
Abstract
The influence of the microenvironment through the various steps of cancer progression is signed by different cytokines and growth factors, that could directly affect cell proliferation and survival, either in cancer and stromal cells. In colon cancer progression, the cooperation between hypoxia, IL-6 and VEGF-A165 could regulate the DNA repair capacity of the cell, whose impairment is the first step of colon cancer development. This cooperation redirects the activity of proteins involved in the metabolic shift and cell death, affecting the cell fate. The pathways triggered by micro environmental factors could modulate cancer-related gene transcription, affecting also small non coding mRNA, microRNAs. MicroRNAs have emerged as key post-transcriptional regulators of gene expression, directly involved in human cancers. The present review will focus first on the intertwined connection between cancer microenvironment and aberrant expression of microRNAs which contribute to carcinogenesis. In particular, the epigenetic mechanisms triggered by tissue microenvironment will be discussed, in view of the recent identification of miRNAs able to directly or indirectly modulate the epigenetic machinery (epi-miRNAs) and that are involved in the epithelial to mesenchimal transition and metastases development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colon cancer develops as a result of the pathologic transformation of normal colonic epithelium to adenomatous polyp, which ultimately leads to invasive cancer [1–3]. Tumor induction and progression are characterized by accumulation of multiple genetic and epigenetic alterations, that confer a selective reproductive advantage to a clone, within a genetically unstable heterogeneous cell population [2, 4, 5]. The survival and the expansion of the neoplastic cell clone are supported by the surrounding tissue that sustains and favours these conditions [6, 7]. Tumor-associated stroma actively fuels the colon cancer progression [6]. Among the tumor-associated cells are endothelial cells and pericytes that form the neo-vasculature. In turn, the newly formed angiogenic blood vessels supply tumor cells with nutrients and oxygen. In addition, mesenchymal cells are present in the stroma at earlier tumor stages, suggesting a co-evolution between stromal and cancer cells that leads to clonal expansion and metastasis [7]. Chronic inflammation, such as inflammatory bowel disease may predispose to malignancy and tumor progression. The chronic inflammatory microenvironment consists of immune, inflammatory and stromal cells, all of which produce cytokines, growth factors and adhesion molecules that may sustain tumor growth, its progression and spreading [8, 9].
The cytokines and growth factors produced by cancer cells function to create optimal growth conditions within the tumor microenvironment, while cytokines secreted by stromal cells may influence the behaviour of malignant cells. Pro-inflammatory cytokines (IL-6, IL-1) [10–12], growth factors (such as VEGF, TGF-β-1, -2, -3) and their cognate receptors [13–16] either in cancer and in stromal cells, influence not only primary gene transcription, but activate several pathways that cooperate to the aberrant clone survival, tumor expansion and metastasis [6, 10, 12]. In addition, alteration of microenvironmental factors induced by hypoxia, represents an hallmark of cancer progression [17, 18].
In this review, we discuss the role played by miRNA in colon cancer initiation and progression, especially in the context of the synergism existing among hypoxic condition, expression of IL-6 pro-inflammatory cytokine and up-regulation of VEGFA165 [19]. We report that this cooperation could act on the neoplastic cell, also by the dysregulation of miRNAs involved in colon cancer progression relevant pathways. Both the over-expression and the silencing of specific miRNAs are associated with the development and progression of colorectal cancer [20].
Recent studies have identified specific miRNAs present in colorectal cancer tissues and blood that may aid the diagnosis of cancer and help to predict disease recurrence [20–22]. Such a role in oncogenesis suggested that miRNAs could represent important targets for gene therapies. Although little is known about the exogenous factors that interfere with the regulation of miRNA expression, the microenvironment seems to play an essential role, affecting the epigenetic mechanisms that modulate miRNA gene expression, including methylation, histone deacetylation, or influencing the function of proteins involved in the maturation processing of microRNAs [23, 24]. The present review will focus on the peculiar relationship between colon cancer cells and microenvironment, that could influence miRNAs dysregulation, the apoptosis escaping and the metabolic shift, leading to invasion and metastasis.
miRNA Dysregulation and Histone Hypo-acetylation: A “Must” of the Metastatic Cell
MicroRNAs (miRNAs) are ~22-nt small noncoding RNAs that are processed from larger (80-nt) precursor hairpins by the RNase III enzyme Dicer into miRNA:miRNA duplexes. One strand of these duplexes associates with the RNAinduced silencing complex (RISC), whereas the other is generally degraded. The miRNA–RISC complex targets messenger RNAs triggering either translational repression or mRNA degradation [25]. The total number of miRNAs that are encoded in the human genome remains to be clarified. Initial estimates suggested there were up to 255 human miRNAs, but cloning and bioinformatic analyses have demonstrated that there are numerous nonconserved human miRNAs and suggest this number may be significantly larger [26]. Thousands of mammalian messenger RNAs are under selective pressure to maintain nucleotide sites matching microRNAs. Usually conserved targets are often highly expressed at developmental stages, before miRNAs expression and their levels tend to fall as the miRNA that targets them begins to accumulate. The phenomenon of the selective avoidance extended to thousand of genes enables temporal and tissue specific miRNA expression pattern, playing an important role in developmental timing [27]. It seems that the acquired differentiation and specialization is profoundly influenced by the impact of microRNA on mRNA repression and evolution. Overall, in a well differentiated tissue miRNAs are destabilizing many target messages to define tissue-specific transcript profile, but other targets could be modulated at translational level, without mRNA degradation. In that regard, changes of microenvironmental factors (as seen during cancer development) could accurately modulate protein expression patterns, acting on miRNA expression. A primary consequence in the transition from differentiated normal to de-differentiated neoplastic cell is the down modulation of miRNAs, involved in tissue specific differentiation. In humans, aberrant expression of miRNAs contributes to carcinogenesis by promoting the expression of proto-oncogenes or by inhibiting the expression of tumor suppressor genes. Such oncomirs have been demonstrated in a variety of haematological and solid tumors [28] and they contribute to cancer development and progression [20–22]. Recently, the altered miRNAs expression in colon cancer has been reported (Table 1). However, the role that miRNAs play in the development of metastasis is more poorly defined [29]. It is worth of note that the same miRNAs could exert tumor suppressor or oncogenic effects, depending on the tumoral context and the stage of neoplastic disease. Recently, Arndt et al. [67] have shown that the re-expression of miR-143 or miR-145 leads to tumor suppressor and oncogenic phenotypes respectively, in a metastatic CRC model. In particular miR-145, reported as a ‘tumor suppressor’ in a non-metastatic context, showed oncogenic effects associated with the down-regulation of the G1/S cell cycle checkpoint and neuregulin pathways in the CRC metastatic setting.
The cancer microenvironment could contribute to miRNAs dysregulation affecting the epigenetic mechanisms (such as DNA methylation or histone acetylation) that regulate miRNA expression or influencing the maturation process of microRNAs [23, 24, 30].
Cancer Microenvironment and Development of Metastases: The Role of VEGF-A165a and IL-6
As previously reported [31], DNA repair failure and cell survival represent the first step in colon cancer expansion. The pro-inflammatory cytokine IL-6 seems to play a role in the inactivation of DNA repair mechanisms and Bax-dependent apoptosis. Although data on the relationship between IL-6 production and tumor progression are still conflicting, recent studies suggest a pathogenetic function of the complexes formed between IL-6 and its soluble receptor (sIL-6R), in colon carcinoma. It seems that an increased formation of IL-6-sIL-6R complexes, able to interact with gp130 on the cell membrane (trans-signaling), leads to an increased expression and nuclear translocation of STAT3, which can cause the induction of anti-apoptotic genes, such as the Bax antagonist Bcl-xL protein [10, 32, 33]. Moreover, it has been observed that in critical conditions (hypoxia, glucose deprivation, oxidative stress), the activation of STAT3, together with increased levels of HIF1α (triggered by the hypoxic condition, 1% pO2) influences the preferential expression of VEGF-A165a isoform [17, 18, 33], that could lead to the inhibition of programmed cell death inducing anti-apoptotic Bcl-2 protein. Two different splicing isoforms of VEGF165 have been identified, exerting pro- or antiangiogenic actions respectively and whose formation is strictly controlled by micro-environmental factors [34]. The production of pro- or anti angiogenic forms of VEGF depends upon splice site choice in the C-terminal. Proxymal splice site selection in exon 8 generates pro-angiogenic isoforms such as VEGF-A165a, and distal splice site selection results in the antagonistic anti-angiogenic isoforms the VEGF-A165b. Cellular choice on splice site selection, strongly influenced by microenviromental factors, depends upon the activity of RNA-binding splice factors, such as ASF/SF2. Recent studies showed a nuclear and cytoplasmic localization of ASF/SF2 splicing factor and correlated the cytoplasmic localization to the inhibition of the proangiogenic form and to a strong increase of the anti-angiogenic isoform b [34, 35].
In colon cancer, it seems reasonable that the cooperative interaction between IL-6 and increased levels of HIF1α favours the shift versus the VEGF-A165 pro-angiogenic isoforms, all these factors contributing ultimately to tumor cell proliferation, apoptotic escaping and cell migration. Along these lines, we found an increase of IL-6 protein levels in human colon cancer tissues and the increase was positively correlated with the tumor stage [36]. IL-6, released as by the tumor itself as by tumor associated macrophages (TAMs), together with IL-6-induced VEGF-A, could influence tumor cell survival interfering with the Ku-CLU-Bax physical interactions. In a colon cancer progression model, we had previously demonstrated that IL-6 increased the expression of s-Clusterin (sCLU), a multifaceted protein strongly involved in cell survival, apoptosis escaping and metastasis [36–39]. In vitro experiments have showed that the Ku-CLU-Bax interactions are modulated by IL-6, and IL-6 together with VEGF-A165 inhibit Bax-dependent cell death increasing the production of the clusterin pro-survival form (sCLU), finally shifting death into survival. In a moderately differentiated colon cancer cell line IL-6 down-modulated Bax expression at mRNA level. Concomitantly, IL-6 exposure influenced Bax at protein level acting on Bax-Ku70-sCLU physical interactions in the cytoplasm. In particular, it affected the acetylation and phosphorylation state of Ku70 [36]. The in vitro treatment with IL-6 and VEGF-A165 modulated the expression of genes involved in tumor invasion and apoptosis, as observed by microarray analysis [36]. In particular, microarray data showed that IL-6 could influence cell proliferation, not only by inducing AP1 formation, but also through the up regulation of RASp21 protein activator 1, myc and NFκB. On the other hand, IL-6 down-modulated the expression of genes involved in the inhibition of metastasis, such as TIMP1 metallopeptidase inhibitor 1 and KISS1 [40, 41]. These still unclear molecular interactions, underline the relevant role of the microenvironmental factors in the complicated cross talk among molecules that could effectively turn the cell fate, by a fine regulation of gene expression, also acting on miRNA expression.
VEGF-A165a and IL-6: A Microenvironment Cooperation Leading to Metastasis Through MicroRNA Dysregulation
Changes of the microenvironment mediators induced by hypoxia, represent an hallmark of cancer progression. In the tumoral context, the transcriptional regulator hypoxia-inducible factor (HIF1α), cooperates with IL-6, TGF-β and VEGF-A165 in the promotion of tumoral growth [10, 15, 17–19].
The activation of hypoxia fosters neo-angiogenesis also regulating the expression of miR-210, and its target Ephrin-A3, involved in tube formation and survival of endothelial cells [42]. A group of candidate miRNAs regulated by hypoxia have been recently identified: miR-16, 20let-7b, miR-17-5p, miR-27, miR-106, miR-107 miR-193, miR-210, miR-320 and miR-361, showing VEGF as potential target [43, 73]. Moreover, using a bioinformatic prediction program, several key genes of proliferation, metabolism, migration and apoptotic response (miR-23, miR-26, miR-27, miR-30, miR-181) were found to be potentially targets of hypoxia-regulated miRNAs [57, 58]. In the colon cancer context, the synergism among hypoxic condition (HIF-1α activation) and IL-6 pro-inflammatory cytokine induces the up-regulation of VEGF-A165, contributing to neoangiogenesis and metastasis formation.
In colon cancer cells (Caco-2), we observed that the synergism of IL-6 and VEGF-A165 influenced the expression of tumor suppressor miRNAs, involved in epigenetic control and epithelial to mesenchymal transition (EMT), which strongly correlated to the malignization of many types of cancers [6]. In particular, the in vitro co-treatment with IL-6 and VEGF-A165 significantly down modulated miR-619, which targets VEGF gene expression determining a positive feed-back loop, finally contributing to cancer growth and spreading [44]. In addition, a significant decrease of miR-200 (12-folds decrease) was found, as compared to untreated colon cancer cells. The target prediction program indicates that highly conserved binding sites for the miRNA200 family are present in the ZEB1 and SIP1 mRNAs, which are repressors of ecadherin, implicated in EMT transition and tumor metastasis. The same down-regulation of miR-200 was obtained in response to TGF-β treatment and this effect was sufficient to induce EMT, in a process requiring up-regulation of ZEB1 and/or SIP1 [45]. In addition, Adam et al. [46] demonstrated that miR-200 controls the sensitivity to the EGFR therapy, which represents an actual issue in the treatment of human highly aggressive and metastatic colorectal cancer. In fact the expression of the miR-200 is sufficient to restore the EGFR dependency in bladder cancer cells, targeting ERRFI-1 which is a novel regulator of EGFR-independent growth.
In the last years, great attention has been given to the development of target anti-cancer therapies, based on the knowledge of transcriptional mechanisms epigenetically regulated in tumoral tissues. Moreover, it has been recently delineated a reciprocal interconnection between microRNAs and epigenetics [30]. The epigenetic modifications, such as DNA methylation or histone acetylation, are able to affect miRNAs expression causing oncomirs dysregulation, but the same microRNA (epi-miRNAs) may directly control the epigenetic machinery targeting its enzymatic components [30].
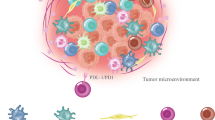
Evidence of the epigenetic influence of the microenvironment on tumoral cells has been observed by co-treating colon cancer cells with IL-6 and VEGF-A165. A strong down-modulation of the growth suppressing miR-449, which targets histone deacetylase 1 molecule (HDAC-1) [47] has been found in our system. This enzyme plays a crucial role in the epigenetic control and display high significance in oncology [48, 49]. Aberrant HDAC level and/or sustained activity has been found in various types of cancer and metastasis [50]. It has known that recruitment and activation of HDACs results in tightening of chromatin structure, thus the transcriptional machinery is prevented from accessing the DNA. Hence, cytokines and growth factors in the microenvironment could modulate gene transcription at epigenetic level, affecting epi-miRNA (as miR-449 [47]), or influencing the activity of protein involved in the regulation of HDAC activity (Fig. 1).

Cancer- stroma interactions in colon cancer progression and spreading. In colon cancer the synergistic action of IL-6, and VEGF-A165a promotes the acquisition of aggressiveness regulating the DNA repair capacity of the cell, whose impairment is the first step of colon cancer development. The cooperative action of these soluble mediators could modulate cancer-related gene transcription (sClusterin, TIMP1), and affect the expression of small non coding mRNA (microRNAs), able to modulate the epigenetic machinery (epi-miRNAs; miR-449) and to induce the epithelial to mesenchimal transition (EMT induction; miR-200 family)
miRNAs Expression and Altered Metabolism in Cancer
The altered metabolism of tumor cells may be a potential means by which these cells evade programmed cell death, favoring survival and tumoral growth. In particular, the metabolism of fatty acids is markedly altered in the tumoral context. It has been demonstrated that the extracellular acidosis in the microenvironment of solid tumors can work in an epigenetic fashion by up-regulating the transcriptional expression of fatty acids synthase (FASN) gene, proposed as the “metabolic oncogene” of cancer cells [51].
Recent reports demonstrated the existence of miRNAs able to recognize and modulate the transcriptional levels of metabolic factors, relevant both in non neoplastic and in cancer cell. In particular, transfection experiments with sense and antisense sequences displayed that the increase of lipogenesis in HepG2 cells (hepatocarcinoma cell line) is directly controlled by miR-122 [52]. This miRNA sequence is up-regulated by miR-370, the last therefore indirectly involved in the upregulation of lipogenic genes. Interestingly the same miR-370 targets the 3′UTR of CPT1, down-regulating the expression of CPT1A gene and the rate of β-oxidation [52].
Carnitine palmitoyl transferase 1 (CPT1) is another limiting factor in the fatty acids metabolism and it is contra-regulated by FASN substrate malonyl-CoA [53]. CPT1 resides physiologically at the mitochondrial membrane transporting long-chain fatty acids for β-oxidation. Recently, a CPT1A transcript splice variant, termed variant 2 (NM_001031847), was identified in colon cancer and breast cancer cells and it was undetectable in the corresponding non neoplastic cell line [53]. This variant codifies for a protein which differs in only 11 aminoacids from CPT1A variant 1, at the C-terminus. The preferential cellular localization of the two CPT1A variants was identified, being the product of transcript variant 2 mainly localized in the nucleus of tumoral cells.
The two transcript variants of CPT1A show great similarity differing only at the 3′-UTR level, where the recognition with miR-370 is located [52]. A different effectiveness of miR-370 in the target recognition could be responsible for the different levels of CPT1A transcripts found in cancer cell, as compared with non neoplastic cell. In fact, miR-370 could preferentially target CPT1 isoform 1 and indirectly favour CPT1 isoform 2, localized in the nucleus of cancer cells.
Experiments performed on human colorectal and breast tumoral tissues and cancer cell lines (Caco-2, HepG2 and MCF-7 cells) showed that the nuclear localization of CPT1A correlated to an increased HDAC activity and a resulting decrease of histone acetylation [53]. Moreover, HDAC-1 protein co-precipitated with CPT1A.
We have evidence of a different binding affinity of the two CPT1A variants for HDAC1. These data are based on the dissimilar CPT1 residues at the interface with HDAC1, for the two complexes. In the case of isoform 2 the interaction with HDAC1 involved a region close to the C-terminus, where the differences with CPT1 isoform 1 are located. Considering the great similarity of the other regions of CPT1 isoforms, this evidence suggested a higher stability of the complex CPT1 isoform2/HDAC1 (unpublished data). HDAC complexes are recruited to specific genomic sites by regulatory proteins. Additional data are needed to confirm if CPT1 (and in particular the nuclear isoform) could function as a novel partner of HDAC-1, stabilizing HDAC complexes at acetylated-histone tails of specific promoters, to silence tumor suppressor genes involved in the control of cancer cell growth and migration.
Altered Metabolism and Cancer Microenvironment
Experimental evidence suggest that, in tumoral context, the modulation of the metabolic factors could be driven by miRNA target recognition [52], but also through the activation of membrane receptors signalling, such as EGF/EGFR family, induced by microenvironmental stimuli [51, 54].
The human epidermal growth factor receptor type 1 (EGFR/HER-1) gene encodes a membrane receptor protein in the epidermal growth factor receptor family (ERBBs) and evidence suggests that EGFR expression and its activation status are associated with hyper-proliferation of colon cancer cells. Increased expression of EGFR is independently associated with poor disease-free survival after adjustment for stage, histological grade, age, and DNA mismatch repair status [55]. Epidermal growth factor and the EGF receptors (known as ERBB1) and ERBB2 have been shown to stimulate fatty acids synthase (FASN), the major enzyme required for the synthesis of fatty acids, although the ultimate mechanisms responsible for tumour-associated FASN overexpression are not completely understood. The effects of growth factors and growth factors receptors on FASN are complex and involve activation and/or cross-talk between multiple signal-transduction pathways [56] The EGFR activation found in colorectal cancer [55] could induce an up-regulation of FASN, determining the down-regulation of CPT1A activity in mitochondria [53].
Overall, in the neoplastic cells the gradient of microenvironmental factors (including EGF/EGFR activation) induces the overexpression of FASN and the resulting inhibition of fatty acids β-oxidation. Thus, a differential expression of CPT1A isoforms, driven by miRNA target recognition [52], could favour the CPT1 nuclear variant that evidence suggest to be implicated in the epigenetic regulation [53].
In conclusion, the metabolic factors altered in cancer could directly regulate the expression of genes implicated in tumor development and metastases, or modulate at epigenetic level the expression of small non coding RNAs. Besides, microRNAs have been identified (and many others remain to be uncovered) that target metabolic factors modulating their expression. Once more this regulatory loop seems to be driven by microenvironmental factors.
Conclusion and Perspectives
Invasion and metastasis are the deadly face of malignant tumors. Considering the high rate of incidence and mortality of colorectal cancer, it is critical to determine the mechanisms of its dissemination. The outcome of this non random process depends, in part, on the interaction of unique tumor cells with a compatible organ microenvironment. On the other hand, overexpression and silencing of specific miRNAs are associated with the development and progression of colorectal cancer. It is worth of note that miRNA dysregulation could play opposite role in the early stages of colon cancer, as compared to a metastatic context. As reported above, the pathway analyses may explain the observed oncogenic effects of miR-145 in metastatic CRC, compared to its reported tumor suppressor effects in the non-metastatic context [67].
The present review attempts to shed light on the fine relationship between altered gene expression profiles implicated in colon cancer progression and regulatory miRNAs, induced by pro-inflammatory partners of tumor microenvironment. Furthermore, in the neoplastic cells the gradient of microenvironmental factors induces the alteration of metabolic enzymes, such as FASN and CPT1A, which act on the epigenome modulating miRNA expression. The other side of the coin is represented by the identification of miRNAs which target the same metabolic factors, modulating their expression levels. Nowadays, the identification of specific miRNAs directly involved in cancer spread and metastasis may signify novel diagnostic tool in the characterization of gene targets.
Abbreviations
- CPT1:
-
Carnitine palmitoyl transferase 1
- CRC:
-
Colorectal cancer
- DNA DSBs:
-
DNA double strand breaks
- EGF:
-
Epidermal growth factor
- EGFR:
-
Epidermal growth factor receptor
- EMT:
-
Epithelial-mesenchymal transition
- FASN:
-
Fatty acids synthase
- HDAC:
-
Histone deacetylase
- HIF-1:
-
Hypoxia inducible factor-1
- HR:
-
Homologous recombination
- IL-1:
-
Interleukin-1
- IL-6:
-
Interleukin-6
- miRNAs:
-
Micro RNAs
- NHEJ:
-
Non homologous end joining
- s-CLU:
-
Secreted clusterin
- sIL-6R:
-
IL-6 soluble receptor
- TAMs:
-
Tumor associated macrophages
- TGF-β:
-
Transforming growth factor-beta
- TIMP-1:
-
Tissue inhibitor of metalloproteinase 1
- VEGF:
-
Vascular endothelia growth factor
References
Jass JR (2006) Colorectal cancer: a multipathway disease. Crit Rev Oncog 12:273–287
Finlay GJ (1993) Genetics, molecular biology and colorectal cancer. Mutat Res 290:3–12
Levin B, Lieberman DA, McFarland B, Smith RA, Brooks D, Andrews KS, Dash C, Giardiello FM, Glick S, Levin TR, Pickhardt P, Rex DK, Thorson A, Winawer SJ (2008) Screening and surveillance for the early detection of colorectal cancer and adenomatous polyps, 2008: a joint guideline from the American Cancer Society, the US Multi-Society Task Force on Colorectal Cancer, and the American College of Radiology. CA Cancer J Clin 58:130–160
Liu B, Farrington SM, Petersen GM, Hamilton SR, Parsons R, Papadopoulos N, Fujiwara T, Jen J, Kinzler KW, Wyllie AH, Vogelstein B, Dunlop MG et al (1995) Genetic instability occurs in the majority of young patients with colorectal cancer. Nat Med 1:348–352
Bousserouel S, Kauntz H, Gossé F, Bouhadjar M, Soler L, Marescaux J, Raul F (2010) Identification of gene expression profiles correlated to tumor progression in a preclinical model of colon carcinogenesis. Int J Oncol 36(6):1485–1490
De Wever O, Mareel MJ (2003) Role of tissue stroma in cancer cell invasion. J Pathol 200:429–447
Bates RC, Mercurio AM (2005) The epithelial-mesenchymal transition (EMT) and colorectal cancer progression. Cancer Biol Ther 4:365–370
Itzkowitz SH (2006) Molecular biology of dysplasia and cancer in inflammatory bowel disease. Gastroenterol Clin North Am 35:553–571
Harpaz N, Polydorides AD (2010) Colorectal dysplasia in chronic inflammatory bowel disease: pathology, clinical implications, and pathogenesis. Arch Pathol Lab Med 134:876–895
Scheller J, Ohnesorge N, Rose-John S (2006) Interleukin-6 trans-signalling in chronic inflammation and cancer. Scand J Immunol 63:321–329
Keller ET, Wanagat J, Ershler WB (1996) Molecular and cellular biology of interleukin-6 and its receptor. Front Biosci 1:340–357
Lewis AM, Varghese S, Xu H, Alexander HR (2006) Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med 4:48
Massaguè J (2008) TGFβ in cancer. Cell 134:215–230
Tian M, Schiemann WP (2009) The TGF-beta paradox in human cancer: an update. Future Oncol 5:259–271
Soufla G, Porichis F, Sourvinos G, Vassilaros S, Spandidos DA, De Wever O, Mareel M (2006) Transcriptional deregulation of VEGF, FGF2, TGF-beta1, 2, 3 and cognate receptors in breast tumorigenesis. Role of tissue stroma in cancer cell invasion. Cancer Lett 235:100–113
Bhowmick NA, Chytil A, Plieth D, Gorska AE, Dumont N, Shappell S, Washington MK, Neilson EG, Moses HL (2004) TGF-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 303:848–851
Semenza GL (2003) Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3:721–732
Lucarini G, Zizzi A, Belvederesi L, Kyriakidou K, Mazzucchelli R, Biagini G (2007) Increased VEGF165 expression in HCT116 colon cancer cells after transient transfection with a GFP vector encoding HIF-1 gene. J Exp Clin Cancer Res 26:515–519
Tokunaga T, Oshika Y, Abe Y, Ozeki Y, Sadahiro S, Kijima H, Tsuchida T, Yamazaki H, Ueyama Y, Tamaoki N, Nakamura M (1998) Vascular endothelial growth factor (VEGF) mRNA isoform expression pattern is correlated with liver metastasis and poor prognosis in colon cancer. Br J Cancer 77:998–1002
Faber C, Kirchner T, Hlubek F (2009) The impact of microRNAs on colorectal cancer. Virchows Arch 454:359–367
Heneghan HM, Miller N, Kerin MJ (2010) Systemic microRNAs: novel biomarkers for colorectal and other cancers? Gut 59:1002–1004
Valeri N, Croce CM, Fabbri M (2009) Pathogenetic and clinical relevance of microRNAs in colorectal cancer. Cancer Genomics Proteomics 6(4):195–204
Bandrés E, Agirre X, Bitarte N, Ramirez N, Zarate R, Roman-Gomez J, Prosper F, Garcia-Foncillas J (2009) Epigenetic regulation of microRNA expression in colorectal cancer. Int J Cancer 125:2737–2743
Balaguer F, Link A, Lozano JJ, Cuatrecasas M, Nagasaka T, Boland CR, Goel A (2010) Epigenetic silencing of miR-137 is an early event in colorectal carcinogenesis. Cancer Res 70(16):6609–6618
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, Sharon E, Spector Y, Bentwich Z (2005) Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37:766–770
Shomron N, Golan D, Hornstein E (2009) An evolutionary perspective of animal microRNAs and their targets. J Biomed Biotechnol 2009:594738
Calin GA, Croce CM (2006) MicroRNA signatures in human cancers. Nat Rev Cancer 6:857–866
Dykxhoorn DM (2010) MicroRNAs and metastasis: little RNAs go a long way. Cancer Res 70(16):6401–6406
Iorio MV, Piovan C, Croce CM (2010) Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta 1799(10–12):694–701
Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE et al (2000) DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 404:510–514
Mitsuyama K, Matsumoto S, Masuda J, Yamasakii H, Kuwaki K, Takedatsu H, Sata M (2007) Therapeutic strategies for targeting the IL-6/STAT3 cytokine signalling pathway in inflammatory bowel disease. Anticancer Res 27:3749–3756
Jarnicki A, Putoczki T, Ernst M (2010) Stat3: linking inflammation to epithelial cancer—more than a “gut” feeling? Cell Div 5:14
Nowak DG, Woolard J, Amin EM, Konopatskaya O, Saleem MA, Churchill AJ, Ladomery MR, Harper SJ, Bates DO (2008) Expression of pro- and anti-angiogenic isoforms of VEGF is differentially regulated by splicing and growth factors. J Cell Sci 121(Pt 20):3487–3495
Qiu Y, Hoareau-Aveilla C, Oltean S, Harper SJ, Bates DO (2009) The anti-angiogenic isoforms of VEGF in health and disease. Biochem Soc Trans 37(Pt 6):1207–1213
Pucci S, Mazzarelli P, Sesti F, Boothman DA, Spagnoli LG (2009) Interleukin-6 affects cell death escaping mechanisms acting on Bax-Ku70-Clusterin interactions in human colon cancer progression. Cell Cycle 8:473–481
Pucci S, Bonanno E, Sesti F, Mazzarelli P, Mauriello A, Ricci F, Zoccai GB, Rulli F, Galatà G, Spagnoli LG (2009) Clusterin in stool: a new biomarker for colon cancer screening? Am J Gastroenterol 104:2807–2815
Pucci S, Mazzarelli P, Nucci C, Ricci F, Spagnoli LG (2009) CLU “in and out”: looking for a link. Adv Cancer Res 105:93–113
Mazzarelli P, Pucci S, Spagnoli LG (2009) CLU and colon cancer. The dual face of CLU: from normal to malignant phenotype. Adv Cancer Res 105:45–61
Gentner B, Wein A, Croner RS, Zeittraeger I, Wirtz RM, Roedel F, Dimmler A, Dorlaque L, Hohenberger W, Hahn EG, Brueckl WM (2009) Differences in the gene expression profile of matrix metalloproteinases (MMPs) and their inhibitors (TIMPs) in primary colorectal tumors and their synchronous liver metastases. Anticancer Res 29:67–74
Nash KT, Phadke PA, Navenot JM, Hurst DR, Accavitti-Loper MA, Sztul E et al (2007) Requirement of KISS1 secretion for multiple organ metastasis suppression and maintenance of tumor dormancy. J Natl Cancer Inst 99:309–321
Pulkkinen K, Malm T, Turunen M, Koistinaho J, Ylä-Herttuala S (2008) Hypoxia induces microRNA miR-210 in vitro and in vivo ephrin-A3 and neuronal pentraxin 1 are potentially regulated by miR-210. FEBS Lett 582:2397–2401
Kulshreshtha R, Davuluri RV, Calin GA, Ivan M (2008) A microRNA component of the hypoxic response. Cell Death Differ 15:667–671
Dumoutier L, Leemans C, Lejeune D, Kotenko SV, Renauld JC (2001) Cutting edge: STAT activation by IL-19, IL-20 and mda-7 through IL-20 receptor complexes of two types. J Immunol 167:3545–3549
Gregory PA, Bracken CP, Bert AG, Goodall GJ (2008) MicroRNAs as regulators of epithelial-mesenchymal transition. Cell Cycle 7:3112–3118
Adam L, Zhong M, Choi W, Qi W, Nicoloso M, Arora A, Calin G, Wang H, Siefker-Radtke A, McConkey D, Bar-Eli M, Dinney C (2009) miR-200 expression regulates epithelial-to-mesenchymal transition in bladder cancer cells and reverses resistance to epidermal growth factor receptor therapy. Clin Cancer Res 15:5060–5072
Noonan EJ, Place RF, Pookot D, Basak S, Whitson JM, Hirata H, Giardina C, Dahiya R (2009) miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 28:1714–1724
Kim DH, Kim M, Kwon HJ (2003) Histone deacetylase in carcinogenesis and its inhibitors as anti-cancer agents. J Biochem Mol Biol 36:110–119
Esteller M, Villar-Garea A (2004) Histone deacetylase inhibitors: understanding a new wave of anticancer agents. Int J Cancer 112:171–178
Mariadason JM (2008) HDACs and HDAC inhibitors in colon cancer. Epigenetics 3:28–37
Menendez JA, Decker JP, Lupu R (2005) In support of fatty acid synthase (FAS) as a metabolic oncogene: extracellular acidosis acts in an epigenetic fashion activating FAS gene expression in cancer cells. J Cell Biochem 94:1–4
Iliopoulos D, Drosatos K, Hiyama Y, Goldberg IJ, Zannis VI (2010) MicroRNA-370 controls the expression of MicroRNA-122 and Cpt1alpha and affects lipid metabolism. J Lipid Res 51(6):1513–1523
Mazzarelli P, Pucci S, Bonanno E, Sesti F, Calvani M, Spagnoli LG (2007) Carnitine palmitoyltransferase I in human carcinomas: a novel role in histone deacetylation? Cancer Biol Ther 6:1606–1613
Wang HQ, Altomare DA, Skele KL, Poulikakos PI, Kuhajda FP, Di Cristofano A, Testa JR (2005) Positive feedback regulation between AKT activation and fatty acid synthase expression in ovarian carcinoma cells. Oncogene 24:3574–3582
Rego RL, Foster NR, Smyrk TC, Le M, O’Connell MJ, Sargent DJ, Windschitl H, Sinicrope FA (2010) Prognostic effect of activated EGFR expression in human colon carcinomas: comparison with EGFR status. Br J Cancer 102:165–172
Menendez JA, Lupu R (2007) Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nature 7:763–777
Kulshreshtha R, Ferracin M, Wojcik SE, Garzon R, Alder H, Agosto-Perez FJ, Davuluri R, Liu CG, Croce CM, Negrini M, Calin GA, Ivan M (2007) A microRNA signature of hypoxia. Mol Cell Biol 27(5):1859–1867
Donker RB, Mouillet JF, Nelson DM, Sadovsky Y (2007) The expression of Argonaute2 and related microRNA biogenesis proteins in normal and hypoxic trophoblasts. Mol Hum Reprod 13(4):273–279
Akao Y, Nakagawa Y, Naoe T (2006) Let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol Pharm Bull 29(5):903–906
Tsuchiya N, Nakagama H (2010) MicroRNA, SND1, and alterations in translational regulation in colon carcinogenesis. Mutat Res 693(1–2):94–100
Michael MZ, O’Connor SM, van Holst Pellekaan NG, Young GP, James RJ (2003) Reduced accumulation of specific microRNAs in colorectal neoplasia. Cancer Res 1(12):882–891
Iliopoulos D, Jaeger SA, Hirsch HA, Bulyk ML, Struhl K (2010) STAT3 activation of miR-21 and miR-181b-1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Mol Cell 39(4):493–506
Asangani IA, Rasheed SA, Nikolova DA, Leupold JH, Colburn NH, Post S, Allgayer H (2008) MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene 27(15):2128–2136
Tili E, Michaille JJ, Alder H, Volinia S, Delmas D, Latruffe N, Croce CM (2010) Resveratrol modulates the levels of microRNAs targeting genes encoding tumor-suppressors and effectors of TGFβ signaling pathway in SW480 cells. Biochem Pharmacol 80(12):2057–2065
Bandrés E, Cubedo E, Agirre X, Malumbres R, Zárate R, Ramirez N, Abajo A, Navarro A, Moreno I, Monzó M, García-Foncillas J (2006) Identification by real-time PCR of 13 mature microRNAs differentially expressed in colorectal cancer and non-tumoral tissues. J Mol Cancer 5:29
Cummins JM, He Y, Leary RJ, Pagliarini R, Diaz LA Jr, Sjoblom T, Barad O, Bentwich Z, Szafranska AE, Labourier E, Raymond CK, Roberts BS, Juhl H, Kinzler KW, Vogelstein B, Velculescu VE (2006) The colorectal microRNAome. Proc Natl Acad Sci USA 103(10):3687–3692
Arndt GM, Dossey L, Cullen LM, Lai A, Druker R, Eisbacher M, Zhang C, Tran N, Fan H, Retzlaff K, Bittner A, Raponi M (2009) Characterization of global microRNA expression reveals oncogenic potential of miR-145 in metastatic colorectal cancer. BMC Cancer 9:374
Xu H, Cheung IY, Guo HF, Cheung NK (2009) MicroRNA miR-29 modulates expression of immunoinhibitory molecule B7-H3: potential implications for immune based therapy of human solid tumors. Cancer Res 69(15):6275–6281
Wang YX, Zhang XY, Zhang BF, Yang CQ, Chen XM, Gao HJ (2010) Initial study of microRNA expression profiles of colonic cancer without lymph node metastasis. J Dig Dis 11:50–54
Volinia S, Galasso M, Costinean S, Tagliavini L, Gamberoni G, Drusco A, Marchesini J, Mascellani N, Sana ME, Abu Jarour R, Desponts C, Teitell M, Baffa R, Aqeilan R, Iorio MV, Taccioli C, Garzon R, Di Leva G, Fabbri M, Catozzi M, Previati M, Ambs S, Palumbo T, Garofalo M, Veronese A, Bottoni A, Gasparini P, Harris CC, Visone R, Pekarsky Y, de la Chapelle A, Bloomston M, Dillhoff M, Rassenti LZ, Kipps TJ, Huebner K, Pichiorri F, Lenze D, Cairo S, Buendia MA, Pineau P, Dejean A, Zanesi N, Rossi S, Calin GA, Liu CG, Palatini J, Negrini M, Vecchione A, Rosenberg A, Croce CM (2010) Reprogramming of miRNA networks in cancer and leukemia. Genome Res 20(5):589–599
Lujambio A, Calin GA, Villanueva A, Ropero S, Sánchez-Céspedes M, Blanco D, Montuenga LM, Rossi S, Nicoloso MS, Faller WJ, Gallagher WM, Eccles SA, Croce CM, Esteller M (2008) A microRNA DNA methylation signature for human cancer metastasis. Proc Natl Acad Sci USA 105(36):13556–13561
Deng S, Calin GA, Croce CM, Coukos G, Zhang L (2008) Mechanisms of microRNA deregulation in human cancer. Cell Cycle 7(17):2643–2646
Hua Z, Lv Q, Ye W, Wong CK, Cai G, Gu D, Ji Y, Zhao C, Wang J, Yang BB, Zhang Y (2006) MiRNA-directed regulation of VEGF and other angiogenic factors under hypoxia. PLoS ONE 1:e116
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License ( https://creativecommons.org/licenses/by-nc/2.0 ), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Pucci, S., Mazzarelli, P. MicroRNA Dysregulation in Colon Cancer Microenvironment Interactions: The Importance of Small Things in Metastases. Cancer Microenvironment 4, 155–162 (2011). https://doi.org/10.1007/s12307-011-0062-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12307-011-0062-y