAA/Cr in the cerebellar vermis and/or hemisphere implying cerebellar dysfunction without overt ataxia. A prospective evaluation of healthy volunteers who have serological evidence of gluten sensitivity may be helpful in better understanding LACA in the context of gluten sensitivity.
Prodromal Stage in Post-infectious Cerebellar Syndrome (PiCS) (José Fidel Baizabal-Carvallo)
Post-infectious cerebellar syndrome (PiCS) is defined as acute cerebellar inflammation induced by immune-mediated mechanisms triggered by a bacterial or viral pathogen, such as mycoplasma pneumoniae, Epstein-Barr virus, varicella zoster virus, cytomegalovirus, Coxsackie B3, or following vaccination [6, 87, 88]. More recently, SARS-CoV2, the cause of COVID19, has been identified as a potential trigger [89, 90]. However, in a substantial proportion of these patients, there is no clear underlying or previous infection. Although this condition is more commonly observed in children, there are reports of adult cases as well [91]. PiCS should be readily differentiated from acute infectious cerebellitis, the latter being caused by direct cerebellar tissue invasion by a specific pathogen.
Antibodies directed against the glutamate receptor delta 2 (anti-GluRδ2) which is highly expressed in Purkinje cells have been identified in the serum and CSF of some patients with cerebellitis following vaccination and diverse infections [92,93,94]. Patients have a variable clinical course ranging from a self-limiting to a fulminant course leading to severe cerebellar damage or death in some instances [95]. MRI may show unilateral or bilateral T2-weighted hyperintensities in the cerebellum [96].
Molecular mimicry is presumed as the pathogenic mechanism, similar to Guillain-Barré syndrome or Sydenham chorea in rheumatic fever [97, 98]. It is unclear how the LACA hypothesis applies to PiCS, owing to the following considerations: (1) the disorder has a self-limited course, (2) the latency between infection and onset of neurological manifestations may be unclear, although it is considered to be short, usually lasting less than 10 days, and (3) prodromal symptoms may be related to the immunological response to the infection. However, the presence of fever, malaise, drowsiness, headache, nausea, vomiting, photophobia, or vertigo, once the infection has seemingly resolved [87, 99], suggests the presence of a smoldering inflammatory process that will eventually reach a threshold for CA.
Biomarkers During Progression of LACA
The signs related to the prodromal stage in IMCAs need to be identified for early interventions. First, we examine how eye movements can be objective markers of prodromal phase prior to onset of immune-mediated CA (section “Eye Movement Abnormalities as Physiological Biomarker (Aasef G. Shaikh)”). In this section, we will further identify nature of movement deficits that can suggest prodromal phase of CA induced by diverse etiologies. Paroxysmal deficits that are at the epicenter of the prodromal phase in CA are discussed. Second, we also discuss possible autoimmune biomarkers for LACA (section “Dynamic Changes of Autoimmune Biomarkers During Progression of LACA (Christiane S Hampe)”). Dynamic changes in some cytokines might reflect the early immune-mediated insults to the cerebellum.
Eye Movement Abnormalities as Physiological Biomarker (Aasef G. Shaikh)
Clinical Features of Subtle and Transient Ocular Motor Deficits
Ocular motor deficits are common in IMCA. They can be seen in its early prodromal phase, prior to onset of CA or gait impairment. The most common form of ocular motor deficit in the neurotology clinic is gaze-evoked nystagmus followed by downbeat nystagmus. Relatively rare deficits such as upbeat nystagmus, slow saccades, and opsoclonus are also reported. Traditional diagnostic algorithm of these deficits includes screening for autoantibodies and neuroimaging. The diagnostic workup frequently reveals subtle cerebellar atrophy affecting vermis or para-vermis cerebellar region. It is not uncommon for such deficits to have negative family history, genetic disorders, or any toxic exposure. In select cases, we may find low titers of anti-GAD Abs, or voltage-gated calcium channel Abs. Hence, it is likely that early, mild cases of dizzy patients, who were found to have downbeat nystagmus and/or gaze-evoked nystagmus with mild cerebellar atrophy have prodromal phase and LACA. In such cases, when the aforementioned Abs are detected, treatment with plasma exchange or IVIg has resulted in mixed responses; i.e., one patient had complete resolution of downbeat nystagmus, while in another case, the response was incomplete.
Paroxysmal ocular motor and vestibular deficits are not uncommon in LACA [31]. These patients have normal inter-ictal neurological examination. Their typical presentation is acute episodes of vertigo with background constant unsteadiness. Clinical examination may reveal provoked nystagmus, typically after hyperventilation. Occasionally, nystagmus is mild and only present in peculiar gaze orientation. Inter-ictal examination of ocular motor system reveals subtle deficits including curved trajectory of saccades, or mild dysmetria. Low-titer anti-GAD Abs are also associated with gravity independent upbeat nystagmus [67]. It is common to find heterogeneous gaze-holding deficit in the syndrome of anti-GAD Ab. These patients have waveform that has mixture of squarewaves, downbeat nystagmus, and opsoclonus. The opsoclonus superimposes upon the downbeat nystagmus, while robust squarewave jerks are present in the axis orthogonal to the downbeat nystagmus [100].
Mechanistic Underpinning of Paroxysmal or Transient Worsening of Ocular Motor Deficits in LACA
Transient ocular motor deficits in LACA can be due to reversible loss of motor control. Such deficits are typical of transient ischemia to the brainstem but can also present with early forms of neurodegenerative or immune disease when loss of cerebellar and brainstem function is not complete and there are residual tissues compensating for the damage that has already happened.
Acute cerebellar inflammatory disorders present with robust cerebellar dysfunction in the acute phase, but treatment with immunomodulation or steroids results in a complete reversal of the abnormality. Ocular motor deficits can be sensitive markers of acute cerebellar dysfunction due to cerebellitis, acute immune reaction, or decompensated neurodegenerative disorders. One of the fundamental ocular motor behaviors included maintenance of the gaze requiring accurate utilization of the gaze-holding network and the brainstem neural integrator (Fig. 4). This network utilizes eye velocity signal from the saccade burst neurons and transforms it into gaze position signal by virtue of mathematical integration of the velocity signal. A fundamental limitation of the neural integrator is that it is always inaccurate. The inaccuracy is seen in the form of drift in eye position during eccentric gaze holding; the latter is typically compensated by the feedback from the visual system or cerebellar Purkinje neurons [101]. This task requiring accurately calibrated cerebellar signal is vulnerable to any form of cerebellar dysfunction. Any abnormality in the cerebellar outflow, either due to transiently inflamed Purkinje neurons sending aberrant output, or dead Purkinje neurons sending no output, leads to lack of calibration and dysfunction of neural integrator. The consequence is drifting eye position toward the central null position and eye-in-orbit position dependence of slow-phase eye velocity, the deficits that characterize the gaze-evoked nystagmus [101]. The gaze-evoked nystagmus is seen in patients with acute cerebellitis, acute autoimmune cerebellar deficit, and it can be treated with prompt medical management. In cases where damage to Purkinje cells is not complete, or it is mild, as seen in LACA, the deficits such as gaze-evoked nystagmus can be triggered by metabolically challenging the brain — hyperventilation-induced nystagmus is one of such examples. The patient with normal ocular motor examination at baseline may produce gaze-evoked or spontaneous nystagmus after 30-s-long hyperventilation.

Schematic presentation of gaze holding network. The pulse of eye velocity signal is integrated by the brainstem neural integrators. This mechanism relies on normal function of the cerebellar Purkinje neurons, visual system, and orbital proprioception. As depicted in panel (A), the three sources of feedback, project to the input of brainstem neural integrators. As depicted in panel (B), the integration fails when one of the sources of feedback is impaired, either by damage of cerebellar Purkinje neurons, visual dysfunction, or disrupted orbital proprioception. The consequence of such abnormality is impaired neural integration. As a consequence, the eyes drift to the central null position, and drifts are followed by correction leading to phenomenology called “nystagmus.” The same sources of feedback, the cerebellum in particular, are also critical for assuring normal amplitude and directional matrix of saccade
The matrix of saccades, such as velocity and amplitude, is tightly controlled by the cerebellar Purkinje neurons. These cells provide critical error feedback and facilitate the enhanced accuracy of ongoing movements [101, 102]. Any impairment in the Purkinje neuronal function due to autoimmune or inflammatory disorders can impair the error feedback. Latter manifests in saccadic dysmetria. Reversal of saccade dysmetria is not uncommon after prompt treatment of autoimmune or inflammatory cerebellar conditions; however, permanent cerebellar damage results in irreversible saccadic dysmetria. Subtle saccade dysmetria is not unusual in compensated degenerative cerebellar disorders. Metabolic challenge due to acute medical illness can transiently affect cerebellar error control mechanism causing transient saccade dysmetria.
Mimics of Ocular Motor Deficits in Prodromal Phase of CA in LACA
Autoimmune or inflammatory deficits or prodromal phase of CA such as LACA are one way of transiently affecting the Purkinje neuron function. The other deficits include transient ischemia in the brain region supplied by the posterior circulation. These transient deficits do not present with diffusion abnormalities in the MRI, suggestive of acute stroke. Frequently, these deficits may resolve spontaneously without further intervention, and in some instances, aggressive hemodynamic management is warranted. The transient nature of brainstem attack due to vascular etiology has clear differences from that due to prodromal phase of CA due to immune or inflammatory etiologies. Immune or inflammatory etiologies can be seen in pediatric population, young adults, adults, and the elderly; in contrast, brainstem attack due to vascular etiology is typically accompanied by vascular risk factors and typically affects older adults. The duration of vascular brainstem attacks may span from several minutes or can last for days and may depend on the blood pressure. On the contrary, the brainstem attack due to inflammatory or immune etiology lasts from days to weeks; blood pressure has no correlation with symptoms. Nevertheless, vascular etiology should be seriously considered in differential to prodromal symptoms of LACA, because if untreated with appropriate anti-lipid and anti-platelet management transient ischemia to the posterior circulation can lead to permanent damage in form of cerebellar stroke.
Dysfunction of the ion channels determining the cell membrane properties can frequently lead to dysfunction of the cerebellar Purkinje neurons. These deficits can be seen in those with genetic channelopathy, leading to episodic ataxias, or intoxication from pharmacological substances, such as antiepileptics. The episodic ataxias (types EA2, EA3, and EA4) present with transient ocular motor dysfunction such as gaze-evoked nystagmus, positional nystagmus, saccade dysmetria, or acute vestibular dysfunction [103, 104]. The deficits can last from hours to days and are self-limiting. There is a familial trend, and there are minimal to no ocular motor dysfunction in the inter-ictal phase. Toxic increased levels of antiepileptics such as phenobarbital, fosphenytoin, lamotrigine, and carbamazepine are known to cause acute ocular motor cerebellar dysfunction and ataxia [105,106,107,108,109,110,111,112]. Typical features are downbeat nystagmus, gaze-evoked nystagmus, axial and appendicular ataxia, and gait instability. The deficits are transient and resolve with normalizing the antiepileptic levels. The mechanism of ocular motor dysfunction in those with toxic antiepileptic levels could be attributed to ion channel dysregulation. Channelopathy closely resembles immune etiologies and prodromal phase of LACA. They can present at any age; however, unlike immune etiologies, there is a family history. On the contrary, drug-induced vestibular and ocular motor symptoms correlate with increased serum concentration of the offending pharmacotherapeutic agent.
Dynamic Changes of Autoimmune Biomarkers During Progression of LACA (Christiane S Hampe)
Latent autoimmune diseases such as LADA are characterized by a slowly progressing pathogenesis, which eventually results in irreversible tissue damage. The disease progression may be linear, or show a remission/relapse pattern, where periods of disease progression are followed by upregulation of anti-inflammatory immune responses, allowing partial recovery. Eventually, the tissue damage is too severe and restoration to normal function can no longer be achieved. The length of the prodromal period varies and may be impacted by genetic susceptibility, environmental factors, or diet. A clear understanding of the molecular events and timing involved in the final breakdown of the immune response is needed to develop intervention therapies. The dynamic nature of the pathogenesis is reflected in changing levels of immune factors, including cytokines and chemokines. In a recent publication [113], disease progression in LADA patients was found to correlate with a decline in levels of cytokines Interleukin-1 receptor agonist (IL-1ra) and interleukin-1 beta (IL-1b). IL-1ra is a receptor antagonist of IL-1 and its direct correlation with C-peptide levels in type 1 DM patients suggests an involvement of the anti-inflammatory cytokine in disease remission [114]. The concomitant decrease in pro-inflammatory cytokine IL-1 beta and anti-inflammatory cytokine IL-1ra in LADA patients demonstrates the complicated, interconnected system of cytokine regulation, where multiple factors are involved in the dynamic changes of cytokine release. While it remains to be determined whether the dynamic changes in cytokine pattern observed in LADA patients are cause or effect of the progressive decline in beta cell function, they may serve as novel biomarkers for disease progression. Further studies are necessary to establish whether changes in cytokine levels or other immune factors can be observed in LACA and can be used as biomarkers for disease progression.
Conclusion (Manto M)
The concept of LACA has implications in terms of prevention and administration of early therapies. Like for LADA, a personalized approach is recommended. The ultimate goal is the preservation of the cerebellar reserve, both functional and structural. In ataxic patients with no manifestly evident autoimmunity, the significance of associated autoantibodies should be carefully assessed for diagnosis of LACA. Patients suspected to present LACA require a close clinical/biological/radiological follow-up with longitudinal observations. The annual progression rate is currently unknown and might be different at a very early stage and later during follow-up, as observed in genetic ataxias where a non-linear progression is found [115]. In preataxic SCA3 patients, elevated levels of neurofilament light (Nfl) are detected already 7.5 years before onset. There might be a similar window in IMCA [116]. The signs related to the prodromal stage need to be identified. IMCAs patients often report fatigue in the months before the ataxia onset. There is a need to obtain data related to cerebellar involvement and extra-cerebellar involvement. The inventory of non-ataxia signs (INAS total count) might be useful in IMCAs also. Regarding the cerebellar aspect, subtle ocular disturbances, motor deficits, and cognitive/affective symptoms need to be scrutinized and correlated with biomarkers.
Data Availability
The concept reported in this manuscript is not associated with raw data.
Abbreviations
- Abs:
-
antibodies
- ANA Ab:
-
anti-nuclear antibodies
- CA:
-
cerebellar ataxia
- CNS:
-
central nervous system
- CSF:
-
cerebrospinal fluid (CSF)
- DM:
-
diabetes mellitus
- EA:
-
episodic ataxia
- ES-RBA:
-
epitope-specific radioligand binding assay
- GA:
-
gluten ataxia
- GAD:
-
glutamate decarboxylase
- ICI:
-
immune checkpoint inhibitors
- IMCAs:
-
immune-mediated cerebellar ataxias
- INAS:
-
inventory of non-ataxia signs
- IVIg:
-
intravenous immunoglobulin
- KLHL11:
-
Kelch-like protein 11
- LACA:
-
latent autoimmune cerebellar ataxia
- LADA:
-
latent autoimmune diabetes in adults
- mRS:
-
modified Rankin Score
- Nfl:
-
neurofilament light
- PACA:
-
primary autoimmune cerebellar ataxia
- PCD:
-
paraneoplastic cerebellar degeneration
- PIC:
-
post-infectious cerebellitis
- PNS:
-
paraneoplastic neurological syndromes
- SCA:
-
spinocerebellar ataxia
- SPIDDM:
-
slowly progressive insulin-dependent DM
- SPS:
-
stiff person syndrome
- SPV-GE nystagmus:
-
slow phase velocity of central and gaze-evoked nystagmus
- TPO:
-
thyroid peroxidase
- SS-A:
-
Sjogren’s-syndrome-related antigen S
- TG6:
-
transglutaminase 6
- Tr/DNER:
-
delta/notch-like epidermal growth factor-related receptor
- TRIM9/67:
-
tripartite motif-containing proteins 9 and 67
References
Hadjivassiliou M. Immune-mediated acquired ataxias. Handb Clin Neurol. 2012;103:189–99.
Mitoma H, Adhikari K, Aeschlimann D, Chattopadhyay P, Hadjivassiliou M, Hampe CS, et al. Consensus paper: neuroimmune mechanisms of cerebellar ataxias. Cerebellum. 2016;15(2):213–32.
Mitoma H, Hadjivassiliou M, Honnorat J. Guidelines for treatment of immune-mediated cerebellar ataxias. Cerebellum Ataxias. 2015;2:14.
Joubert B, Rotásky J, Honnorat J. Nonparaneoplastic autoimmune cerebellar ataxia. Handb Clin Neurol. 2018;155:313–32.
Hadjivassiliou M, Mitoma H, Manto M. Autoimmune ataxia. In: Mitoma H, Manto M, editors. Neuroimmune diseases; from cells to the living brain. Springer Nature; 2019. p. 599–620.
Mitoma H, Manto M, Hadjivassiliou M. Immune-mediated cerebellar ataxias: clinical diagnosis and treatment based on immunological and physiological mechanisms. J Mov Disord. 2021;14(1):10–28.
Hadjivassiliou M, Graus F, Honnorat J, Jarius S, Titulaer M, Manto M, Hoggard N, Sarrigiannis P, Mitoma H. Diagnostic criteria for primary autoimmune cerbellar ataxia (PACA)-guidlines from an International Task Force on Immune Mediated Cerbellar Ataxia. Cerebellum. 2020;19(4):605–10.
Nanri K, Niwa H, Mitoma H, Takei A, Ikeda J, Harada T, et al. Low-titer anti-GAD-antibody-positive cerebellar ataxia. Cerebellum. 2013;12(2):171–5.
Baizabal-Carvallo JF, Alonso-Juarez M. Cerebellar disease associated with anti-glutamic acid decarboxylase antibodies: review. A J Neural Transm (Vienna). 2017;124(10):1171–82.
Petrijan T, Menih M. Low-Titre GAD Antibody-associated late-onset cerebellar ataxia with a significant clinical response to intravenous immunoglobulin treatment. Cerebellum. 2017;16(4):868–71.
Muñiz-Castrillo S, Vogrig A, Joubert B, Pinto AL, Gonçalves D, Chaumont H, et al. Transient neurological symptoms preceding cerebellar ataxia with glutamic acid decarboxylase antibodies. Cerebellum. 2020;19(5):715–21.
Tuomi T, Groop LC, Zimmet PZ, Rowley MJ, Knowles W, Mackay IR. Antibodies to glutamic acid decarboxylase reveal latent autoimmune diabetes mellitus in adults with a non-insulin-dependent onset of disease. Diabetes. 1993;42(2):359–62.
Stenström G, Gottsäter A, Bakhtadze E, Berger B, Sundkvist G. Latent autoimmune diabetes in adults: definition, prevalence, beta-cell function, and treatment. Diabetes. 2005;54(Suppl 2):S68–72.
Yasui J, Kawasaki E, Tanaka S, Awata T, Ikegami H, Imagawa A, et al. Clinical and genetic characteristics of non-insulin-requiring glutamic acid decarboxylase (GAD) autoantibody-positive diabetes: a nationwide survey in Japan. PLoS One. 2016;11:e0155643.
Huang G, Yin M, Xiang Y, Li X, Shen W, Luo S, et al. Persistence of glutamic acid decarboxylase antibody (GADA) is associated with clinical characteristics of latent autoimmune diabetes in adults: a prospective study with 3-year follow-up. Diabetes Metab Res Rev. 2016;32(6):615–22.
Carlsson S. Etiology and pathogenesis of latent autoimmune diabetes in adults (LADA) compared to type 2 diabetes. Front Physiol. 2019;10:320.
Mitoma H, Buffo A, Gelfo F, Guell X, Fucà E, Kakei S, et al. Consensus paper. Cerebellar reserve: from cerebellar physiology to cerebellar disorders. Cerebellum. 2019;19(1):131–53.
Graus F, Saiz A, Dalmau J. GAD antibodies in neurological disorders-insights and challenges. Nat Rev Neurol. 2020;16:353–65.
Kikuchi A, Yoneda M, Hasegawa T, Matsunaga A, Ikawa M, Nakamura T, et al. High prevalence of serum anti-NH2-terminal α-enolase antibodies un patients with multiple system atrophy and corticobasal syndrome. J Neurol. 268(11):4291–5.
Siddiqui TG, Whitfield T, Praharaju SJ, Sadiq D, Kazmi H, Ben-Joseph A, et al. Magnetic resonance imaging in stable mild cognitive impairment, prodromal Alzheimer’s disease, and prodromal dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2020;49(6):583–8.
Lavan O, Apter A, Benaroya-Milshtein N, Fennig S. The prodrome of psychotic disorders. Harefuah. 2021;160(2):104–9.
Chen CC, Yao NW, Lin CW, Su WS, Wu CT, Chang C, et al. Neuroimaging spectrum at pre-, early, and late symptomatic stages of SCA 17 mice. Cerebellum. 2020;19(4):487–500.
Velázquez-Pérez L, Rodriguez-Labrada R, González-Garcés Y, Arrufat-Pie E, Torres-Vega R, Medrano-Montero J, et al. Prodromal spinocerebellar ataxia type 2 subjects have quantifiable gait and postural sway deficits. Mov Disord. 2021;36(2):471–80.
de Oliveira CM, Leotti VB, Bolzan G, Cappelli AH, Rocha AG, Ecco G, et al. Pre-ataxic changes of clinical scales and eye movement in Machado-Joseph disease: BIGPRO study. Mov Disord. 2021;36(4):985–94.
Jacobi H, du Montcel ST, Romanzetti S, Harmuth F, Mariotti C, Nanetti L, et al. Conversion of individuals at risk for spinocerebellar ataxia type 1,2,3,and 6 to manifest ataxia (RISCA): longitudinal cohort study. Lancet Neurol. 2020;19(9):738–47.
Kobayashi T, Tamemoto K, Nakanishi K, Kato N, Okubo M, Kajio H, et al. Immunogenetic and clinical characterization of slowly progressive IDDM. Diabetes Care. 1993;16(5):780–8.
Zimmet PZ, Tuomi T, Mackay IR, Rowley MJ, Knowles W, Cohen M, et al. Latent autoimmune diabetes mellitus in adults (LADA): the role of antibodies to glutamic acid decarboxylase in diagnosis and prediction of insulin dependency. Diabet Med. 1994;118(3):299–303.
Nociti V, Frisullo G, Tartaglione T, Patanella AK, Iorio R, Tonali PA, et al. Refractory generalized seizures and cerebellar ataxia associated with anti-GAD antibodies responsive to immunosuppressive treatment. Eur J Neurol. 2010;17(1):e5.
Virgilio R, Corti S, Agazzi P, Santoro D, Lanfranconi S, Candelise L, et al. Effect of steroid treatment in cerebellar ataxia associated with anti-glutamic acid decarboxylase antibodies. J Neurol Neurosurg Psychiatry. 2009;80(1):95–6.
Sunwoo JS, Chu K, Byun JI, Moon J, Lim JA, Kim TJ, Lee ST, Jung KH, Park KI, Jeon D, Jung KY, Kim M, Lee SK. Intrathecal-specific glutamic acid decarboxylase antibodies at low titers in autoimmune neurological disorders. J Neuroimmunol. 2016;290:15–21.
Ariño H, Gresa-Arribas N, Blanco Y, Martínez-Hernández E, Sabater L, Petit-Pedrol M, Rouco I, Bataller L, Dalmau JO, Saiz A, Graus F. Cerebellar ataxia and glutamic acid decarboxylase antibodies: immunologic profile and long-term effect of immunotherapy. JAMA Neurol. 2014;71(8):1009–16.
Mitoma H, Manto M, Hampe CS. Pathogenic roles of glutamic acid decarboxylase 65 autoantibodies in cerebellar ataxias. J Immunol Res. 2017;2017:2913297.
Baizabal-Carvallo JF. The neurological syndromes associated with glutamic acid decarboxylase antibodies. J Autoimmun. 2019;101:35–47.
Matsumoto S, Kusuhara T, Nakajima M, Ouma S, Takahashi M, Yamada T. Acute attacks and brain stem signs in a patient with glutamic acid decarboxylase autoantibodies. J Neurol Neurosurg Psychiatry. 2002;73(3):345–6.
Muñiz-Castrillo S, Vogrig A, Montagnac C, Joubert B, Benaiteau M, Casez O, et al. Familial autoimmunity in neurological patients with GAD65 antibodies: an interview-based study. J Neurol. 2021; https://doi.org/10.1007/s00415-021-10424-w.
Baekkeskov S, Aanstoot HJ, Christgau S, Reetz A, Solimena M, Cascalho M, Folli F, Richter-Olesen H, De Camilli P. Identification of the 64K autoantigen in insulin-dependent diabetes as the GABA-synthesizing enzyme glutamic acid decarboxylase. Nature. 1990;347(6289):151–6.
Daw K, Powers AC. Two distinct glutamic acid decarboxylase auto-antibody specificities in IDDM target different epitopes. Diabetes. 1995;44(2):216–20.
Powers AC, Bavik K, Tremble J, Daw K, Scherbaum WA, Banga JP. Comparative analysis of epitope recognition of glutamic acid decarboxylase (GAD) by autoantibodies from different autoimmune disorders. Clin Exp Immunol. 1999;118(3):349–56.
Hagopian WA, Michelsen B, Karlsen AE, Larsen F, Moody A, Grubin CE, Rowe R, Petersen J, McEvoy R, Lernmark A. Autoantibodies in IDDM primarily recognize the 65,000-Mr rather than the 67,000-Mr isoform of glutamic acid decarboxylase. Diabetes. 1993;42(4):631–6.
Manto M, Honnorat J, Hampe CS, Guerra-Narbona R, López-Ramos JC, Delgado-García JM, Saitow F, Suzuki H, Yanagawa Y, Mizusawa H, Mitoma H. Disease-specific monoclonal antibodies targeting glutamate decarboxylase impair GABAergic neurotransmission. Front Behav Neurosci. 2015;9:78.
Liimatainen S, Honnorat J, Pittock SJ, McKeon A, Manto M, Radtke JR, T1D Exchange Biobank, Hampe CS. GAD65 autoantibody characteristics in patients with co-occurring type 1 diabetes and epilepsy may help identify underlying epilepsy etiologies. Orphanet J Rare Dis. 2018;13(1):55.
Fouka P, Alexopoulos H, Akrivou S, Trohatou O, Politis PK, Dalakas MC. GAD65 epitope mapping and search for novel autoantibodies in GAD-associated neurological disorders. J Neuroimmunol. 2015;281:73–7.
Gresa-Arribas N, Ariño H, Martínez-Hernández E, Petit-Pedrol M, Sabater L, Saiz A, Dalmau J, Graus F. Antibodies to inhibitory synaptic proteins in neurological syndromes associated with glutamic acid decarboxylase autoimmunity. PLoS One. 2015;10(3):e0121364.
Graus F. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75(8):1135–40.
Graus F, Vogrig A, Muñiz-Castrillo S, Antoine JG, Desestret V, Dubey D, et al. Updated diagnostic criteria for paraneoplastic neurological syndromes. Neurol Neuroimmunol Neuroinflamm. 2021;8(4):e1014.
Giometto B. Paraneoplastic neurologic syndrome in the PNS Euronetwork database: a European study from 20 centers. Arch Neurol. 2010;67(3):330.
Kanikannan MA, Sirisha Y, Uppin MS, Jabeen SA, M Kandadai R, Sundaram C, et al. Incidence and spectrum of paraneoplastic neurological syndromes: single center study. J Neuro-Oncol. 2015;125(1):197–206.
Vogrig A, Gigli GL, Segatti S, Corazza E, Marini A, Bernardini A, et al. Epidemiology of paraneoplastic neurological syndromes: a population-based study. J Neurol. 2020;267(1):26–35.
Hébert J, Riche B, Vogrig A, Muñiz-Castrillo S, Joubert B, Picard G, et al. Epidemiology of paraneoplastic neurologic syndromes and autoimmune encephalitides in France. Neurol Neuroimmunol Neuroinflamm. 2020;7(6):e883.
Yshii L, Bost C, Liblau R. Immunological bases of paraneoplastic cerebellar degeneration and therapeutic implications. Front Immunol. 2020;11:991.
Shams’ili S, Grefkens J, de Leeuw B, van den Bent M, Hooijkaas H, van der Holt B, et al. Paraneoplastic cerebellar degeneration associated with antineuronal antibodies: analysis of 50 patients. Brain. 2003;126(Pt6):1409–18.
Ducray F, Demarquay G, Graus F, Decullier E, Antoine J-C, Giometto B, et al. Seronegative paraneoplastic cerebellar degeneration: the PNS Euronetwork experience. Eur J Neurol. 2014;21(5):731–5.
Bernal F, Shams’ili S, Rojas I, Sanchez-Valle R, Saiz A, Dalmau J, et al. Anti-Tr antibodies as markers of paraneoplastic cerebellar degeneration and Hodgkin’s disease. Neurology. 2003;60(2):230–4.
de Graaff E, Maat P, Hulsenboom E, van den Berg R, van den Bent M, Demmers J, et al. Identification of delta/notch-like epidermal growth factor-related receptor as the Tr antigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2012;71(6):815–24.
Graus F. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124(Pt6):1138–48.
Pittock SJ, Lucchinetti CF, Lennon VA. Anti-neuronal nuclear autoantibody type 2: paraneoplastic accompaniments. Ann Neurol. 2003;53(5):580–7.
Simard C, Vogrig A, Joubert B, Muñiz-Castrillo S, Picard G, Rogemond V, et al. Clinical spectrum and diagnostic pitfalls of neurologic syndromes with Ri antibodies. Neurol Neuroimmunol Neuroinflamma. 2020;7(3):e699.
Dalmau J. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127(Pt8):1831–44.
Dubey D, Wilson MR, Clarkson B, Giannini C, Gandhi M, Cheville J, et al. Expanded clinical phenotype, oncological associations, and immunopathologic insights of paraneoplastic Kelch-like protein-11 encephalitis. JAMA Neurol. 2020;77(11):1420–9.
Do LD, Gupton SL, Tanji K, Bastien J, Brugière S, Couté Y, et al. TRIM9 and TRIM67 are new targets in paraneoplastic cerebellar degeneration. Cerebellum. 2019;18(2):245–54.
Ruiz-García R, Martínez-Hernández E, Joubert B, Petit-Pedrol M, Pajarón-Boix E, Fernández V, et al. Paraneoplastic cerebellar ataxia and antibodies to metabotropic glutamate receptor 2. Neurol Neuroimmunol Neuroinflamm. 2019;7(2):e658.
Peterson K, Rosenblum MK, Kotanides H, Posner JB. Paraneoplastic cerebellar degeneration. I. A clinical analysis of 55 anti-Yo antibody-positive patients. Neurology. 1992;42(10):1931–7.
Vogrig A, Bernardini A, Gigli GL, Corazza E, Marini A, Segatti S, et al. Stroke-like presentation of paraneoplastic cerebellar degeneration: a single-center experience and review of the literature. Cerebellum. 2019;18(5):976–82.
Lucchinetti CF, Kimmel DW, Lennon VA. Paraneoplastic and oncologic profiles of patients seropositive for type 1 antineuronal nuclear autoantibodies. Neurology. 1998;50(3):652–7.
Vogrig A, Fouret M, Joubert B, Picard G, Rogemond V, Pinto A-L, et al. Increased frequency of anti-Ma2 encephalitis associated with immune checkpoint inhibitors. Neurol Neuroimmunol NeuroInflamm. 2019;6:e604.
Baizabal-Carvallo JF, Alonso-Juarez M. Vertical nystagmus associated with glutamic acid decarboxylase antibodies responding to cyclophosphamide. J Neuroimmunol. 2018;317:5–7.
Feldman D, Otero-Millan J, Shaikh AG. Gravity-independent upbeat nystagmus in syndrome of anti-GAD antibodies. Cerebellum. 2019;18(2):287–90.
Aguiar TS, Fragoso A, Albuquerque CR, Teixeira PF, Souza MV, Zajdenverg L, et al. Clinical characteristics of patients with cerebellar ataxia associated with anti-GAD antibodies. Arq Neuropsiquiatr. 2017;75(3):142–6.
Yilmaz FM, Little D, Gallagher M, Colcher A. Anti-glutamate dehydrogenase antibody positive cerebellar ataxia and stiff person syndrome responding to dual treatment with steroids and intravenous immunoglobulin: a case presentation and literature review. Cureus. 2019;11(6):e4851.
Giometto B, Miotto D, Faresin F, Argentiero V, Scaravilli T, Tavolato B. Anti-gabaergic neuron autoantibodies in a patient with stiff-man syndrome and ataxia. J Neurol Sci. 1996;143(1-2):57–9.
Hadjivassiliou M, Aeschlimann D, Grünewald RA, Sanders DS, Sharrack B, Woodroofe N. GAD antibody-associated neurological illness and its relationship to gluten sensitivity. Acta Neurol Scand. 2011;123(3):175–80.
Hadjivassiliou M, Sarrigiannis PG, Shanmugarajah PD, Sanders DS, Grünewald RA, Zis P, Hoggard N. Clinical characteristics and management of 50 patients with anti-GAD ataxia: gluten-free diet has a major impact. Cerebellum. 2021;20(2):179–85.
Honnorat J, Saiz A, Giometto B, Vincent A, Brieva L, de Andres C. Cerebellar ataxia with anti-glutamic acid decarboxylase antibodies: study of 14 patients. Arch Neurol. 2001;58(2):225–30.
Dagklis IE, Papagiannopoulos S, Theodoridou V, Kazis D, Argyropoulou O, Bostantjopoulou S. Miller-Fisher syndrome: are anti-GAD antibodies implicated in its pathophysiology? Case Rep Neurol Med. 2016;2016:3431849.
Pedroso JL, Braga-Neto P, Dutra LA, Barsottini OG. Cerebellar ataxia associated to anti-glutamic acid decarboxylase autoantibody (anti-GAD): partial improvement with intravenous immunoglobulin therapy. Arq Neuropsiquiatr. 2011;69(6):993.
Nanri K, Okita M, Takeguchi M, Taguchi T, Ishiko T, Saito H, et al. Intravenous immunoglobulin therapy for autoantibody-positive cerebellar ataxia. Intern Med. 2009;48(10):783–90.
McKeon A. Purkinje cell cytoplasmic autoantibody type 1 accompaniments: the cerebellum and beyond. Arch Neurol. 2011;68(10):1282–9.
Hoche F, Guell X, Vangel MG, Sherman JC, Schmahmann JD. The cerebellar cognitive affective/Schmahmann syndrome scale. Brain J Neurol. 2018;141(1):248–70.
Dalmau J, Gonzalez RG, Lerwill MF. A 56-year-old woman with rapidly progressive vertigo and ataxia. N Engl J Med. 2007;356(6):612–20.
Ogawa E, Sakakibara R, Kawashima K, Yoshida T, Kishi M, Tateno F, et al. VGCC antibody-positive paraneoplastic cerebellar degeneration presenting with positioning vertigo. Neurol Sci. 2011;32:1209–12.
Vogrig A, Péricart S, Pinto AL, Rogemond V, Muñiz-Castrillo S, Picard G, et al. Immunopathogenesis and proposed clinical score for identifying Kelch-like protein-11 encephalitis. Brain Commun. 2021;3(3):fcab185.
Small M, Treilleux I, Couillault C, Pissaloux D, Picard G, Paindavoine S, et al. Genetic alterations and tumor immune attack in Yo paraneoplasctic cerebellar degeneration. Acta Neuropathol. 2018;135(4):569–79.
Hadjivassiliou M, Grünewald RA, Chattopadhyay AK, Davies-Jones GA, Gibson A, Jarratt JA, et al. Clinical, radiological, neurophysiological and neuropathological characteristics of gluten ataxia. Lancet. 1998;352(9140):1582–5.
Hadjivassiliou M, Sanders DS, Grunewald RA, Woodroofe N, Boscolo S, Aeschlimann D. Gluten sensitivity: from gut to brain. Lancet Neurol. 2010;9(3):318–30.
Hadjivassiliou M, Aeschlimann P, Sanders DS, Mäki M, Kaukinen K, Grünewald RA, et al. Transglutaminase 6 antibodies in the diagnosis of gluten ataxia. Neurology. 2013;80(19):1–6.
Hadjivassiliou M, Grünewald RA, Sanders DS, Shanmugarajah P, Hoggard N. Effect of gluten-free diet on cerebellar MR spectroscopy in gluten ataxia. Neurolgy. 2017;89(7):705–6.
Connolly AM, Dodson WE, Prensky AL, Rust RS. Course and outcome of acute cerebellar ataxia. Ann Neurol. 1994;35(6):673–9.
Kornreich L, Shkalim-Zemer V, Levinsky Y, Abdallah W, Ganelin-Cohen E, Straussberg R. Acute cerebellitis in children: a many-faceted disease. J Child Neurol. 2016;31(8):991–7.
Fadakar N, Ghaemmaghami S, Masoompour SM, Shirazi Yeganeh B, Akbari A, Hooshmandi S, Ostovan VR. A first case of acute cerebellitis associated with coronavirus disease (COVID-19): a case report and literature review. Cerebellum. 2020;19(6):911–4.
Povlow A, Auerbach AJ. Acute cerebellar ataxia in COVID-19 infection: a case report. J Emerg Med. 2021;60(1):73–6.
van Samkar A, Poulsen MNF, Bienfait HP, Van Leeuwen RB. Acute cerebellitis in adults: a case report and review of the literature. BMC Res Notes. 2017;10(1):610.
Kubota M, Takahashi Y. Steroid-responsive chronic cerebellitis with positive glutamate receptor delta 2 antibody. J Child Neurol. 2008;23(2):228–30.
Murakami H, Iijima S, Kawamura M, Takahashi Y, Ichikawa H. A case of acute cerebellar ataxia following infectious mononucleosis accompanied by intrathecal anti-glutamate receptor δ2 antibody. Rinsho Shinkeigaku. 2013;53(7):555–8. Japanese
Ichikawa K, Kikuchi M, Takeshita S, Nezu A. A case of chronic recurrent cerebellar ataxia responding to steroid therapy. Brain and Development. 2009;31(1):83–5.
Papavasiliou AS, Kotsalis C, Trakadas S. Transient cerebellar mutism in the course of acute cerebellitis. Pediatr Neurol. 2004;30(1):71–4.
Sawaishi Y, Takada G. Acute cerebellitis. Cerebellum. 2002;1(3):223–8.
Baizabal-Carvallo JF, Jankovic J. Autoimmune and paraneoplastic movement disorders: an update. J Neurol Sci. 2018;385:175–84.
Baizabal-Carvallo JF, Cardoso F. Chorea in children: etiology, diagnostic approach and management. J Neural Transm (Vienna). 2020;127(10):1323–42.
Al-Shokri SD, Karumannil SA, Mohammed SS, Sadek MS. Post-Epstein-Barr virus acute cerebellitis in an adult. Am J Case Rep. 2020;21:e918567.
Theeranaew W, Wang F, Ghasia FF, Wilmot G, Shaikh AG. Gaze-holding and anti-GAD antibody: prototypic heterogeneous motor dysfunction in immune disease. Cerebellum. 2021; https://doi.org/10.1007/s12311-021-01272-5.
Zee DS, Yee RD, Cogan DG, Robinson DA, Engel WK. Ocular motor abnormalities in hereditary cerebellar ataxia. Brain. 1976;99(2):207–34.
Ritchie L. Effects of cerebellar lesions on saccadic eye movements. J Neurophysiol. 1976;39(6):1246–56.
Strupp M, Thurtell MJ, Shaikh AG, Brandt T, Zee DS, Leigh RJ. Pharmacotherapy of vestibular and ocular motor disorders, including nystagmus. J Neurol. 2011;258(7):1207–22.
Leigh RJ, Zee DS. The neurology of eye movements. Oxford University Press, NY, NY; 2016. Edition 5
Kim HA, Oh EH, Choi SY, Choi JH, Park JY, Lee H, Choi KD. Transient vestibular symptoms preceding posterior circulation stroke: a prospective nulticenter study. Stroke. 2021;52(6):e224–8.
Wirfs L, Whitworth K, Kellar J. Nystagmus associated with carbamazepine toxicity. Clin Pract Cases Emerg Med. 2017;1(4):441–2.
Adamec I, Nanković S, Zadro I, Hajnšek S, Habek M. Oxcarbazepine-induced jerky see-saw nystagmus. Neurol Sci. 2013;34(10):1839–40.
Alkawi A, Kattah JC, Wyman K. Downbeat nystagmus as a result of lamotrigine toxicity. Epilepsy Res. 2005;63(2-3):85–8.
Lifshitz M, Gavrilov V, Sofer S. Signs and symptoms of carbamazepine overdose in young children. Pediatr Emerg Care. 2000;16(1):26–7.
Chrousos GA, Cowdry R, Schuelein M, Abdul-Rahim AS, Matsuo V, Currie JN. Two cases of downbeat nystagmus and oscillopsia associated with carbamazepine. Am J Ophthalmol. 1987;103(2):221–4.
Alpert JN. Downbeat nystagmus due to anticonvulsant toxicity. Ann Neurol. 1978;4(5):471–3.
Landy PJ. “Dilatin” overdosage. Med J Aust. 1968;2(15):639.
Hals IK, Björklund A, Fiskvik Fleiner H, Grill V. Time-dependent effects on circulating cytokines in patients with LADA: a decrease in IL1-ra and IL-1 beta is associated with progressive disease. Cytokine. 2022;151:155792.
Pfleger C, Mortensen HB, Hansen L, Herder C, Roep BO, Hoey H, Aanstoot HJ, Kocova M, Schloot NC, Hvidøre Study Group on Childhood Diabetes. Association of IL-1ra and adiponectin with C-peptide and remission in patients with type 1 diabetes. Diabetes. 2008;57(4):929–37.
Nigri A, Sarro L, Mongelli A, Pinardi C, Porcu L, Castaldo A, et al. Progression of cerebellar atrophy in spinocerebellar ataxia type 2 gene carriers: a longitudinal MRI study in preclinical and early disease stages. Front Neurol. 2020;11:616419.
Wilke C, Haas E, Reetz K, Faber J, Garcia-Moreno H, Santana MM, et al. Neurofilaments in spinocerebellar ataxia type 3: blood biomarkers at the preataxic and ataxic stage in human and mice. EMBO Mol Med. 2020;12(7):e11803.
Gadoth A, Kryzer TJ, Fryer J, McKeon A, Lennon VA, Pittock SJ. Microtubule-associated protein 1B: novel paraneoplastic biomarker: MAP 1B IgG. Ann Neurol. 2017;81(2):266–77.
Yu Z, Kryzer TJ, Griesmann GE, Kim K, Benarroch EE, Lennon VA. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49(2):146–54.
Honnorat J, Cartalat-Carel S, Ricard D, Camdessanche JP, Carpentier AF, Rogemond V, et al. Onco-neural antibodies and tumour type determine survival and neurological symptoms in paraneoplastic neurological syndromes with Hu or CV2/CRMP5 antibodies. J Neurol Neurosurg Psychiatry. 2008;80(4):412–6.
Graus F, Vincent A, Pozo-Rosich P, Sabater L, Saiz A, Lang B, et al. Anti-glial nuclear antibody: marker of lung cancer-related paraneoplastic neurological syndromes. J Neuroimmunol. 2005;165(1-2):166–71.
Pittock SJ, Lucchinetti CF, Parisi JE, Benarroch EE, Mokri B, Stephan CL, et al. Amphiphysin autoimmunity: paraneoplastic accompaniments. Ann Neurol. 2005;58(1):96–107.
Graus F, Lang B, Pozo-Rosich P, Saiz A, Casamitjana R, Vincent A. P/Q type calcium-channel antibodies in paraneoplastic cerebellar degeneration with lung cancer. Neurology. 2002;59(5):764–6.
Zalewski NL, Lennon VA, Lachance DH, Klein CJ, Pittock SJ, Mckeon A. P/Q- and N-type calcium-channel antibodies: oncological, neurological, and serological accompaniments: neuronal Calcium-Channel Abs. Muscle Nerve. 2016;54(2):220–7.
Sabater L, Bataller L, Carpentier AF, Aguirre-Cruz ML, Saiz A, Benyahia B, et al. Protein kinase Cgamma autoimmunity in paraneoplastic cerebellar degeneration and non-small-cell lung cancer. J Neurol Neurosurg Psychiatry. 2006;77(12):1359–62.
Höftberger R, Kovacs GG, Sabater L, Nagy P, Racz G, Miquel R, et al. Protein kinase Cγ antibodies and paraneoplastic cerebellar degeneration. J Neuroimmunol. 2013;256(1-2):91–3.
Bataller L, Sabater L, Saiz A, Serra C, Claramonte B, Graus F. Carbonic anhydrase-related protein VIII: autoantigen in paraneoplastic cerebellar degeneration. Ann Neurol. 2004;56(4):575–9.
Höftberger R, Sabater L, Velasco F, Ciordia R, Dalmau J, Graus F. Carbonic anhydrase-related protein VIII antibodies and paraneoplastic cerebellar degeneration. Neuropathol Appl Neurobiol. 2014;40(5):650–3.
Prevezianou A, Tzartos JS, Dagklis IE, Bentenidi E, Angelopoulos P, Bostantjopoulou S. Paraneoplastic cerebellar degeneration in a patient with breast cancer associated with carbonic anhydrase-related protein VIII autoantibodies. J Neuroimmunol. 2020;344:577242.
Bartels F, Prüss H, Finke C. Anti-ARHGAP26 autoantibodies are associated with isolated cognitive impairment. Front Neurol. 2018;9:656.
Basal E, Zalewski N, Kryzer TJ, Hinson SR, Guo Y, Dubey D, et al. Paraneoplastic neuronal intermediate filament autoimmunity. Neurology. 2018;91(18):e1677–89.
Berzero G, Hacohen Y, Komorowski L, Scharf M, Dehais C, Leclercq D, et al. Paraneoplastic cerebellar degeneration associated with anti-ITPR1 antibodies. Neurol Neuroimmunol Neuroinflamm. 2017;4(2):e326.
Alfugham N, Gadoth A, Lennon VA, Komorowski L, Scharf M, Hinson S, et al. ITPR1 autoimmunity: frequency, neurologic phenotype, and cancer association. Neurol Neuroimmunol Neuroinflamm. 2018;5(1):e418.
Chan KH, Vernino S, Lennon VA. ANNA-3 anti-neuronal nuclear antibody: Marker of lung cancer-related autoimmunity. Ann Neurol. 2001;50(3):301–11.
Code Availability
There is no software application or custom code used in this paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Committee Approval
Not applicable
Consent to Participate
All subjects provided consent to participate in the study.
Consent for Publication
All subjects provided consent to publication of the findings in medical journal.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Manto, M., Hadjivassiliou, M., Baizabal-Carvallo, J.F. et al. Consensus Paper: Latent Autoimmune Cerebellar Ataxia (LACA). Cerebellum 23, 838–855 (2024). https://doi.org/10.1007/s12311-023-01550-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-023-01550-4