
Abstract
Cerebellar hypoplasia and dysplasia encompass a group of clinically and genetically heterogeneous disorders frequently associated with neurodevelopmental impairment. The Neuron Navigator 2 (NAV2) gene (MIM: 607,026) encodes a member of the Neuron Navigator protein family, widely expressed within the central nervous system (CNS), and particularly abundant in the developing cerebellum. Evidence across different species supports a pivotal function of NAV2 in cytoskeletal dynamics and neurite outgrowth. Specifically, deficiency of Nav2 in mice leads to cerebellar hypoplasia with abnormal foliation due to impaired axonal outgrowth. However, little is known about the involvement of the NAV2 gene in human disease phenotypes. In this study, we identified a female affected with neurodevelopmental impairment and a complex brain and cardiac malformations in which clinical exome sequencing led to the identification of NAV2 biallelic truncating variants. Through protein expression analysis and cell migration assay in patient-derived fibroblasts, we provide evidence linking NAV2 deficiency to cellular migration deficits. In model organisms, the overall CNS histopathology of the Nav2 hypomorphic mouse revealed developmental anomalies including cerebellar hypoplasia and dysplasia, corpus callosum hypo-dysgenesis, and agenesis of the olfactory bulbs. Lastly, we show that the NAV2 ortholog in Drosophila, sickie (sick) is widely expressed in the fly brain, and sick mutants are mostly lethal with surviving escapers showing neurobehavioral phenotypes. In summary, our results unveil a novel human neurodevelopmental disorder due to genetic loss of NAV2, highlighting a critical conserved role of the NAV2 gene in brain and cerebellar development across species.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
In humans, development of the cerebellum begins around the ninth week of gestation and continues postnatally following highly orchestrated processes that involve a series of complex morphogenic events [1]. These events are tightly regulated by intra- and extra-cellular molecular pathways, which promote and regulate neuronal proliferation, differentiation, and migration, resulting in the formation of a foliated and lobulated structure that plays a key role in motor and cognitive functions [2]. Acquired or genetic disruptions that impair the complex regulatory machinery of cerebellar development may result in a broad array of diverse congenital anomalies frequently associated with neurodevelopmental disorders (NDDs) [3]. Among these, cerebellar vermis hypoplasia is the most common, often representing a non-specific finding in a large proportion of individuals affected with intellectual disability [4]. In contrast, cerebellar dysplasia is very rare and is often part of a more complex brain malformation [5]. Despite the significant advances in our understanding of the molecular basis of cerebellar malformations, roughly half of individuals with cerebellar hypoplasia remain genetically undiagnosed [6].
The NAV2 gene (MIM: 607,026) encodes the Neuron Navigator 2 protein, a member of the Neuron Navigator protein family that is abundantly expressed in the developing central nervous system (CNS) [7, 8] and known to affect cytoskeletal dynamics [9]. NAV2 was first identified as an all-trans retinoic acid responsive gene in human neuroblastoma SH-SY5Y cell line [10, 11], having pivotal functions in neurite outgrowth and axon elongation [9]. Homologs to NAV2 are present in animal models such as Drosophila (sick) and Caenorhabditis elegans (unc-53) and are well known to regulate cell migration, neurite outgrowth, and axon elongation [12, 13]. Notably, transgene expression of the human full-length NAV2 was able to rescue the mechanosensory neuron axon elongation defects in the unc-53 mutant [9]. The critical role of NAV2 in neuronal migration and axon elongation has also been observed in the hypomorphic Nav2 mutant mice that display ataxia due to defects in the development of cerebellar vermis, characterized by reduced cerebellar granule cell migration and impaired axonal outgrowth [14]. Despite robust evidence underscoring the importance of NAV2 in CNS development and function in both vertebrate and invertebrate models, no patients have been reported with mutations in NAV2. To explore whether Neuron Navigator 2 affects human brain and cerebellum development and other phenotypes, we screened (whole or clinical) exome sequencing (ES) data from individuals affected with (sporadic) molecularly undetermined cerebellar dysplasia for de novo or biallelic variants in the NAV2 gene. This led to the identification of compound heterozygous truncating variants in NAV2 in a female individual affected with developmental delay and a complex brain malformation including vermian hypoplasia and cerebellar cortical dysplasia. Cellular studies in this patient fibroblasts showed decreased and aberrant NAV2 transcripts and proteins. Cell migration assays on patient cells link NAV2 deficiency to perturbed migration processes. The comparison of Nav2 hypomorphic mouse histopathology and patient neuroimaging features revealed strikingly overlapping brain and cerebellar findings in humans and mice. Moreover, the NAV2 ortholog in flies, sick, is required for proper mushroom body development as well as proper motor and neurobehavioral functions, thus unveiling a critical role of NAV2 for brain development across different species.
Materials and Methods
Discovery Cohort
We screened for biallelic and/or de novo variants in NAV2 genomic datasets part of the GIGA (Gaslini IIT Genomic Alliance) and the SYNaPS study group consortia (which are involved in the genetic investigation of rare undiagnosed pediatric neurodevelopmental disorders) that include ES data of about 30,000 families; furthermore, we interrogated publicly available databases, including DECIPHER (https://www.deciphergenomics.org/), LOVD (https://www.lovd.nl/), and the Matchmaker Exchange platform GeneMatcher[15]. Written informed consent for patients who underwent ES was obtained under protocols approved by local institutional review boards. We only included cases with detailed clinical phenotyping and available brain imaging. We also excluded cases with a prior genetic diagnosis or an established candidate disease-causing variant. No de novo variants were identified in NAV2 and a fully segregating biallelic variant was found in a patient that was genetically investigated at Baylor Genetics Laboratories and previously submitted to GeneMatcher. In this patient, no pathogenic or candidate variants in any of the known disease genes were found, and clinical trio ES led to the identification of compound heterozygous variants that should cause early truncations in the NAV2 transcripts/proteins that were initially classified of uncertain significance. Since this gene was not associated with human disease, the family was subsequently enrolled in the Undiagnosed Diseases Network (UDN) to further explore the molecular etiology of her symptoms.
Exome Sequencing and Variant Analysis
Clinical ES was performed on DNA isolated from peripheral blood of affected patients and their parents when available, as previously described [16, 17]. Sequencing data were processed using commercial tools for the execution of the GATK Best Practices pipeline for ES variant analysis. Exon-level read counts, removal of duplicate reads, mean coverage of coding sequence regions, alignment, and variant annotation were performed using analytical pipelines that include publicly available tools and custom scripts. We looked at non synonymous-exonic and splicing variants with a minor allele frequency ≤ 0.001 in gnomAD database. Validation, parental origin of the resulting variants, and family segregation studies were performed by Sanger sequencing. Variants were interpreted according to the ACMG criteria.
Histopathological Analysis of Mutant Nav2 Mice
To assess the role of NAV2 in both brain and cerebellar development, we conducted a detailed re-evaluation of the histopathological features of the Nav2 hypomorphic mutant mice that were previously published [14]. For this purpose, we analyzed cerebellum as well as all other brain regions in Nissl-stained Sects. (30 μm) from two litter matched pairs of wild-type and hypomorphic Nav2 mice at 8 weeks of age that had been backcrossed 20 times into a C57BL/6 background. All animal studies were performed under an approved IACUC animal protocol according to institutional guidelines at the University of Wisconsin-Madison.
Western Blot Analysis
The fibroblast cell lysates were prepared using lysis buffer (50 mM Tris–Cl pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 5% β-mercaptoethanol) supplemented with a protease inhibitor cocktail (Roche). Protein concentration of each sample was determined using the Bradford method (Bio-Rad). In total, 40 µg of total protein was denatured at 95 °C and separated on 4–15% Mini-PROTEAN gel (Bio-Rad). The gel was transferred on to a nitrocellulose membrane using Trans-Blot Turbo Transfer System and Trans-Blot Turbo Mini 0.2 µm Nitrocellulose Transfer Pack (Bio-Rad). NAV2 protein was detected using NAV2 Polyclonal Antibody (1:1000, PA5103968—ThermoFisher Scientific) and rabbit secondary antibody (1:10,000, Millipore). Clarity™ Western ECL Substrate (Bio-Rad) was used for the detection of the signals. Image was acquired by Uvitec Mini HD9 (Uvitec).
RT-PCR
Total RNA was extracted from fibroblast cells by using miRNeasy Mini Kit (QIAGEN) according to the manufacturer’s protocol. About 250 ng of total RNA was used to synthetize the first-strand cDNA using iScript cDNA Synthesis Kit (Bio-Rad). The gene expression levels were detected by using EvaGreen qPCR Mastermix (Bio-Rad) and performing real-time PCR on the CFX96 C1000 Touch Real-time PCR system (Bio-Rad) with the following PCR conditions: 98 °C for 30 s, followed by 39 cycles of 98 °C for 2 s and 60 °C for 5 s, and then heating from 70 to 95 °C with either 0.5 °C increments, 5 s/step. Primers for Nav2 (PrimerBank ID 350276221c1 forward: 5'-ACTGGGCCAATCATTACCTAGC-3', reverse: 5'-CGCCATCTGTCACATCTTGCT-3' and PrimerBank ID 350276221c3 forward: 5'-GGTCCTACCGCGAGGGTAT-3', reverse: 5'-TGGCTGCGTCGGTTGTTAG-3') were obtained from the public database PrimerBank (https://pga.mgh.harvard.edu/primerbank/). Differential expression was determined by the 2−DDCT method using GAPDH and ATP5F1 as the internal control.
For flies, RT-PCR was performed as previously described [18] with the following changes. All-In-One 5X RT MasterMix (abm #G592), iTaq Universal SYBR Green Master Mix (BioRad #1,725,120), and a BioRad C1000 Touch Cycler were used. Primers for sick: forward: 5'- CACAATTTCCGATGGGTGCTC-3', reverse: 5'- CCTCGGCCCAATGGTTACAT-3'; for rp49 a housekeeping gene: forward: 5'-TGTCCTTCCAGCTTCAAGATGACCATC-3', reverse: 5'-CTTGGGCTTGCGCATTTGTG-3'.
Wound Healing Assay
Fibroblasts were seeded in a culture-insert (ibidi culture-insert 2 well, IBIDI) at a density of 2 × 104 cells per well. After allowing the cells to attach and reach confluence, the culture-insert was removed and provided with fresh medium. Migration was documented by taking sequential digital photographs of the gap using an automated microscope (Nikon TiE). Wound area closure was then quantified with ImageJ software, by applying fine edges and sharpen processing tools and analyze particles tool.
Cell Morphology Analysis
To assess the morphology of cells, fibroblasts were fixed by adding 200 μl of 10% neutral buffered formalin (05‐01005Q, Bio‐Optica) for 5 min at room temperature. After three washings in phosphate‐buffered saline (PBS), cells were permeabilized with Triton X‐100 0.3% in PBS for 5 min, blocked with 1% BSA in PBS for 2 h and then incubated with 0.17 µM Alexa Fluor® 555 Phalloidin (ThermoFisher Scientific) in PBS + 1% BSA for 30 min. Cells were rinsed 3 × with PBS and mounted with Fluoroshield with 4′,6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich) to stain cell nuclei before imaging. Image acquisition was performed using a laser scanning confocal microscope Leica SP8 (Leica Microsystems). Image analysis was performed using Leica and ImageJ software to detect and count cells with filopodia-like cell protrusions. In total, 200–250 cells of the donor and controls were analyzed. Data from control cells were pooled together for statistical analysis.
Drosophila Immunostaining
Immunostaining of fly larval and adult brains was conducted as described [19]. In short, the dissected samples were fixed in 4% paraformaldehyde (PFA) followed by blocking in 0.2% PBST with 5% normal goat serum. Primary antibodies used: Mouse anti-Repo (DSHB: 8D12) 1:50, Rat anti-Elav (DSHB: 7E8A10) 1:500, Secondary antibodies used: Anti-Rat-647 (Jackson ImmunoResearch, 112–605-003) 1:1000, Anti-Mouse-488 (Jackson ImmunoResearch, 115–545-062) 1:1000. Samples were thoroughly washed with 0.2% PBST and mounted on a glass slide using Fluoromount-G (Southernbiotech, 0100–20). The samples were scanned using a laser confocal microscope (Zeiss LSM 880), and images were processed using ImageJ.
Drosophila Behavioral Assessment
Twelve-day-old flies were used for behavioral assessment. Climbing (negative geotaxis) assays were performed essentially as previously described [20]. Flies were transferred to a clean, empty vial and given 5–15 min to habituate before being tapped to the bottom of the vial and assessed for a negative geotaxis response. Climbing distances were measured at 15 cm in a given time (20 s as maximum). All flies were reared at 25 °C. Flies were transferred into a fresh vial every 3 days.
Heat-induced seizure assays were performed as previously described [21]. Flies were transferred to a clean vial and allowed to habituate for 5–15 min before the vial was immersed in a 42 °C water bath for 30 s. Seizures were defined as failure to maintain an upright posture combined with wing fluttering, leg twitching, and sometimes abdominal curling. The percentage of seizing flies at 30 s was calculated. After immersion for 30 s, the vial was taken out of the water to allow recovery. The recovery time of individual flies to an upright posture was measured.
Drosophila stocks
The following stocks were used in this study:
Fly line | Genotype | Source |
|---|---|---|
sickT2A−GAL4 | y1 w*; Mi{Trojan-GAL4.0}sickMI08398-TG4.0/SM6a | BDSC #76,195 |
sick-Df | w[1118]; Df(2L)ED1303, P{w[+ mW.Scer\FRT.hs3] = 3'.RS5 + 3.3'}ED1303/SM6a | BDSC #8679 |
UAS-mCherry.NLS | w[*];; P{w[+ mC] = UAS-mCherry.NLS}3 | BDSC #38,424 |
UAS-mCD8-RFP | w[*]; P{y[+ t7.7] w[+ mC] = 10XUAS-IVS-mCD8::RFP}attP40; | BDSC #32,219 |
Canton S | Wild type | Bellen Lab |
Data Presentation and Analysis
Statistical analysis was performed using GraphPad software (GraphPad Prism v9.0; GraphPad Software, USA). Data were presented as representative images or as mean ± SEM. A statistical analysis of data was performed with Student’s t-test and/or ANOVA.
Results
Identification of NAV2 Variants and Bioinformatic Analyses
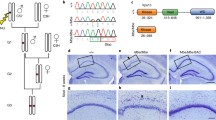
Stepwise filtering of ES analysis retained two compound heterozygous variants in NAV2 (NM_001244963.2): c.5179_5180delAG, p.(Leu1728Trpfs*2) and c.6757delA,p.(Ile2253*) in a female with developmental delay and a diagnosis of cerebellar hypoplasia and dysplasia. Sanger sequencing confirmed co-segregation of the variants with the disease within the family. The unaffected parents and a healthy sibling all carry one of the two variants (Fig. 1A). Both variants were absent from the gnomAD database (https://gnomad.broadinstitute.org) and classified as of unknown significance according to the ACMG criteria. Remarkably, NAV2 is a loss-of-function intolerant gene (pLI = 1) as no homozygous loss-of-function variants are reported in the gnomAD database (last accessed 05 May 2021; Supplemental Table 1) [22]. Furthermore, NAV2 is predicted to be potentially associated with a recessive condition according to a linear discriminant analysis (LDA) score of − 0.003 (< 0.5 corresponds to a “likely recessive” class) by the DOMINO algorithm [23]. Moreover, NAV2 is abundantly expressed in the human CNS, especially in the cerebellum (Fig. 1D), and it is conserved across different species (Fig. 1F), suggesting that loss-of-function in humans may have detrimental effects similar to the Drosophila, Caenorhabditis elegans, and mouse.

Clinical and genetic findings of the NAV2-related neurodevelopmental disorder in the family of the proband. (A) Pedigree of the family showing the affected individual (shaded). + represents the reference allele. (B) Craniofacial dysmorphism of the affected subject (at the age of 3 years left panel and 7 years right panel) including deep set eyes, upslanting palpebral fissures, bulbous nasal tip, thin upper lip, and dimple and broad chin. (C) Retinal fundus photograph of the right and left eyes illustrating aberrant retinal vasculature with a subclinical retinal detachment in the right eye and retinal neovascularization left eye. Ultra-widefield fluorescein angiography of the right and left eyes demonstrating retinal ischemia and retinal neovascularization (inset). (D) RNA-seq tissue data generated by the Genotype-Tissue Expression (GTEx) project and reported as mean pTPM (protein coding transcripts per million), corresponding to the values of the different individual samples for respective subregion. Cerebellum has the highest expression (pTPM 20.4). (E) Depictions of the pathogenic variants p.(L1728Wfs*2) and p.(Ile2253*) and protein domains (CH, calponin homology domain; cytoskeletal interacting domain or CSID; CC, coiled coil domain; AAA, AAA-ATPase domain; purple indicates poly-Proline, Serine, and Lysine regions). (F) NAV2 protein sequences of different species based on the constraint-based alignment tool COBALT. The red colour indicates highly conserved protein regions among species and blue indicates less conserved ones. Percent of identity (indicating the percentage of the orthologous sequence matching the Human sequence according to the Ensembl database) and the percent of similarlity have been calculated using the EMBOSS Needle tool
Interrogation of GeneMatcher and additional Matchmaker platforms for NAV2 failed to identify additional cases with biallelic loss-of-function variants and/or similar phenotypes. Finally, no biallelic loss-of-function variants in this gene were found in the genomic datasets of the 100 K Genome Project.
ES from the affected individual did not reveal pathogenic or likely pathogenic variants in any known disease gene or other gene. The biallelic loss-of-function variants in NAV2 therefore emerged as the most likely cause for the disease, given the severity of the biallelic variants (predicted to undergo nonsense mediated decay), and the known role of Neuron Navigator 2 across different species.
Clinical and Neuroradiological Features Associated with Biallelic NAV2 Variants
The affected individual was the second child to healthy and nonconsanguineous parents of Caucasian ancestry (Fig. 1A, II.2). Family history was unremarkable. Mother’s pregnancy was complicated by oligohydramnios and gestational diabetes treated with diet. Antenatal history was remarkable for frequent atrial ectopy, polyvalvular heart disease with atrioventricular valve regurgitation, and pericardial effusion in the fetus. She was treated with digoxin for suspected supraventricular tachycardia. She was born by cesarean section at 38 weeks gestation. Growth parameters at birth indicated weight 2915 g (− 0.96SDs), length 47.6 cm (− 1.21SDs), and occipital frontal circumference (OFC) 33 cm (− 1.19SDs). Postnatal echocardiogram showed a common atrium, an unicommisural aortic valve with mild stenosis, bicuspid pulmonary valve with mild stenosis, and mildly thickened tricuspid and mitral valves without stenosis. Head ultrasound showed thin corpus callosum with suspected vermian dysplasia. Brain MRI at birth showed marked hypoplasia of the corpus callosum with small cerebellar vermis and dysmorphic appearance of the pons. Renal ultrasound and EEG were unremarkable.

Neuroimaging features of the NAV2-related neurodevelopmental disorder in the proband. Brain MRI of the patient performed at 6 years of age (A–D) and of an age-matched control subject (E–F). A) Axial reformatted 3D T1-weighted images reveal abnormal cerebellar foliation fissuration, more pronounced at the level of the inferior cerebellar hemispheres (empty arrows). The pons is flattened and slightly asymmetric (dotted arrows). The superior cerebellar peduncles are thinned and splayed (arrows) with associated narrowing of the isthmic region (arrowhead) leading to a molar tooth-like appearance of the midbrain. Note the diffuse cortical dysgyria, with prevalent insular involvement (thick arrows), and the asymmetric enlargement of the lateral ventricles (asterisks). (B) Sagittal reformatted 3D T1-weighted image shows the hypoplasia and dysplasia with mild upward rotation of the cerebellar vermis, and prevalent involvement of the anterior and superior posterior lobes (empty arrows). There are also corpus callosum hypodysgenesis (arrow), agenesis of the anterior commissure (dotted arrow), narrow isthmus (arrowhead), and small pons (thick arrow). Note the small optic nerve chiasm. (C) Coronal T2-weighted image demonstrates the agenesis of the olfactory bulbs (arrows) and mild hypoplasia of the optic nerves (arrowheads). (D) Diffusion tensor imaging, axial color-coded fractional anisotropy (FA) maps reveal small asymmetric corticospinal tracts at the level of the pons (dotted arrows) and horizontal course of the superior cerebellar peduncles (arrows) with preservation of their decussation at the midbrain level (arrowhead). (E) Axial reformatted 3D T1-weighted images show the normal size and morphology of the cerebellar hemispheres. The cerebellar folia run parallel to the calvarium (onion-like configuration; thick arrows). (F) Axial color-coded FA maps, magnified view at 2 brainstem levels (middle pons and middle midbrain). Conventional color scheme: blue (inferior-superior), green (anteroposterior), and red (left–right). CST, corticospinal tract; MCP, middle cerebellar peduncles; SCP, superior cerebellar peduncles; SCPD, superior cerebellar peduncles’ decussation; SAF, somatosensory ascending fibers; TPF, transverse pontocerebellar fibers
At 4 weeks of life, she presented with cardiogenic shock requiring cardioversion and intubation. She had severe global ventricular dysfunction and atrial flutter. At 5 weeks of age, she underwent mitral and tricuspid valve repair, open aortic valvotomy, and pericardial patch closure of secundum atrial septal defect. Her initial ophthalmological evaluation indicated possible motor apraxia and mild optic atrophy. Brain MRI repeated at 3 and 6 years of age revealed marked cerebellar vermis hypoplasia, bilateral cerebellar foliation defects, pontine hypo-dysplasia, splayed thin superior cerebellar peduncles with a molar tooth-like configuration, corpus callosum hypodysgenesis, absent anterior commissure, diffuse dysgyria, agenesis of the olfactory bulbs, mild optic nerve hypoplasia, and enlarged dysmorphic lateral ventricles (Fig. 2). Cerebellar morphometry data of the patient were compared with normal values derived from an in-house database of controls [24]. The transverse cerebellar diameter (89 mm), the cranio-caudal diameter of the vermis (27.5 mm), and the antero-posterior diameter of the vermis (20 mm) were below 2SD compared with age-matched controls. The results of volumetric comparison of the cerebellum of the patient with an age-matched healthy subject performed with the SUIT toolbox of SPM 12 [25] are displayed in Fig. 3. She had significant motor delays; however, her cognitive functioning was assessed to be normal when assessed using Wechsler Preschool and Primary Scale of Intelligence (WPPSI-IV) at 3 years of age. She struggled with motor delays and, despite some improvements with physiotherapy and occupational therapy, continued to need assistance with most activities of daily living. Her formal developmental assessment at the age of 6 years and 8 months revealed gross and fine motor delay and both receptive and expressive language impairment. She subsequently developed decreased vision in both eyes and was found to have mild optic atrophy, atypical chorioretinal scarring, retinal ischemia, retinal neovascularization, and retinal detachment (Fig. 1C). She subsequently had panretinal photocoagulation laser of both eyes. Her physical examination at 7 years of age showed microcephaly with OFC of 48 cm (− 2.8 SDs) with normal weight and height. Mild dysmorphic features were seen including deep-set eyes, upslanting palpebral fissures, bulbous nasal tip, thin upper lip vermilion, and dimpled and broad chin (Fig. 1B). She also had hypotonia and impairment of voluntary, saccadic eye movements (Supplemental material Videos 1, 2).

Comparison of the cerebellar volumes of the patient with an age-matched healthy subject. (A–C) Segmentation of the cerebellar volumes of the patient (green maps) and of an aged-matched control subject (red maps) overlayed on sagittal (A), coronal (B), and axial-reformatted (C) 3DT1-weighted images. (D–F) Volumetric reconstructions of cerebellar segmentations of the patient (green cerebellum, D), of the control subject (red cerebellum, E), and of the fusion of both cerebellar volumes (F) overlayed on 3D T1-weighted images. Note that the volume of both the cerebellar hemispheres and vermis of the patient is smaller compared to the age-matched control
Histopathological Features of Nav2 Mutant Mice and Comparison with Human Phenotype
Analysis of the mouse model confirmed an overall reduction in cerebellar size, abnormal foliation in the I-V region along with impaired development of VIa and VIb/VII lobes (Fig. 4, lower panels, Supplemental Fig. 1D). Previously unreported abnormalities in other brain regions included thinning of the corpus callosum and a reduction in the size of the thalamus and hypothalamus. There were no major abnormalities of the cerebral cortex, brainstem, anterior commissure, and olfactory bulbs (Fig. 3). A detailed comparison between the human and mouse phenotype is summarized in Table 1 and depicted in Supplemental Fig. 1.

Brain and cerebellar abnormalities in the Nav2 hypomorphic mutant mice. Medial sagittal sections (panels a and b; panel b closest to midline) from a wild-type (WT) and homozygous hypomorph (HOM) show an overall reduction in cerebellar size, and a reduction in overall development of VIa,VIb/VII with absence of the intercrural fissue (arrowhead) in the HOM. In other brain regions, abnormalities noted in the HOM include thinning of the corpus callosum and a reduction in the size of the regions encompassing the thalamus/hypothalamus (Th/Hyp). The pons and medulla appear normal in size in both genotypes, with the pontine nucleus (PN) and inferior olive (IO) shown for reference. The cortex and tectum, anterior commissure (AC), and olfactory bulb showed no obvious dysmorphogenesis. Scale bar: 1 mm
NAV2 Deficiency Perturbs Cell Migration and Cytoskeleton Organization
We first assessed the expression of the mutant NAV2 by western blot analysis on patient and control fibroblasts. As expected, we found an intense band corresponding to a molecular weight of ~ 283 kDa in total lysates from four different control fibroblasts that was almost totally absent in lysates from patient fibroblasts (Fig. 5A, B). Some faint and one intense bands of lower molecular weights (< 55 kDa) appeared in patient lysates, most likely representing protein fragments resulting from degradation of the truncated NAV2 protein (Fig. 5A). This finding was consistent with the mRNA quantification by real-time RT-PCR that showed a significant reduction compared to controls, indicating that mutant NAV2 mRNA undergoes nonsense-mediated decay (Fig. 5C).

Cellular phenotype associated with the genetic loss of Neuron Navigator 2 in the NAV2 compound mutant patient. (A–C) Analysis of NAV2 protein and mRNA expression in fibroblasts obtained from 4 healthy individuals and the “NAV2 patient.” (A) Representative Western blot experiment showing immunodetection of NAV2 (top) and GAPDH (bottom) in the indicated fibroblasts lysates. (B) Bar graph showing the densitometric analysis of the upper band, corresponding to the full-length protein (2830 KDa); data are normalized for GAPDH expression. N = 3, *** p < 0.001 (ANOVA with Tukey’s post hoc test). (C) Bar graph showing NAV2 mRNA quantification by real time PCR. N = 3, ** p < 0.01 (ANOVA with Tukey’s post hoc test). (D and E) Analysis of cell migration by wound healing assay. Representative images (D) and analysis (E) of wounded areas of confluent fibroblasts at different time points. Wound edges, detected by image segmentation analysis, are outlined in green. N = 3; * p < 0.05, ** p < 0.01. (F) Representative image and summary graph showing results from cell morphology analysis. Fibroblasts were fixed, stained with phalloidin (red) and Hoechst 33,342 (blue) and analyzed by confocal microscopy. Scale bar = 10 µm. 200–250 cells/donor were analyzed. * p < 0.05
We next sought to assess the effect of this severe NAV2 deficiency on cell migration. In the wound healing assay, patient fibroblasts showed perturbed migration compared to controls, as demonstrated by the increased quantification of the wound area closure at different time points (Fig. 5D, E). In addition, we noticed that patient-derived fibroblasts also displayed a peculiar morphology compared to controls, characterized by the presence of several cell protrusions. Indeed, phalloidin staining and confocal microscopy imaging and analysis revealed an increased proportion of lateral filopodia-rich cells (Fig. 5F), a phenotype likely caused by abnormal cytoskeleton dynamics due to NAV2 genetic deficiency.
The Drosophila NAV2 ortholog, sick, is enriched in the developing and adult CNS and mutants are semi-lethal
To determine the role of NAV in development and neuronal function, we explored the phenotypes associated with loss of the NAV orthologue in flies. The fruit fly has a single Neuron Navigator gene called sick, orthologous to all three NAV(1/2/3) family members in humans. Based on multiple orthology prediction26, sick is most orthologous to NAV2 (DIOPT score is 9/16, Supplemental Fig. 2A). Moreover, both the CH and AAA domains are highly conserved in both Sick and NAV2 (Supplemental Fig. 2B). We previously generated sick mutants as part of the Gene Disruption Project27. This allele, sickT2A−GAL4, was generated by introduction of an artificial exon containing a splice acceptor-T2A-GAL4-polyA cassette between exons 10 and 11 and is predicted to act as strong loss-of-function mutation because of the presence of a poly-A tail (Fig. 6A, Supplemental Fig. 2C). The sickT2A−GAL4 allele also produces a GAL4 in the same spatial and temporal pattern as the sick gene reflecting the endogenous expression. The sickT2A−GAL4 mutants are homozygous lethal and but are semi-lethal (only 19.2% of flies eclose) when in trans to deficiency, Df(2L)ED1303 (Fig. 6B), suggesting that the sickT2A−GAL4 chromosome may carry a modifier. In the sickT2A−GAL4/ Df flies, the transcript levels are decreased to 15.2% of wild-type controls (Canton S) (Supplemental Fig. 2 D).

The NAV2 ortholog in Drosophila, sick, is expressed in brain and mutants are semi-lethal with motor defects and heat-sensitive seizure-like behavior. (A) Schematics of sickT2A−GAL4 acting as a gene trap: the insertion of SA-T2A-GAL4-polyA cassette leads to generation of truncated Sick and expression of GAL4 under the control of the regulatory sequences of the sick. (B) Complementation tests of sickT2A−GAL4 with a corresponding deficiency (Df(2L)ED1303). (C–E) Gene expression of sick based on sickT2A−GAL4; UAS-mCD8::RFP flies. Whole third instar larva (C), third instar larval brain (D), and adult brain (E) are shown. Note expression in the mushroom bodies (upper panel, dashed lines), olfactory glomeruli (upper panel, dashed ellipse), and antennal mechanosensory and motor center (upper panel, dashed ellipse). Scale bars = 100 µm. (F) Climbing assessment of sickT2A−GAL4/Df flies in a negative geotaxis assay reveals motor deficits in sick mutants that are 12 days old. N are shown within the bars. Unpaired t tests, **** p < 0.0001. (G) sick mutants display heat-induced seizures in a 42 °C water bath (30 s). (H) Time to recover. N are shown within the bars. Unpaired t tests, *** p < 0.001, **** p < 0.0001
In order to determine the expression pattern of sick, we generated sickT2A−GAL4; UAS-mCherry.NLS flies. The GAL4 drives the expression of nuclear mCherry. Third instar larvae exhibit robust expression of the reporter in the CNS with sparse expression in the trachea (Fig. 6C). To reveal the projections of the neurons that express sick, we crossed the sickT2A−GAL4 mutants to a membrane bound reporter (UAS-mCD8::RFP). In the third instar larvae, sick is enriched in motor neurons of the ventral nerve cord as well as the mushroom body neurons (Fig. 6D). In the adult brain, sick is widely expressed, but it shows notable enrichment in the optic lobes, mushroom body, and antennal mechanosensory and motor center of the fly brain (Fig. 6E). To determine if sick is restricted to neurons or is also present in the glia, we crossed the sickT2A−GAL4 mutants to UAS-mCherry.NLS and examined third instar larva brain and adult brains that are co-stained with nuclear markers for neurons (Elav) and glia (Repo). As shown in Supplemental Fig. 3, sick is expressed in both neurons and glia in the developing CNS (Supplemental Fig. 3A) and adult brain (Supplemental Fig.3 B).
Surviving Sick Mutants Show Climbing and Heat-Induced Seizures
A previous study documented that sick mutants exhibit axonal growth defects during development [13], but nothing was reported in adult flies. To determine the functional consequences of loss of sick in adult flies, we conducted neurobehavioral assessments. We found that the climbing ability of 12-day-old sickT2A−GAL4/Df flies is severely impaired compared to control flies (Fig. 6F). These data indicate that the motor function of sick mutant flies is impaired.
Hyperthermia increases the intrinsic excitability of both excitatory and inhibitory neurons, and the dysregulated synaptic activity can induce febrile seizures [21, 26, 27]. To assess if loss of sick also leads to altered synaptic activity, we performed heat induced seizures by subjecting flies to 42 °C for 30 s [21]. We observed that sickT2A−GAL4/Df flies exhibit a rapid onset of seizure like behavior (Supplemental material Videos 3, 4). Over 75% of sickT2A−GAL4/Df flies exhibit heat induced seizures and they require about 15 s to recover once returned to room temperature (Fig. 6G, H). Hence, the data indicate dysregulated synaptic activity in sick mutants.
Discussion
In this study, we report biallelic truncating variants in NAV2 associated with a novel human neurodevelopmental phenotype characterized by vermis hypoplasia and cerebellar cortical dysplasia as well as other brain malformation. The NAV2 gene encodes multiple transcripts and proteins based on alternate promoter usage and splicing. The NAV2 transcript variant 5 encodes the largest isoform of 2488 amino acids. Full-length NAV2 proteins contain several putative functional domains, including a calponin-homology (CH) domain at the N-terminus, several coiled-coil regions, a cytoskeletal interacting domain (or CSID), and an ATP/GTP nucleotide-binding site (AAA-domain) at the C-terminus (Fig. 1E). We functionally investigated the impact of compound heterozygous variants identified in this study [NM_001244963.2: p.(L1728Wfs*2) and p.(Ile2253*)] on a cellular level, and showed that the variants cause an almost total absence of mRNA production and protein expression in patient-derived fibroblasts, suggesting that they cause a severe loss of function.
The brain and cerebellar phenotype reported in this study is partially overlapping to the one observed in the Nav2 mouse model. Indeed, hypomorphic mutant mice lacking the full-length Nav2 transcript exhibit ataxia with reduced volume and abnormal foliation of the vermis mostly affecting folia VI-VII [14]. Nav2 hypomorphic mutants show the same cerebellar malformations after 20 backcrosses as described previously by McNeill et al. (2011). In both the earlier and present study, hypoplasia of folia VI-VII and loss of the intercrural fissure were found in all Nav2 mutants and none of the wild-type controls (Fig. 3, Supplementary Fig. 1 and McNeill et al., 2011). In the earlier study, an effect on anterior folia was observed in 67% of Nav2 mutants but was not observed in wild-type mice. The malformations seen in the Nav2 mutant are distinct from spontaneous malformations in the cerebellar vermis of wild-type C57BL/6 mice which largely affect folia VIII-IX [28, 29]. Similar to the Nav2 mutant mouse, our patient displays hypoplasia and dysplasia of the vermis with prevalent involvement of the anterior and superior posterior lobes. In Nav2 mutant mice, there is a delay in the disappearance of the external germinal layer resulting from impaired granule cell migration toward the interior of the cerebellum during development. The inability of granule cells to extend neurites/parallel axon fibers and migrate properly was observed in cultured explants and dissociated granule cell cultures from mutants [14]. This finding could explain the abnormal foliation observed in the patient, suggesting that abnormal granule cell migration and axonal outgrowth defects lead to cerebellar cortical dysplasia.
Overall, the cerebellar features due to NAV2 genetic deficiency are further corroborated by our comparison between the histopathological analysis of the mutant mice and the neuroimaging features of our patient, underscoring an impairment of cerebellar development predominantly affecting the vermis and the correct folia orientation. Moreover, the presence of oculomotor apraxia in the patient suggests disruption of cerebro-cerebellar circuits that are crucial for the control of voluntary eye movements. This is not surprising given the broad expression of NAV2 across the entire CNS [8] and its role in the development of other brain structures such as cranial nerves [30]. A novel finding emerged from the re-evaluation of the mouse brain histopathological features including corpus callosum hypodysgenesis similar to the patient; this suggests migration and axon elongation defects of midline neuronal circuits/networks that are affected by NAV2. Interestingly, the patient also displayed mild optic nerve hypoplasia and agenesis of the olfactory bulbs and both optic nerve hypoplasia and impaired integrity of olfactory sensory systems have also been observed in the hypomorphic Nav2 mutant mouse [31]. Moreover, it is known that retinal ganglion cell axons guide the formation of an astrocytic network that in turn dictate the pattern of developing retinal vasculature [32, 33]. Therefore, the aberrant retinal vasculature found in the patient may also be related to inappropriate ganglion cell migration during retinal development leading to subsequent retinal neovascularization and detachment later in life.
The wound healing assay in patient fibroblast cultures revealed a cellular migration defect. This is consistent with the increased staining of filopodia observed with confocal microscopy suggesting an alteration of cytoskeletal architecture that affects migration process. Filopodia are thin membrane protrusions that sense the extracellular environment at their tips using cell surface receptors and promote retrograde flow of actin upon binding with external targets, eventually leading to different cell migration processes such as wound healing and neurite outgrowth [34]. However, substantial differences in terms of migration dynamics and regulatory networks occur between fibroblasts and cerebellar granule cells [35].
It is worth mentioning that impaired proliferation may account for wound healing defect [36]. Since we did not specifically look at proliferation in our wound healing assay, we cannot rule out a possible contribution of abnormal proliferation for the reduced size of the cerebellum as previously suggested [14].
NAV2 may act by facilitating interactions between microtubules and other proteins such as neurofilaments that are key players in the formation and stability of growing neurites, and the aberrant cytoskeleton architecture derived from the loss of NAV2 might lead to secondary microtubule dysfunction [9]. This hypothesis is supported by several imaging features observed in the context of NAV2 deficiency that can be also found in individuals affected with rare tubulinopathies, such as cerebellar dysplasia with foliation defects, brainstem abnormalities, diffuse cortical dysgyria, olfactory bulb agenesis, and asymmetric lateral ventricles [37]. Importantly, only the absence of the characteristic basal ganglia anomalies differentiates these two conditions. This feature might be explained by the different expression profile of the NAV2 in the CNS. Indeed, brain regions with the most abundant expression included the developing cortex, hippocampus, thalamus, olfactory bulb, and granule cells of the cerebellum. In contrast, expression of NAV2 in basal ganglia and white matter expression was largely undetectable [8]. Noteworthy, cerebellar cortical dysplasia is also found in a few other NDDs that are frequently autosomal recessive, such as Joubert syndrome and related disorders (JSRDs), Poretti–Boltshauser syndrome, muscular dystrophy (dystroglycanopathies), and Chudley-McCullough syndrome [4, 5]. In these conditions, the association with other medical issues and additional brainstem and/or cerebral imaging features may help the differential diagnosis, as presence of hearing loss, callosal agenesis, fronto-mesial polymicrogyria, and intracranial cysts in Chudley–McCullough syndrome or the molar tooth sign and progressive retinal, kidney, and liver disease in JSRDs.
Studies of the NAV2 ortholog, sick, in Drosophila show that it is present in both the mushroom body and the antennal lobes as well as some other neurons. The mushroom body is widely known as the learning and memory center of the flies[38], whereas the antennal lobes integrate olfactory information [39]. A previous study showed that Sick is required for axonal growth of mushroom body neurons and that its loss caused defective mushroom body and ellipsoid bodies [13]. It is worth noticing that the core architecture of mushroom body circuit is strikingly similar to that of the vertebrate cerebellum [40, 41]. Moreover, the sick mutants display motor deficits indicating Sick function is important for proper motor output in flies. Taken together, these findings further underscore the conserved function of NAV2/Sick in brain and cerebellar development throughout several species.
While the affected subject did not display seizure, the sick mutant developed heat-induced seizure. Since the homeostatic plasticity of the brain requires proper wiring of circuits containing excitatory and inhibitory neurons, dysregulated synaptic activity can cause susceptibility to febrile seizures [21, 26, 27]. Specifically in the sick mutant flies, the inhibitory GABAergic neurons in the mushroom body may not reach their target neurons to inhibit their excitabilities properly [13, 42]. This may be one of the reasons why they are vulnerable to heat-induced seizures. Further reports of affected subjects will elucidate whether epilepsy may be part of the NAV2-related phenotypic spectrum.
Interestingly, the finding of congenital heart defects in the affected individual reported in this study may indicate a potential role of NAV2 also in the cytoskeleton dynamics of cardiac myocytes. This may be consistent with the observation of wide migration defects in the Caenorhabditis elegans unc-53 mutant, with abnormal developing myoblasts and excretory cells in addition to cells from the ventral nerve cord45. Notably, congenital anomalies beyond the CNS have not been assessed before in the hypomorphic Nav2 mouse model. However, additional (unidentified) genetic causes underlying the congenital heart defects in our patient cannot be excluded, as clinical ES would not cover intronic variants or complex genomic rearrangement.
Conclusion
In conclusion, we report a novel neurodevelopmental disorder characterized by severe impairment of brain and cerebellar development and found its association with biallelic loss-of-function variants in NAV2. The neuroradiological phenotype of the affected individual is characterized by a complex brain malformation with a peculiar combination of cerebellar and brainstem malformations including vermian hypoplasia, extensive foliation defects, pontine hypo-dysplasia, and splayed thin superior cerebellar peduncles with a molar tooth-like configuration. Through functional analyses in human cells, the Nav2 hypomorphic mouse, and the NAV2 ortholog in flies (sick), our study highlights a potentially conserved role of NAV2 in regulating neuronal migration and CNS development across different species. The identification of other affected individuals carrying biallelic variants in NAV2 will be fundamental to confirm the implication of this gene in the phenotype we observed and to characterize the clinical spectrum of the disease.
Consortia
Members of The Undiagnosed Diseases Network (UDN) and SYNaPS are listed below.
Members of the Undiagnosed Diseases Network
Mahshid S. Azamian21, Carlos A. Bacino21, Ashok Balasubramanyam21, Lindsay C. Burrage21, Hsiao-Tuan Chao21, Gary D. Clark21, William J. Craigen21, Hongzheng Dai21, Fariha Jamal21,
Lefkothea Karaviti21, Shamika Ketkar21, Brendan H. Lee21, Richard A. Lewis21, Ronit Marom21, Paolo M. Moretti21, Sarah K. Nicholas21, James P. Orengo21, Jennifer E. Posey21, Lorraine Potocki21, Daryl A. Scott21, Alyssa A. Tran21, Tiphanie P. Vogel21, Monika Weisz Hubshman21, Kim Worley21, Michael F. Wangler21, Shinya Yamamoto21, Christine M. Eng21, Pengfei Liu21, Patricia A. Ward21, Edward Behrens22, Kosuke Izumi22, Marni Falk22, Kelly Hassey22, Kathleen Sullivan22, Anna Raper22, Gonench Kilich22, Zhe Zhang22, Adeline Vanderver22, David B. Goldstein23, Heidi Cope24, Allyn McConkie-Rosell24, Kelly Schoch24, Vandana Shashi24, Edward C. Smith24, Rebecca C. Spillmann24, Jennifer A. Sullivan24, Queenie K.-G. Tan24, Nicole M. Walley24,
Pankaj B. Agrawal25, Alan H. 25, Gerard T. Berry25, LaurenC. Briere25, Laurel A. Cobban25, Matthew Coggins25, Cynthia M. Cooper25, Elizabeth L. Fieg25, Frances High25, Ingrid A. Holm25, Susan Korrick25, Joel B. Krier25, Sharyn A. Lincoln25, Joseph Loscalzo25, Richard L. Maas25, Calum A. MacRae25, J. Carl Pallais25, Deepak A. Rao25, Lance H. Rodan25, Edwin K. Silverman25, Joan M. Stoler25, David A. Sweetser25, Melissa Walker25, Chris A. Walsh25, Cecilia Esteves25, Isaac S. Kohane25, Kimberly LeBlanc25, Alexa T. McCray25, Shilpa N. Kobren25, Amelia L. M. Tan25, Rachel Mahoney25, Surendra Dasari26, Brendan C.Lanpher26, Ian R. Lanza26, Eva Morava26, Devin Oglesbee26, Guney Bademci27, Deborah Barbouth27, Stephanie Bivona27, Olveen Carrasquillo27, Ta Chen Pete Chang27, Irman Forghani27, Alana Grajewski27, Rosario Isasi27, Byron Lam, 27 Roy Levitt27, Xue Zhong Liu, Jacob McCauley27, Ralph Sacco27, Mario Saporta27, Judy Schaechter27, Mustafa Tekin27, Fred Telischi27, Willa Thorson27, Stephan Zuchner27, Heather A. Colley28, Jyoti G. Dayal28, David J. Eckstein28, Laurie C. Findley28, Donna M. Krasnewich28, Laura A. Mamounas28, Teri A. Manolio28, John J. Mulvihill28, Grace L. LaMoure28, Madison P. Goldrich28, Tiina K. Urv28, Argenia L. Doss28, Maria T. Acosta28, Carsten Bonnenmann28, Precilla D'Souza28, David D. Draper28, Carlos Ferreira28, Rena A. Godfreyv, Catherine A. Groden28, Ellen F. Macnamara28, Valerie V. Maduro, 28 Thomas C. Markello28, Avi Nath28, Donna Novacic28, Barbara N. Pusey28, Camilo Toro28, ColleenE. Wahl28, Eva Baker28, Elizabeth A. Burke28, David R. Adams, William A. Gahl28, May Christine V. Malicdan28, Cynthia J. Tifft28, Lynne A. Wolfe28, John Yang28, Bradley Power28, Bernadette Gochuico28, Laryssa Huryn28, Lea Latham28, Joie Davis28, Deborah Mosbrook-Davis28, Francis Rossignol28, Ben Solomon28, John MacDowall28, Audrey Thurm28, Wadih Zein28, Muhammad Yousef28, Margaret Adam29, Laura Amendola29, Michael Bamshad29, Anita Beck29, Jimmy Bennett29, Beverly Berg-Rood29, Elizabeth Blue29, Brenna Boyd29, PeterByers29, Sirisak Chanprasert29, Michae Cunningham29, Katrina Dipple29, Daniel Doherty29, Dawn Earl, Ian Glass29, Katie Golden-Grant29, Sihoun Hahn29, Anne Hing, Fuki M. Hisama29, Martha Horike-Pyne29, Gail P. Jarvik29, Jeffrey Jarvik29, Suman Jayadev29, Christina Lam29, Kenneth Maravilla29, Heather Mefford29, J. Lawrence Merritt29, Ghayda Mirzaa29, Deborah Nickerson29, Wendy Raskind29, Natalie Rosenwasser29, C. RonScott29, Angela Sun29, Virginia Sybert29, Stephanie Wallace29, Mark Wener29, Tara Wenger29, Euan A. Ashley30, Gill Bejerano30, Jonathan A. Bernstein30, Devon Bonner30, Terra R. Coakley30, Liliana Fernandez30, Paul G. Fisher30, Jason Hom30, Yong Huang30, Jennefer N.Kohler30, ElijahKravets30, Beth A. Martin30, Shruti Marwaha30, Archana N. Raja30, Chloe M. Reuter30, Maura Ruzhnikov30, Jacinda B. Sampson30, Kevin S. Smith30, Shirley Sutton30, Holly K. Tabor30, Brianna M Tucker30, Matthew T. Wheeler30, Diane B. Zastrow30, Chunli Zhao30, William E. Byrd31, Andrew B. Crouse31, Matthew Might31, Mariko Nakano-Okuno31, Jordan Whitlock31, Gabrielle Brown32, Manish J. Butte32, Esteban C. Dell'Angelica32, Naghmeh Dorrani32, Emilie D. Douine32, Brent L. Fogel32, Irma Gutierrez32, Alden Huang32, Deborah Krakow32, Hane Lee32, Sandra K. Loo32, Bryan C. Mak32, Martin G. Martin32, Julian A.Martínez-Agosto32, Elisabeth McGee32, Stanley F. Nelson32, Shirley Nieves-Rodriguez32, Christina G.S. Palmer32, Jeanette C. Papp32, Neil H. Parker32, Genecee Renteria32, Rebecca H. Signer32, Janet S. Sinsheimer32, Jijun Wan32, Lee-kai Wang32, Katherine Wesseling Perry32, Jeremy D. Woods32, Justin Alvey33, Ashley Andrews33, Jim Bale33, John Bohnsack33, Lorenzo Botto33, John Carey33, Laura Pace33, Nicola Longo33, Gabor Marth33, Paolo Moretti33, Aaron Quinlan33, Matt Velinder33, Dave Viskochil33, Pinar Bayrak-Toydemir33, Rong Mao33, Monte Westerfield34, Anna Bican35, Elly Brokamp35, Laura Duncan35, Rizwan Hamid35, Jennifer Kennedy35, Mary Kozuira35, John H. Newman35, John A. Phillips III35, Lynette Rives35, Amy K. Robertson35, Emily Solem35, Joy D.Cogan35, F. Sessions Cole36, Nichole Hayes36, Dana Kiley36, Kathy Sisco36, Jennifer Wambach36, Daniel Wegner36, Dustin Baldridge36, Stephen Pak36, Timothy Schedl36, Jimann Shin36, Lilianna Solnica-Krezel36.
Study Group Members Affiliations – Undiagnosed Disease Network
21 Baylor College of Medicine, TX, TX 77,030, USA.
22 Children’s Hospital of Philadelphia, PA 19,104, USA.
23 Columbia University, NY, NY 10,027, USA.
24 Duke University, NC 27,708, USA.
25 Harvard University, Cambridge, MA, USA.
26 Mayo Clinic (Rochester, MN), USA.
27 University of Miami, FL 33,146, USA.
28 National Institutes of Health, MD 20,814, USA.
29 University of Washington and/or Seattle Children’s Hospital, WA 98,195, USA.
30 Stanford University, CA 94,305, USA.
31 University of Alabama Birmingham, AL 35,294, USA.
32 University of California, Los Angeles (UCLA), CA 90,095, USA.
33 University of Utah, UT 84,112, USA.
34 University of Oregon, OR 97,403, USA.
35 Vanderbilt University, TN 37,235, USA.
36 Washington University, St. Louis, MO 63,130, USA.
The Syn aptopathies a nd P aroxysmal S yndromes (SYNaPS) Study Group ( http://neurogenetics.co.uk/synaptopathies-synaps/ ).
Study Group Members:
Stephanie Efthymiou17, Reza Maroofian17, Mhammed Aguennouz19, Yamna Kriouile37, Mohamed El Khorassani38, Blagovesta Karashova38, Daniela Avdjieva38, Hadil Kathom38, Radka Tincheva38, Lionel Van Maldergem39, Wolfgang Nachbauer40, Sylvia Boesch40, Larissa Arning41, Dagmar Timmann42, Vincenzo Leuzzi43, Belen Pérez-Dueñas44, Gabriella Di Rosa45, Erica Pironti45, Jatinder S. Goraya46, Tipu Sultan47, Salman Kirmani48, Shahnaz Ibrahim49, Farida Jan49, Jun Mine50, Selina Banu51, Pierangelo Veggiotti52, Michel D. Ferrari53, Arn M J M van den Maagdenberg53, Alberto Verrotti54, Gian Luigi Marseglia55, Salvatore Savasta55, Gian Vincenzo Zuccotti56, Carmela Scuderi57, Eugenia Borgione57, Valeria Dipasquale58, Maria Concetta Cutrupi58, Rosaria Nardello59, Benigno Monteagudo Sanchez60, Mercedes Pineda-Marfa’61, Francina Munell61, Alfons Macaya61, Richard Boles62, Gali Heimer63, Salvatore Leonardi64, Martino Ruggieri64, Rosalba De-Sarro19, Nancy Malintan17, Oscar D. Bello17, Maria Natalia Zanetti17, Henry Houlden17.
Study Group Members Affiliations—SYNaPS
17 Department of Neuromuscular Diseases, UCL Institute of Neurology, London WC1N 3BG, UK.
37 Children's Hospital of Rabat, University of Rabat, Rabat 6527, Morocco.
38 Department of Paediatrics, Medical University of Sofia, Sofia 1431, Bulgaria.
39 Centre of Human Genetics, University Hospital Liege, Liege 4000, Belgium.
40 Department of Neurology, Medical University Innsbruck, Anichstrasse 35, Innsbruck 6020, Austria.
41 Department of Human Genetics, Ruhr-University Bochum, Bochum 44,801, Germany.
42 Braun Neurologische Universitätsklinik Universität Essen, Hufelandstr 55, Essen D-45122, Germany.
43 Department of Human Neuroscience, Unit of Child Neurology and Psychiatry, University La Sapienza, 00,185, Rome, Italy.
44 Hospital Sant Joan de Deu, Esplugues de Llobregat 08,950, Barcelona, Spain.
45 Department of Pediatrics, University of Messina, Messina 98,123, Italy.
46 Division of Paediatric Neurology, Dayanand Medical College & Hospital, Ludhiana, Punjab 141,001, India.
47 Department of Paediatric Neurology, Children's Hospital of Lahore, Lahore 381-D/2, Pakistan.
48 Department of Medical Genetics, Aga Khan University Hospital, Karachi, Karachi City, Sindh 74,800, Pakistan.
49 Department of Paediatric Neurology, Aga Khan University Hospital, Karachi, Karachi City, Sindh 74,800, Pakistan.
50 Department of Pediatrics, Shimane University School of Medicine, 89‐1 Enya, Izumo, Shimane 693‐8501, Japan.
51 Institute of Child Health and Shishu Shastho Foundation Hospital, Mirpur, Dhaka 1216, Bangladesh.
52 Pediatric Neurology Unit, V. Buzzi Children's Hospital, Via Castelvetro 32, 20,154 Milan, Italy.
53 Leiden University Medical Center, Albinusdreef 2, Leiden 2333, Netherlands.
54 Paediatric Department, San Salvatore Hospital, University of L'Aquila, L'Aquila, Italy.
55 Department of Pediatrics, University of Pavia, IRCCS Policlinico "San Matteo", Pavia 27,100, Italy.
56 Department of Biomedical and Clinical Sciences "L. Sacco", University of Milan; Paediatric Clinical Research Centre Fondazione "Romeo ed Enrica Invernizzi", University of Milano; Department of Paediatrics, Children's Hospital "V. Buzzi", Milano, Italy.
57 Laboratorio di Neuropatologia Clinica, U.O.S. Malattie, Neuromuscolari Associazione OASI Maria SS. ONLUS – IRCCS, Via Conte Ruggero 73, 94,018 Troina, Italy.
58 Department of Pediatrics, University Hospital “Gaetano Martino”, University of Messina, Messina 98,123, Italy.
59 Child Neuropsychiatry Unit, University of Palermo, Italy.
60 Hospital Arquitecto Marcide, Avenida de la Residencia S/N, Ferrol (A Coruña), 15,401 Spain.
61 Neuropediatrics Unit, University Hospital Vall d'Hebron, Barcelona 08,035, Spain.
62 Courtagen Life Sciences, 12 Gill Street Suite 3700, Woburn, MA 01,801 USA.
63 Division of Pediatric Neurology, Edmond and Lily Children's Hospital, Chaim Sheba Medical Center, 52,621 Ramat Gan, Israel.
64 Department of Clinical and Experimental Medicine, University of Catania, Catania, Italy.
Data availability
Additional information regarding the genetic and functional studies are available upon request to the corresponding author.
References
Hibi M, Shimizu T. Development of the cerebellum and cerebellar neural circuits. Dev Neurobiol. 2012;72(3):282–301. https://doi.org/10.1002/dneu.20875.
Koziol LF, Budding D, Andreasen N, D’Arrigo S, Bulgheroni S, Imamizu H, et al. Consensus paper: the cerebellum’s role in movement and cognition. Cerebellum. 2014;13(1):151–77. https://doi.org/10.1007/s12311-013-0511-x.
Pinchefsky EF, Accogli A, Shevell MI, Saint-Martin C, Srour M. Developmental outcomes in children with congenital cerebellar malformations. Dev Med Child Neurol. 2019;61(3):350–8. https://doi.org/10.1111/dmcn.14059.
Accogli A, Addour-Boudrahem N, Srour M. Diagnostic Approach to Cerebellar Hypoplasia. Cerebellum. 2021;20(4):631–58. https://doi.org/10.1007/s12311-020-01224-5.
Severino M, Huisman TAGM. Posterior fossa malformations. Neuroimaging Clin N Am. 2019;29(3):367–83. https://doi.org/10.1016/j.nic.2019.03.008.
Aldinger KA, Timms AE, Thomson Z, Mirzaa GM, Bennett JT, Rosenberg AB, et al. Redefining the etiologic landscape of cerebellar malformations. Am J Hum Genet. 2019;105(3):606–15. https://doi.org/10.1016/j.ajhg.2019.07.019.
Maes T, Barceló A, Buesa C. Neuron navigator: a human gene family with homology to unc-53, a cell guidance gene from Caenorhabditis elegans. Genomics. 2002;80(1):21–30. https://doi.org/10.1006/geno.2002.6799.
Pook C, Ahrens JM, Clagett-Dame M. Expression pattern of Nav2 in the murine CNS with development. Gene Expr Patterns. 2020;35:119099. https://doi.org/10.1016/j.gep.2020.119099.
Muley PD, McNeill EM, Marzinke MA, Knobel KM, Barr MM, Clagett-Dame M. The atRA-responsive gene neuron navigator 2 functions in neurite outgrowth and axonal elongation. Dev Neurobiol. 2008;68:1441–53. https://doi.org/10.1002/dneu.20670.
Merrill RA, Plum LA, Kaiser ME, Clagett-Dame M. A mammalian homolog of unc-53 is regulated by all-trans retinoic acid in neuroblastoma cells and embryos. Proc Natl Acad Sci U S A. 2002;99(6):3422–7. https://doi.org/10.1073/pnas.052017399.
Clagett-Dame M, McNeill EM. Muley PD Role of all-trans retinoic acid in neurite outgrowth and axonal elongation. J Neurobiol. 2006;66(7):739–56. https://doi.org/10.1002/neu.20241.
Hedgecock EM, Culotti JG, Hall DH, Stern BD. Genetics of cell and axon migrations in Caenorhabditis elegans. Development. 1987;100(3):365–82.
Abe T, Yamazaki D, Murakami S, Hiroi M, Nitta Y, Maeyama Y, Tabata T. The NAV2 homolog Sickie regulates F-actin-mediated axonal growth in Drosophila mushroom body neurons via the non-canonical Rac-Cofilin pathway. Development. 2014;141(24):4716–28. https://doi.org/10.1242/dev.113308.
McNeill EM, Klöckner-Bormann M, Roesler EC, Talton LE, Moechars D, Clagett-Dame M. Nav2 hypomorphic mutant mice are ataxic and exhibit abnormalities in cerebellar development. Dev Biol. 2011;353(2):331–43. https://doi.org/10.1016/j.ydbio.2011.03.008.
Azzariti DR, Hamosh A. Genomic data sharing for novel Mendelian disease gene discovery: the matchmaker exchange. Annu Rev Genomics Hum Genet. 2020;21:305–26. https://doi.org/10.1146/annurev-genom-083118-014915.
Yang Y, Muzny DM, Xia F, Niu Z, Person R, Ding Y, et al. Molecular findings among patients referred for clinical whole-exome sequencing. JAMA. 2014;312(18):1870–9. https://doi.org/10.1001/jama.2014.14601.
Iacomino M, Baldassari S, Tochigi Y, Kośla K, Buffelli F, Torella A, et al. Loss of Wwox perturbs neuronal migration and impairs early cortical development. Front Neurosci. 2020;14:644. https://doi.org/10.3389/fnins.2020.00644.
Goodman LD, Cope H, Nil Z, Ravenscroft TA, Charng WL, Lu S, et al. TNPO2 variants associate with human developmental delays, neurologic deficits, and dysmorphic features and alter TNPO2 activity in Drosophila. Am J Hum Genet. 2021;108(9):1669–91. https://doi.org/10.1016/j.ajhg.2021.06.019.
Dutta D, Briere LC, Kanca O, Marcogliese PC, Walker MA, High FA, et al. De novo mutations in TOMM70, a receptor of the mitochondrial import translocase, cause neurological impairment. Hum Mol Genet. 2020;29:1568–79. https://doi.org/10.1093/hmg/ddaa081.
Madabattula ST, Strautman JC, Bysice AM, O’Sullivan JA, Androschuk A, Rosenfelt C, et al. Quantitative analysis of climbing defects in a Drosophila model of neurodegenerative disorders. J Vis Exp. 2015;100:e52741. https://doi.org/10.3791/52741.
Sun L, Gilligan J, Staber C, Schutte RJ, Nguyen V, O’Dowd DK, Reenan R. A knock-in model of human epilepsy in Drosophila reveals a novel cellular mechanism associated with heat-induced seizure. J Neurosci. 2012;32(41):14145–55. https://doi.org/10.1523/JNEUROSCI.2932-12.2012.
Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–91. https://doi.org/10.1038/nature19057.
Quinodoz M, Royer-Bertrand B, Cisarova K, Di Gioia SA, Superti-Furga A, Rivolta C. DOMINO: Using Machine Learning to Predict Genes Associated with Dominant Disorders. Am J Hum Genet. 2017;101(4):623–9. https://doi.org/10.1016/j.ajhg.2017.09.001.
Severino M, Tortora D, Pistorio A, Ramenghi LA, Napoli F, Mancardi MM, et al. Expanding the spectrum of congenital anomalies of the diencephalic-mesencephalic junction. Neuroradiology. 2016;58(1):33–44. https://doi.org/10.1007/s00234-015-1601-x.
Diedrichsen, J. A spatially unbiased atlas template of the human cerebellum. 2006. Neuroimage, 33, 1, p. 127–138. doi: https://doi.org/10.1016/j.neuroimage.2009.01.045.
Kim JA, Connors BW. High temperatures alter physiological properties of pyramidal cells and inhibitory interneurons in hippocampus. Front Cell Neurosci. 2012;6:1–12. https://doi.org/10.3389/fncel.2012.00027.
Warner TA, Liu Z, Macdonald RL, Kang JQ. Heat induced temperature dysregulation and seizures in Dravet syndrome/GEFS+ Gabrg2+/Q390X mice. Epilepsy Res. 2017;134:1–8. https://doi.org/10.1016/j.eplepsyres.2017.04.020.
Mangaru Z, Salem E, Sherman M, Van Dine SE, Bhambri A, Brumberg JC, et al. Neuronal migration defect of the developing cerebellar vermis in substrains of C57BL/6 mice: cytoarchitecture and prevalence of molecular layer heterotopia. Dev Neurosci. 2013;35(1):28–39. https://doi.org/10.1159/000346368.
Van Dine SE, Siu NY, Toia A, et al. Cuoco JA1, Betz AJ, Bolivar VJ, Spontaneous malformations of the cerebellar vermis prevalence inheritance and relationship to lobule/fissure organization in the C57BL/6 lineage. Neuroscience. 2015;310:242–51. https://doi.org/10.1016/j.neuroscience.2015.09.025.
McNeill EM, Roos KP, Moechars D, Clagett-Dame M. Nav2 is necessary for cranial nerve development and blood pressure regulation. Neural Dev. 2010;5:6. https://doi.org/10.1186/1749-8104-5-6.
Peeters PJ, Baker A, Goris I, Daneels G, Verhasselt P, Luyten WHML, et al. Sensory deficits in mice hypomorphic for a mammalian homologue of unc-53. Brain Res Dev Brain Res. 2004;150(2):89–101. https://doi.org/10.1016/j.devbrainres.2004.03.004.
Paisley CE, Kay JN. Seeing stars: development and function of retinal astrocytes. Dev Biol. 2021;478:144–54. https://doi.org/10.1016/j.ydbio.2021.07.007.
O’Sullivan ML, Puñal VM, Kerstein PC, Brzezinski JA 4th, Glaser T, Wright KM, Kay JN. Astrocytes follow ganglion cell axons to establish an angiogenic template during retinal development. Glia. 2017;65(10):1697–716. https://doi.org/10.1002/glia.23189.
Lundquist EA. The Finer Points of Filopodia. PLoS Biol. 2009;7(6):e1000142. https://doi.org/10.1371/journal.pbio.1000142.
Cooper JA. Cell biology in neuroscience: mechanisms of cell migration in the nervous system. J Cell Biol. 201; 202(5):725–34 https://doi.org/10.1083/jcb.201305021
Latifi-Pupovci H, Kuçi Z, Wehner S, Bönig H, Lieberz R, Klingebiel T, Bader P, Kuçi S. In vitro migration and proliferation (“wound healing”) potential of mesenchymal stromal cells generated from human CD271(+) bone marrow mononuclear cells. J Transl Med. 2015;13:315. https://doi.org/10.1186/s12967-015-0676-9.
Mutch CA, Poduri A, Sahin M, Barry B, Walsh CA, Barkovich AJ. Disorders of Microtubule Function in Neurons: Imaging Correlates. AJNR Am J Neuroradiol. 2016;37(3):528–35. https://doi.org/10.3174/ajnr.A4552.
McGuire SE, Le PT, Davis RL. The role of Drosophila mushroom body signaling in olfactory memory Science. 2001; 293(5533):1330–3. https://doi.org/10.1126/science.1062622
Hsu CT, Bhandawat V. Organization of descending neurons in Drosophila melanogaster. Sci Rep. 2016;6:20259. https://doi.org/10.1038/srep20259.
Farris SM. Are mushroom bodies cerebellum-like structures? Arthropod Struct Dev. 2011;40(4):368–79. https://doi.org/10.1016/j.asd.2011.02.004.
Li F, Lindsey JW, Marin EC, Otto N, Dreher M, Dempsey G, et al. The connectome of the adult Drosophila mushroom body provides insights into function. Elife. 2020;9:e62576. https://doi.org/10.7554/eLife.62576.
Apostolopoulou AA, Lin AC. Mechanisms underlying homeostatic plasticity in the Drosophila mushroom body in vivo. Proc Natl Acad Sci USA. 2020;117:16606–15. https://doi.org/10.1073/pnas.1921294117.
Wang J, Liu Z, Bellen HJ, Yamamoto S. Navigating MARRVEL, a Web-based tool that integrates human genomics and model organism genetics information. J Vis Exp. 2019;150:e59542. https://doi.org/10.3791/59542.
Lee P-T, Zirin J, Kanca O, Lin W-W, Schulze KL, Li-Kroeger D, et al. A gene-specific T2A-GAL4 library for Drosophila. Elife. 2018;7:e35574. https://doi.org/10.7554/eLife.35574.
Stringham E, Pujol N, Vandekerckhove J, Bogaert T. unc-53 controls longitudinal migration in C elegans. Development. 2002;129(14):3367–79.
Acknowledgements
We thank the study family for their enthusiastic participation.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary file7 (MP4 5117 KB)
Supplementary file8 (MP4 9747 KB)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Accogli, A., Lu, S., Musante, I. et al. Loss of Neuron Navigator 2 Impairs Brain and Cerebellar Development. Cerebellum 22, 206–222 (2023). https://doi.org/10.1007/s12311-022-01379-3
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12311-022-01379-3