
Abstract
Parkinson’s disease (PD) is a common neurodegenerative disorder caused by the loss of dopamine neurons in the substantia nigra and the formation of Lewy bodies, which are mainly composed of alpha-synuclein fibrils. Alpha-synuclein plays a vital role in the neuroinflammation mediated by the nucleotide-binding oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing protein 3 (NLRP3) inflammasome in PD. A better understanding of the NLRP3 inflammasome-mediated neuroinflammation and the related mitochondrial impairment during PD progression may facilitate the development of promising therapies for PD. This review focuses on the molecular mechanisms underlying NLRP3 inflammasome activation, comprising priming and protein complex assembly, as well as the role of mitochondrial impairment and its subsequent inflammatory effects on the progression of neurodegeneration in PD. In addition, the therapeutic strategies targeting the NLRP3 inflammasome for PD treatment are discussed, including the inhibitors of NLRP3 inflammatory pathways, mitochondria-focused treatments, microRNAs, and other therapeutic compounds.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is the most common age-related disorder worldwide after Alzheimer’s disease [1]. PD is characterized by the rapid growth of neurodegenerative conditions [2]. Patients with PD show both motor symptoms, such as resting tremor, stiffness, bradykinesia, and muscular rigidity, and non-motor symptoms, such as olfactory impairment, depression, anxiety, fatigue, orthostatic hypotension, gastrointestinal dysfunction, and circadian rhythm dysregulation [3,4,5,6]. The pathological hallmarks of PD are the selective loss of midbrain dopamine (DA) neurons and the accumulation of Lewy bodies formed by alpha-synuclein protein in different brain compartments [7,8,9]. Various treatments such as DA replacement are only partially or transiently effective [3]. As these therapies cannot restore DA neurons to curb the progression of PD, it is critical to find new targets and effective therapies for PD.
Neuroinflammation is one of the most critical factors that contribute to the onset and progression of PD [10]. Both pathological and genetic studies indicate the involvement of neuroinflammation in PD. Postmortem reports and experimental studies have also revealed the roles of both innate and adaptive immunity in the degenerative process [11]. Inflammasomes are important elements associated with neuroinflammation and neurodegenerative disorders [12]. Inflammasomes are multiprotein complexes found in neural cells, microglia, and astrocytes that respond to danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs); they induce the release of pro-inflammatory cytokines into the extracellular space. Various inflammasomes play essential roles in neurodegenerative diseases, primarily the nucleotide-binding oligomerization domain (NOD)-, leucine-rich repeat (LRR)-, and pyrin domain (PYD)-containing protein 3 (NLRP3) inflammasome [12]. As the primary immunological cell of the brain, microglia are a major cellular mediator during neuroinflammatory processes, and microglial activation is vital in the neurodegenerative process [13,14,15]. Activation of the microglial NLRP3 inflammasome and the release of pro-inflammatory cytokines from microglia contribute to PD progression. Impaired DA neurons directly lead to microglial activation, first producing large amounts of pro-inflammatory cytokines and then contributing to the apoptosis of DA neurons. Thus, microglial activation-induced neuroinflammation aggravates the pathological process of ongoing neurodegeneration in PD [8].
In addition, PD progression is associated with mitochondrial impairment, such as disruption of mitochondrial fusion and mitophagy [16]. On the other hand, the impairment of mitochondrial function augments NLRP3 inflammasome activation, triggering systemic or local inflammation [17, 18]. It has been documented that NLRP3 inflammasome-mediated neuroinflammation actively participates in PD progression [19, 20]. However, the relationship between mitochondrial dysfunction and the NLRP3 inflammasome is not well understood. Thus, achieving a better understanding of the interaction between mitochondrial impairment and NLRP3 inflammasome-mediated neuroinflammation in PD progression may facilitate the development of novel therapies for PD. In addition, the therapeutic strategies targeting the NLRP3 inflammasome for PD treatments are discussed in this review, including the application of NLRP3 inflammatory pathway inhibitors, mitochondria-focused treatments, microRNAs, and other therapeutic compounds.
The NLRP3 Inflammasome
Formation of the NLRP3 Inflammasome
The components of inflammasomes consist of the apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC) protein, pattern recognition receptors (PRRs), and procaspase-1. There are three types of intracellular PRR: nucleotide-binding domain and leucine-rich repeat-containing receptors (NLRs), absent in melanoma (AIM)-like receptors (ALRs), and the tripartite motif-containing (TRIM) protein pyrin/TRIM20. They are also known to be immunological sensors that can detect DAMPs, PAMPs, and double-stranded DNA in the cytosol [21]. ASC connects the caspase recruitment domain (CARD) of procaspase-1 to the PYD of NLR, ALR, or pyrin [22]. The NLR family includes NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRP12, and NLR family CARD domain-containing 4 (NLRC4) [23]. NLRP3 is activated by PAMPs or DAMPs to form a multimeric protein complex called the NLRP3 inflammasome, a crucial component of the innate immune system.
Activation of the NLRP3 Inflammasome
Two signaling pathways contribute to NLRP3 inflammasome activation, comprising the non-canonical and canonical pathways [24]. The non-canonical pathway mainly depends on caspase-11 and is mediated by caspase-4, caspase-5, and caspase-8; the canonical pathway predominantly depends on caspase-1. Non-canonical signaling pathways are activated explicitly by Gram-negative bacteria or cytosolic lipopolysaccharides (LPS), inducing the cleavage of gasdermin D (GSDMD) and promoting the activation of the NLRP3 inflammasome and the production of interleukin-1beta (IL-1β) and IL-18, leading to pyroptosis [25, 26].
Generally, activation of the NLRP3 inflammasome in the canonical signaling pathway noted above requires two steps [27]: priming (Step 1) and protein complex assembly (Step 2). Step 1 is first triggered by PRR signals, such as tumor necrosis factor receptor (TNFR) or Toll-like receptor (TLR) activation, and then inducing activation of the nuclear factor-kappa B (NF-kB)-dependent pathway to upregulate the transcription and expression of NLRP3, pro-IL-1β, and pro-IL-18 [28]. Numerous studies have suggested that Step 1 is very complicated, involves transcriptional and post-translational mechanisms, and requires many protein-binding partners. Step 2 is induced by a variety of PAMPs and DAMPs, including extracellular ATP and pore-forming toxins, as well as cellular events, including mitochondrial impairment and the production of reactive oxygen species (ROS), ion flux (K+/Cl− efflux and Ca2+ influx), and cathepsin B from lysosomal damage [29,30,31]. For example, there is evidence that K+ efflux, known as a reduction of the intracellular K+ concentration, is one of the critical cellular events that activate NLRP3 [32]. The activation of NLRP3 can be prevented by maintaining a high extracellular K+ state or by inhibiting K+ efflux [32]. Many events can induce K+ efflux [33]. For instance, after binding extracellular ATP, the ligand-gated ion channel P2X7R opens, allowing K+ to pass through. In addition, bacterial toxins, such as valinomycin, nigericin, and gramicidin, form membrane pores in the cell membrane to stimulate K+ efflux. In response to K+ efflux, NIMA-related kinase 7 (NEK7), a downstream modulator, acts as an important mediator of NLRP3 inflammasome activation through the catalytic domain of NEK7 and the LRR domain of NLRP3 [34,35,36,37].
Finally, the inflammasome complex results in the activation of caspase-1, consequently prompting the cleavage of the pro-inflammatory cytokines IL-1β and IL-18 into mature forms and turning the pyroptotic substrate GSDMD into the N-terminus of GSDMD (N-GSDMD) and the C-terminus of GSDMD (C-GSDMD) [38, 39]. The oligomerization of N-GSDMD forms pores in the plasma membrane following cleavage of GSDMD, then mediates the programmed cell death known as pyroptosis, and finally results in the secretion of mature IL-1β and IL-18. It should be noted that, although pyroptosis [40] is similar to apoptosis [41] and necroptosis [42] it, is a type of programmed cell death, and they all differ in morphology [43, 44] and mechanism [45,46,47]. However, during the apoptosis process, if apoptotic cells cannot be cleared by macrophages, the process changes to caspase-3-mediated pyroptosis [48,49,50]. Activation of the NLRP3 inflammasome is involved in the pathogenesis of many neurodegenerative disorders, such as PD [51], Huntington’s disease [52], Alzheimer’s disease [53], amyotrophic lateral sclerosis [54], multiple sclerosis [54], and prion diseases [52]. This review focuses on the molecular mechanisms underlying the activation of the NLRP3 inflammasome and its subsequent inflammatory effects on the progression of neurodegeneration in PD.
NLRP3 Inflammasome-mediated Neuroinflammation in the Pathogenesis of PD
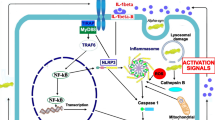
An animal model study revealed that the NLRP3 inflammasome, not the NLRP1, NLRP2, NLRC4, and AIM2 inflammasomes, might be the pivotal complex that boosts 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced pathogenesis. MCC950, an NLRP3 inhibitor, reverses MPTP-induced nigrostriatal damage [55]. There is evidence that abnormal aggregation of alpha-synuclein protein activates microglial cells and stimulates the NLRP3 pathway [23]. Activation of the NLRP3 inflammasome prompts the maturation of caspase-1, which causes the release of pro-inflammatory cytokines such as IL-1β and IL-18, thus inducing pyroptosis [56]. The expression level of oligomerized and phosphorylated alpha-synuclein, as well as IL-1β, in the peripheral blood of PD patients, is remarkably elevated [57]. Besides, researchers have reported upregulated gene expression of NLRP3, ASC, and caspase-1 and elevated protein expression of NLRP3, caspase-1, and IL-1β in peripheral blood mononuclear cells from PD patients [58]. Similarly, another study demonstrated that PD patients have significantly higher cerebrospinal fluid (CSF) concentrations of IL-1β and IL-18 than healthy controls [59]. Thus, NLRP3 inflammasome-mediated neuroinflammation plays an important role in PD progression. As the activation of the NLRP3 inflammasome requires a priming process (Step 1) and a protein complex assembly process (Step 2), we present the mechanisms underlying NLRP3 inflammasome activation in PD during these two steps, as shown in Fig. 1.

The mechanism underlying the mitochondrial impairment and NLRP3 inflammasome-mediated neuroinflammation in PD and the therapeutic approaches (green frames). In the priming process, alpha-synuclein is regulated by Fyn kinase in conjunction with CD36 to promote PKCδ-mediated NF-kB activation. Besides, alpha-synuclein can also interact with TLR1/2, leading to the nuclear translocation of NF-kB in a MyD88-dependent manner. In addition, TNFR, IL-1R, and other TLRs on microglia recognize TNF, IL-18, or DAMPs released from damaged neurons, inducing activation of the NF-kB pathway, subsequently leading to upregulated expression of NLRP3, pro-IL-1β, and pro-IL-18. In the protein complex assembly process, the alpha-synuclein is taken up by microglial cells in the CD36 or TLR2-mediated pathway. The mitochondrial impairment by excessive ROS and mtDNA, disruption of mitophagy, and Drp1-mediated mitochondrial fission promote activation of the NLRP3 inflammasome. ROS and alpha-synuclein fibrils stimulate the deubiquitination of NLRP3 through BRCC3 deubiquitinase, resulting in the formation of the NLRP3-ASC-procaspase-1 complex. After that, the inflammasome complex activates caspase-1, consequently prompting the production of mature IL-1β and IL-18 and the formation of plasma membrane pores that mediate pyroptosis and result in the secretion of IL-1β and IL-18, which exert inflammatory effects that damage DA neurons. Impaired DA neurons directly lead to microglial activation, producing large amounts of pro-inflammatory cytokines, then conversely contributing to the apoptosis of DA neurons. NLRP3, nucleotide-binding oligomerization domain-, leucine-rich repeat-, and pyrin domain-containing protein 3; PD, Parkinson's disease; PKCδ, protein kinase C-delta; NF-kB, nuclear factor-kappa B; TLR, Toll-like receptor; TNFR, tumor necrosis factor receptor; IL-1R, Interleukin-1 receptor; TNF, tumor necrosis factor; IL-18, interleukin-18; DAMPs, danger-associated molecular patterns; IL-1β, interleukin-1beta; ROS, reactive oxygen species; Drp1, dynamin-related protein 1; BRCC3, BRCA1-BRCA2-containing complex subunit 3; ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; DA, dopamine; GSDMD, gasdermin D.
Priming Process in PD
The pathological hallmark of PD is Lewy bodies, constituted by fibrillar alpha-synuclein [60]. Under normal circumstances, alpha-synuclein is expressed in synaptic terminals and participates in synaptic function [61]. There is evidence that alpha-synuclein is strikingly expressed in the neurons of the brain regions affected by PD, such as the substantia nigra pars compacta, dorsal motor nucleus of the vagus, and olfactory bulb [62]. The levels of both oligomers and phosphorylated alpha-synuclein are significantly elevated in the peripheral plasma of PD patients [57]. Extracellular alpha-synuclein that is released from neuronal cells can activate the inflammatory responses in microglia, contributing to PD progression [63]. The use of alpha-synuclein in the development of animal models of PD has become popular in recent years, including the injection of alpha-synuclein pre-formed fibrils model and the recombinant adeno-associated virus vector-mediated alpha-synuclein overexpression model [64]. In the clinic, alpha-synuclein may be helpful as a biomarker for PD, given its role in pathogenesis [65]. Total alpha-synuclein, as well as its phosphorylated and oligomeric forms in CSF, plasma, and saliva, can be detected by ELISA, mass spectrometry, or western blot [66, 67]. Extracellular alpha-synuclein may act as a DAMP for microglia. Alpha-synuclein can be regulated by Fyn kinase, a non-receptor Src family tyrosine kinase, in conjunction with the class B scavenger receptor CD36, to promote protein kinase C-delta-mediated NF-kB activation, which contributes to NLRP3 inflammasome priming [68]. Alpha-synuclein can also interact with the heterodimer Toll-like receptor 1 and 2 (TLR1/2) at the cell membrane, leading to the nuclear translocation of NF-kB in a MyD88-dependent manner [69]. In addition, the TLR2 or other TLRs on microglia can recognize the extracellular DAMPs released from damaged neurons, inducing activation of the NF-kB pathway [51], thus resulting in persistent neuroinflammation. Although the DAMPs may not be initiating factors in PD, they appear to be co-factors in its progression [70]. In addition, the receptors TNFR and IL-1 receptor (IL-1R) induce the priming process once stimulated by their ligands TNF or IL-1β, leading to activation of the NF-kB pathway [51]. NF-kB appears to play an important role in activating and regulating neuroinflammation in PD [71]. Activation of the NF-kB pathway subsequently leads to upregulated expression of NLRP3, pro-IL-1β, and pro-IL-18 [28].
Protein Complex Assembly Process in PD
The priming process leads to the production of NLRP3, pro-IL-1β, and pro-IL-18. However, as ubiquitination of NLRP3 prevents its oligomerization with ASC, it is preactivated in this state. The ROS and neurotoxic alpha-synuclein fibrils called the second signal of the protein complex assembly process, stimulate the deubiquitination of NLRP3 through BRCA1-BRCA2-containing complex subunit 3 (BRCC3) deubiquitinase, as well as activate the nucleation of the inflammasome with ASC, resulting in the formation of the NLRP3–ASC–procaspase-1 complex [51]. A study found that BRCC3 expression is increased in PD models, whereas knocking it down with shRNA lentivirus decreases the NLRP3 inflammasome activity. Thus, BRCC3 may contribute to regulating the NLRP3 inflammasome in PD [72]. Alpha-synuclein can be taken up by microglial cells in a TLR2-mediated endocytosis-dependent manner [63] or via the CD36 and Fyn kinase-mediated pathway [68]. Afterward, aggregated alpha-synuclein can act as an endogenous danger signal to induce NLRP3 inflammasome activation. The alpha-synuclein in microglia induces the propagation of mitochondrial ROS and consequently leads to the activation of the NLRP3 inflammasome [68]. In addition to ROS production, the phagocytosis of alpha-synuclein also promotes the release of cathepsin B into the cytosol, leading to NLRP3 inflammasome activation [73]. Then, the inflammasome complex results in the activation of caspase-1, consequently prompting the production of mature forms of IL-1β and IL-18 [38, 39]. The plasma membrane pores formed by the oligomerization of N-GSDMD mediate pyroptosis and result in the secretion of mature IL-1β and IL-18, which exert inflammatory effects that damage DA neurons. It has been reported that primary human microglia exposed to alpha-synuclein fibrils significantly induce inflammasome assembly and secretion of IL-1β, demonstrating the similar mechanisms in primary human and mouse microglia in the activation of the NLRP3 inflammasome by alpha-synuclein [74]. Microglial NLRP3 inflammasome activation plays an essential role in the dopaminergic neurodegeneration process [75].
However, studies have shown that DA neurons can inhibit NLRP3 inflammasome activation via the dopamine D1 receptor (DRD1) [76,77,78]. The administration of A68930, a DRD1-specific agonist, decreases the expression of NLRP3, caspase 1, and IL-1β and inhibits microglial activation [79]. Moreover, studies have found that DRD1 signaling inhibits NLRP3 inflammasome activation through cyclic adenosine monophosphate, which binds to NLRP3, leading to the ubiquitination and degradation of NLRP3 [80]. In addition to DRD1, the D2 receptor (DRD2) may also be involved in the DA-induced inhibition of the NLRP3 inflammasome [81]. The study indicates that DA may negatively regulate the K+ efflux-induced activation of the NLRP3 inflammasome, which contributes to the DA neuron degeneration in PD [81]. Thus, there is a mutual regulation between DA neurons and the NLRP3 inflammasome. Not only is the NLRP3 inflammasome able to damage DA neurons, but DA neurons can inhibit NLRP3 inflammasome activation. Therefore, microglial NLRP3 inflammasome-mediated neuroinflammation is a crucial element in the pathogenesis of PD.
Mitochondrial Impairment in the NLRP3 Inflammasome-mediated Neuroinflammation in PD
Mitochondria comprise various proteins, a small number of which are encoded by the mitochondrial genome, called mtDNA, located in the matrix [82]. New mitochondria are produced through mitochondrial binary fission [83, 84]. The clearance of damaged or unwanted mitochondria, called mitophagy, is also required to maintain mitochondrial and cellular homeostasis [85]. The essential function of mitochondria is to produce energy [86]. They also produce many biosynthetic intermediates to mediate autophagy and apoptosis, store Ca2+ for cell signaling activities, and regulate cell growth. Thus, mitochondrial impairment plays a role in many diseases, including neurodegenerative diseases [87]. Studies are increasingly showing that the impairment of mitochondrial function augments NLRP3 inflammasome activation, triggering systemic or local inflammation [17, 18]. Autophagy negatively regulates NLRP3 inflammasome activity, while ROS positively regulates it [17]. After sensing mitochondrial impairment, the NLRP3 inflammasome promotes an inflammatory process, consequently augmenting mitochondrial damage [17]. There is evidence indicating that mitochondrial dysfunction aggravates the pro-inflammatory cascade mediated by the microglial NLRP3 inflammasome, contributing to the neurodegenerative process in DA neurons [88]. Thus, mitochondrial impairment in NLRP3 inflammasome-mediated neuroinflammation is of importance in PD.
Evidence indicates that mitochondria participate in immune system activation, inflammation, and the pathogenesis of inflammatory diseases through endogenous mitochondrial damage-associated molecular patterns (mtDAMPs) that include ATP, mtDNA, mitochondrial ROS, mitochondrial N-formyl peptides, and mitochondrial cardiolipin, which are released to recognize PRRs during mitochondrial damage [89]. Accumulating studies in PD patients and animal PD models indicate that mitochondrial impairment participates in PD progression. In both sporadic and familial forms of PD, mitochondrial dysfunction includes electron transport chain impairment, mitochondrial morphology, alterations in dynamics, disruption of mitochondrial homeostasis and biogenesis, deficiency of mitochondrial oxidative phosphorylation, defective mitophagy, mtDNA mutations, and Ca2+ imbalance, resulting in a decline in energy generation and ROS production, as well as increased apoptosis [90,91,92,93]. The mitochondrial impairment, oxidative stress, and protein turnover defect induce DA neuron death [16]. A study indicates that MPTP causes NLRP3 inflammasome activation in PD [94]. MPTP is a neurotoxin that induces mitochondria damage, which might activate the NLRP3 inflammasome by mitochondrial DAMPs, such as excessive ROS and mtDNA. In addition, a study revealed that rotenone augments dynamin-related protein 1 (Drp1)-mediated mitochondrial fission, which induces mitochondrial dysfunction. Moreover, Drp1-mediated mitochondrial fission promotes the nuclear translocation of NF-kB and activation of the NLRP3 inflammasome [95]. Similarly, another study demonstrated that rotenone aggravates NLRP3 inflammasome activation through mitochondrial dysfunction. The researchers found that mitochondrial dysfunction in microglia triggers NLRP3 inflammasome activation, aggravating the process of DA neuron degeneration [88]. Activation of the NLRP3 inflammasome may require a signal from mitochondria damage. Furthermore, pharmacological attenuation of mitochondria damage inhibits microglial activation and the NLRP3 inflammasome pathway, promoting neuronal survival in PD [96]. In addition, mitophagy is involved in the quality control of mitochondria. Disruption of mitophagy can lead to NLRP3 inflammasome activation in microglia and neurodegeneration in PD [97]. Thus, mitochondrial impairment is critically involved in NLRP3 inflammasome-mediated neuroinflammation in PD.
The role of the alpha-synuclein/TLRs/NF-kB/NLRP3 inflammasome axis and microglial activation in PD has been elucidated in a recent review [98]. The review revealed that alpha-synuclein interacts with TLR1/2, TLR2, and TLR4, triggering NF-kB-dependent upregulation of NLRP3, activation of the NLRP3 inflammasome, and the production of pro-inflammatory cytokines [98]. In this present review, regarding the initiation of the priming process, we do not only focus on the TLR signaling pathway but also CD36, as well as the TNFR and IL-1R signaling pathways. Taken together, as shown in Fig. 1, in the priming process, the CD36, TLR1/2, TNFR, IL-1R, and other TLRs on microglia can recognize the alpha-synuclein, TNF, IL-18, or other DAMPs released from damaged neurons, inducing activation of the NF-kB pathway, subsequently leading to upregulated expression of NLRP3, pro-IL-1β, and pro-IL-18. In the protein complex assembly process, the alpha-synuclein is taken up by microglial cells in the CD36 or TLR2-mediated pathway, activating the nucleation of the inflammasome. Mitochondrial impairment, including excessive ROS and mtDNA, disruption of mitophagy, and Drp1-mediated mitochondrial fission, promotes activation of the NLRP3 inflammasome. After that, the inflammasome complex activates caspase-1, consequently prompting the cleavage of the pro-inflammatory cytokines IL-1β and IL-18 and the formation of plasma membrane pores that mediate pyroptosis and result in the secretion of IL-1β and IL-18, which exert inflammatory effects to damage DA neurons. Impaired DA neurons directly lead to microglial activation, producing large amounts of pro-inflammatory cytokines, then contributing to the apoptosis of DA neurons.
The NLRP3 Inflammasome: A Target for Novel Therapeutic Approaches in PD
Currently, drugs that treat PD clinically are divided into three categories: direct supplementation of exogenous DA precursors such as levodopa, DA receptor agonists such as pramipexole, and monoamine oxidase B inhibitors such as selegiline. Among these, levodopa is the best option [99,100,101]. However, although these treatments are initially effective, they present many problems, such as low tolerance and long-term effects. Currently, no therapy can efficiently curb the progression of PD [2]. Informed by new insights into the involvement of mitochondrial impairment and NLRP3 inflammasome-mediated neuroinflammation in this disease, the therapeutic strategies targeting the NLRP3 inflammasome for PD treatment are discussed next, including the NLRP3 inflammatory pathway inhibitors, mitochondria-focused treatments, microRNA, and other therapeutic compounds.
Inhibitors Targeting the NLRP3 Inflammasome
As the NLRP3 inflammasome plays a vital role in PD progression, inhibitors of NLRP3 inflammatory pathways that may hamper NLRP3 inflammasome activation offer novel therapeutic avenues for PD therapy. A study revealed that MCC950, an NLRP3 inhibitor, inhibits NLRP3 inflammasome activation and alleviates behavioral dysfunctions and DA neuronal degeneration in MPTP-administered mice [55]. Glibenclamide, another inhibitor of the NLRP3 inflammasome, blocks NLRP3 inflammasome activation, as demonstrated by decreased expression of NLRP3, activated caspase-1, and mature IL-1β in paraquat- and maneb-administered mice. Moreover, glibenclamide relieves the paraquat and maneb-induced microglial M1 pro-inflammatory reaction and NF-kB activation to protect DA neurons in PD mice [102]. In addition, Ac-YVAD-CMK, a caspase-1 inhibitor, suppresses the downstream pathway of the NLRP3–caspase-1–IL-1β axis in LPS- and 6-hydroxydopamine-induced PD rats, providing a new basis for PD therapy [103]. ZYVAD, another caspase-1 inhibitor, inhibits the caspase-7/poly (ADP-ribose) polymerase 1/apoptosis-inducing factor-mediated DA neuronal apoptosis pathway in PD mice [104]. Similarly, VX765, a potent caspase-1 inhibitor, decreases the truncation of alpha-synuclein into its highly aggregation-prone form, playing a neuroprotective role in a neuronal cell model of PD [105]. In addition, anakinra, an IL-1 receptor antagonist, inhibits the binding of IL-1β and its receptor to relieve the inflammatory effect of IL-1β and curb the progression of PD [106]. Thus, these inhibitors and antagonists are potential therapeutic drugs targeting the NLRP3 inflammasome in the treatment of PD. The structures of these inhibitors are shown in Fig. 2A–F.

The structures of inhibitors of the NLRP3 inflammatory pathway (A–F) and mitochondria-focused therapeutic compounds G–I that target the NLRP3 inflammasome for the treatment of PD. A MCC950, B Glibenclamide, C Ac-YVAD-CMK, D Z-YVAD, E VX765, F Anakinra, G Mdivi-1, H Perillyl alcohol, I Urolithin A.
Mitochondria-Focused Treatments to Attenuate NLRP3 Inflammasome-mediated Neurodegeneration
It is known that mitochondrial dysfunction is critically involved in NLRP3 inflammasome-mediated neuroinflammation in PD, so it is a promising target for PD treatment [107]. As noted above, a study has revealed that rotenone augments Drp1-mediated mitochondrial fission, and this, in turn, induces mitochondrial dysfunction, promoting the nuclear translocation of NF-kB and activation of the NLRP3 inflammasome [95]. Mdivi-1, a selective Drp1 inhibitor, significantly attenuates the morphological disruption of mitochondria and Drp1 translocation, negatively regulates the nuclear translocation of NF-kB, and eventually curbs activation of the NLRP3 inflammasome [95]. NLRP3 inflammasome activation may require a signal from mitochondria damage. Moreover, pharmacological treatment, such as perillyl alcohol, inhibits the nuclear translocation of NF-kB, promotes the translocation of PARKIN into mitochondria, and maintains cellular redox homeostasis, inhibiting microglial activation and the NLRP3 inflammasome pathway and promoting neuronal survival in PD [96]. Mitophagy contributes to mitochondrial quality control, and its disruption leads to activation of the microglial NLRP3 inflammasome and neurodegeneration in PD. Urolithin A, a natural compound produced by gut bacteria, increases mitophagy. A study has shown that urolithin A restores mitochondrial function and inhibits NLRP3 inflammasome activation, protecting against dopaminergic neurodegeneration in PD [97]. Thus, these potential mitochondria-focused treatments may be promising strategies to influence the activation of the NLRP3 inflammasome in PD. The structures of the mitochondria-focused therapeutic compounds that target the NLRP3 inflammasome for PD treatment are shown in Fig. 2G–I.
MicroRNA and Other Therapeutic Compounds Targeting the NLRP3 Inflammasome
Recently, accumulating evidence has indicated that microRNA is involved in inflammatory responses. A study has shown that miR-188-3p-enriched exosome treatment inhibits autophagy and pyroptosis by targeting NLRP3 and CDK5 in MPTP-treated mice and MN9D cells [108]. Besides, a study found that miR-124-3p is the target of the long non-coding RNA (lncRNA) HOXA11-AS and follistatin-like protein 1 (FSTL1). Inhibiting HOXA11-AS suppresses neuroinflammation and neuronal apoptosis via the miR-124-3p–FSTL1–NF-kB–NLRP3 inflammasome axis in PD mice [109]. In addition, researchers have found that miR-135b mimics suppresses MPP(+)-induced pyroptosis and the upregulation of thioredoxin-interacting protein (TXNIP), NLRP3, caspase-1, ASC, N-GSDMD, and IL-1β by targeting forkhead box 1 [110]. Moreover, miR-190 alleviates neuronal damage and inhibits inflammation by inhibiting the expression and activation of the NLRP3 inflammasome in MPTP-induced PD mice [111]. In addition, the miR-30e agomir significantly suppresses the expression and activation of the NLRP3 inflammasome, attenuates the release of inflammatory cytokines, inhibits DA neuron loss, and improves behavioral deficits in MPTP-induced PD mice [112]. Similarly, miR-29c-3p negatively regulates NLRP3 inflammasome activation by targeting the nuclear factor of activated T cells 5 (NFAT5) to exert anti-inflammatory effects in PD animals and neuronal models [113]. In addition, microRNA-7 mimics inhibit the neuroinflammation mediated by microglial NLRP3 inflammasome activation in MPTP-induced PD mice [114]. Lastly, targeting miR-326 [115] or miR-1301-3p [116] may inhibit NLRP3 inflammasome activation in PD. Thus, these microRNAs are potential targets for PD therapy.
In addition, some studies have revealed that other exogenous compounds indirectly attenuate NLRP3 inflammasome-mediated neuroinflammation in PD. For example, GW501516, a peroxisome proliferator-activated receptor β/δ agonist, inhibits NLRP3-mediated neuroinflammation in MPTP model mice [117]. Besides, melatonin remarkably suppresses microglial activation and NLRP3 inflammasome activity, reduces DA neuron damage, and alleviates behavioral dysfunction via a silence information regulator 1-dependent pathway in MPTP-induced PD models [118]. Moreover, N-methyl-4-isoleucine-cyclosporine (NIM811), a derivative of the immunosuppressant cyclosporin A, passes through the blood-brain barrier (BBB). The researchers found that NIM811 inhibits rotenone-induced NLRP3 inflammasome activation and pyroptosis [119]. Therefore, these compounds are potential therapeutic drugs in PD treatment. In addition, it should be noted that a recent review demonstrated that microglial autophagy plays a role in maintaining brain homeostasis and negatively regulates NLRP3 inflammasome-mediated neuroinflammation [120]. Autophagy, a lysosomal degradation pathway, keeps cells functioning normally by removing misfolded or aggregated proteins, such as alpha-synuclein, and clearing damaged organelles, such as mitochondria, endoplasmic reticulum, and peroxisomes, as well as eliminating intracellular pathogens [121, 122]. Studies have shown that disruption of autophagy is a contributor to age-related pathologies and cognitive and motor dysfunction [123]. By stimulating microglial autophagy, autophagy inducers eliminate the misfolded proteins, remove damaged mitochondria, and degrade the NLRP3 inflammasome and its components. The previously noted review laid a foundation for using NLRP3 inflammasome inhibitors in combination with autophagy inducers. And this combination therapy may be more effective than a single treatment in treating neurodegenerative diseases [120]. Thus, combination therapy may inhibit the NLRP3 inflammasome-mediated neuroinflammation in a more efficient manner, which requires further research.
In this review, the therapeutic strategies targeting NLRP3 inflammasome for the treatment of PD are discussed, including the inhibitors of NLRP3 inflammatory pathways, mitochondria-focused treatments, microRNAs, and other therapeutic molecules. The pharmacological activities of the therapeutic compounds that target the NLRP3 inflammasome for PD treatment are summarized in Table 1.
Research Progress in Clinical Trials Targeting the NLRP3 Inflammasome
In 2015, a study published in Nature Medicine revealed that MCC950, a small-molecule inhibitor of the NLRP3 inflammasome, can be used to treat inflammatory diseases [124]. Afterward, the research on NLRP3 inhibitors entered the clinical stage. Inflazome, IFM Tre, and Olatec are the three representative companies. Inflazome, acquired by Roche, generated three kinds of the compound with distinctive tissue distribution properties based on MCC950 [125]: inzomelid, a central nervous system (CNS) penetrant NLRP3 inhibitor, is being investigated for Alzheimer's disease, PD, cryopyrin-associated periodic syndrome, and amyotrophic lateral sclerosis; Somalix, a peripherally restricted NLRP3 inhibitor, is for cryopyrin-associated periodic syndrome; in addition, gut-restricted compounds are in development. IFM Tre, acquired by Novartis, developed IFM-2747, which is somewhat differentiated from MCC950, to treat gout, coronary artery disease, non-alcoholic fatty liver disease, and Crohn’s disease [125]. Olatec developed dapansutrile, a simple β-sulfonyl nitrile and the most advanced NLRP3 inhibitor in clinical trials, to treat acute gout [126]. NLRP3 seems to be a promising drug target; however, it faces some problems. As the NLRP3 inflammasome plays a vital role in defense against various infections, its inhibitors may disturb the host response. Evidence has shown that inhibition of NLRP3 disrupts the host response to influenza A infection [127], Streptococcus pneumoniae infection [128], and fungal infection [129]. Besides, since PD is a degenerative disorder of the CNS, it is necessary to improve the BBB penetration of NLRP3 inhibitors. Small molecules can be structurally modified to increase their BBB penetration by improving diffusion (increasing lipophilicity, reducing hydrogen bond donor capacity, reducing topological polar surface area, enhancing rigidity, reducing pKa, and controlling multiple parameters), reducing efflux (multiple drug resistance 1 and breast cancer resistance protein), and activating carrier transporters (L-type amino-acid transporter and glucose transporter 1) [130]. Future small molecule CNS drug development can refer to these strategies. Taken together, NLRP3 possesses a complex activation mechanism, thus NLRP3 inhibitors need to be further explored.
Conclusion
Alpha-synuclein participates in NLRP3 inflammasome-mediated neuroinflammation in microglia and is a crucial element in the pathogenesis of PD. It has been shown that several risk factors in PD modulate immune function, such as Leucine-rich repeat kinase 2 (LRRK2), Synuclein Alpha (SNCA), glucocerebrosidase (GBA), Parkin RBR E3 Ubiquitin Protein Ligase (PRKN), and PTEN-induced kinase 1 (PINK1) [8, 131]. These risk factors contribute to PD pathogenesis. For example, LRRK2 and SNCA are involved in vesicular trafficking, LRRK2 and GBA are involved in autophagy, LRRK2 is involved in mitochondrial function, and GBA is involved in the lysosomal process [132]. Our previous review reported that, as a non-motor manifestation of PD, intestinal dysfunction might affect the intestinal microenvironment to influence the CNS through alpha-synuclein pathology and systemic inflammation [133]. Specifically, the NLRP3 gene may be one of the mechanisms linking intestinal inflammation and PD [133]. It should be noted that NLRP3 inflammasome-mediated neuroinflammation in microglia occurs in the early stages of PD [134]. Positron emission tomography (PET) examination has also revealed that microglial activation appears at very early or preclinical stages of PD [7]. In addition to microglia, NLRP3 inflammasome-mediated neuroinflammation may also occur in mesencephalic neurons. A study demonstrated that, in PD patients, the expression of NLRP3 is elevated in mesencephalic neurons [135]. Besides, it has been shown that NLRP3 rs7525979, a synonymous single-nucleotide polymorphism, significantly reduces PD risk by altering the translation efficiency of NLRP3 [135]. Furthermore, evidence indicates that mitochondrial dysfunction aggravates the microglial NLRP3 inflammasome-mediated pro-inflammatory cascade, contributing to the DA neuron neurodegenerative process in PD [88]. In summary, the microglial NLRP3 inflammasome and the related mitochondrial impairment are crucial elements in the pathogenesis of PD and might be promising targets for PD therapy. The therapeutic strategies targeting the NLRP3 inflammasome for PD treatment include inhibitors of the NLRP3 inflammatory pathways, mitochondria-focused treatments, microRNAs, and other therapeutic compounds. However, even although inhibiting NLRP3 inflammasome activation has been shown to have therapeutic effects in PD animal models, the safety and effectiveness of these potential drugs need to be confirmed in PD patients in clinical trials. We should accelerate the progress of transforming basic research into clinical applications to achieve novel therapeutic strategies for PD.
References
Di Stefano A, Marinelli L. Advances in Parkinson’s disease drugs. Biomolecules 2021, 11: 1640.
Bloem BR, Okun MS, Klein C. Parkinson’s disease. Lancet 2021, 397: 2284–2303.
Pingale T, Gupta GL. Current and emerging therapeutic targets for Parkinson’s disease. Metab Brain Dis 2021, 36: 13–27.
Pfeiffer RF. Non-motor symptoms in Parkinson’s disease. Park Relat Disord 2016, 22: S119–S122.
Liu Y, Niu L, Liu X, Cheng C, Le W. Recent progress in non-motor features of parkinson’s disease with a focus on circadian rhythm dysregulation. Neurosci Bull 2021, 37: 1010–1024.
Li S, Wang Y, Wang F, Hu LF, Liu CF. A new perspective for parkinson’s disease: Circadian rhythm. Neurosci Bull 2017, 33: 62–72.
Lai TT, Kim YJ, Ma HI, Kim YE. Evidence of inflammation in parkinson’s disease and its contribution to synucleinopathy. J Mov Disord 2022, 15: 1–14.
Yao L, Wu J, Koc S, Lu G. Genetic imaging of neuroinflammation in parkinson’s disease: Recent advancements. Front Cell Dev Biol 2021, 9: 655819.
Teng JS, Ooi YY, Chye SM, Ling APK, Koh RY. Immunotherapies for parkinson’s disease: Progression of clinical development. CNS Neurol Disord Drug Targets 2021, 20: 802–813.
Rasheed M, Liang J, Wang C, Deng Y, Chen Z. Epigenetic regulation of neuroinflammation in parkinson’s disease. Int J Mol Sci 2021, 22: 4956.
Hirsch EC, Standaert DG. Ten unsolved questions about neuroinflammation in parkinson’s disease. Mov Disord 2021, 36: 16–24.
Heneka MT, McManus RM, Latz E. Inflammasome signalling in brain function and neurodegenerative disease. Nat Rev Neurosci 2018, 19: 610–621.
Streit WJ, Mrak RE, Griffin WS. Microglia and neuroinflammation: A pathological perspective. J Neuroinflammation 2004, 1: 14.
Morillas AG, Besson VC, Lerouet D. Microglia and neuroinflammation: What place for P2RY12? Int J Mol Sci 2021, 22: 1636.
Liu GJ, Middleton RJ, Hatty CR, Kam WWY, Chan R, Pham T. The 18 kDa translocator protein, microglia and neuroinflammation. Brain Pathol 2014, 24: 631–653.
Mullin S, Schapira A. α-synuclein and mitochondrial dysfunction in parkinson’s disease. Mol Neurobiol 2013, 47: 587–597.
Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469: 221–225.
Liu Q, Zhang D, Hu D, Zhou X, Zhou Y. The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol 2018, 103: 115–124.
Wang S, Yuan YH, Chen NH, Wang HB. The mechanisms of NLRP3 inflammasome/pyroptosis activation and their role in Parkinson’s disease. Int Immunopharmacol 2019, 67: 458–464.
Si XL, Fang YJ, Li LF, Gu LY, Yin XZ, Jun-Tian, et al. From inflammasome to Parkinson’s disease: Does the NLRP3 inflammasome facilitate exosome secretion and exosomal alpha-synuclein transmission in Parkinson’s disease? Exp Neurol 2021, 336: 113525.
Dubois H, Wullaert A, Lamkanfi M. General strategies in inflammasome biology. Curr Top Microbiol Immunol 2016, 397: 1–22.
Voet S, Srinivasan S, Lamkanfi M, van Loo G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 2019, 11: e10248.
Guan Y, Han F. Key mechanisms and potential targets of the NLRP3 inflammasome in neurodegenerative diseases. Front Integr Neurosci 2020, 14: 37.
Xiang H, Zhu F, Xu Z, Xiong J. Role of inflammasomes in kidney diseases via both canonical and non-canonical pathways. Front Cell Dev Biol 2020, 8: 106.
Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell 2014, 157: 1013–1022.
Lamkanfi M, Dixit VM. Inflammasomes and their roles in health and disease. Annu Rev Cell Dev Biol 2012, 28: 137–161.
Paik S, Kim JK, Silwal P, Sasakawa C, Jo EK. An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol 2021, 18: 1141–1160.
McKee CM, Coll RC. NLRP3 inflammasome priming: A riddle wrapped in a mystery inside an enigma. J Leukoc Biol 2020, 108: 937–952.
Kelley N, Jeltema D, Duan Y, He Y. The NLRP3 inflammasome: An overview of mechanisms of activation and regulation. Int J Mol Sci 2019, 20: 3328.
Swanson KV, Deng M, Ting JPY. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat Rev Immunol 2019, 19: 477–489.
Cullen SP, Kearney CJ, Clancy DM, Martin SJ. Diverse activators of the NLRP3 inflammasome promote IL-1β secretion by triggering necrosis. Cell Rep 2015, 11: 1535–1548.
Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K+ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38: 1142–1153.
Próchnicki T, Mangan MS, Latz E. Recent insights into the molecular mechanisms of the NLRP3 inflammasome activation. F1000Research 2016, 5: F1000FacultyRev–F1000Faculty1469.
Chen Y, Meng J, Bi F, Li H, Chang C, Ji C, et al. EK7 regulates NLRP3 inflammasome activation and neuroinflammation post-traumatic brain injury. Front Mol Neurosci 2019, 12: 202.
He Y, Zeng MY, Yang D, Motro B, Núñez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 2016, 530: 354–357.
Liu H, Gu C, Liu M, Liu G, Wang Y. NEK7 mediated assembly and activation of NLRP3 inflammasome downstream of potassium efflux in ventilator-induced lung injury. Biochem Pharmacol 2020, 177: 113998.
Liu R, Liu Y, Liu C, Gao A, Wang L, Tang H, et al. NEK7-mediated activation of NLRP3 inflammasome is coordinated by potassium efflux/syk/JNK signaling during Staphylococcus aureus infection. Front Immunol 2021, 12: 747370.
Rashidi M, Wicks IP, Vince JE. Inflammasomes and cell death: Common pathways in microparticle diseases. Trends Mol Med 2020, 26: 1003–1020.
Bai B, Yang Y, Wang Q, Li M, Tian C, Liu Y, et al. NLRP3 inflammasome in endothelial dysfunction. Cell Death Dis 2020, 11: 776.
Gaidt MM, Hornung V. Pore formation by GSDMD is the effector mechanism of pyroptosis. EMBO J 2016, 35: 2167–2169.
Taylor RC, Cullen SP, Martin SJ. Apoptosis: Controlled demolition at the cellular level. Nat Rev Mol Cell Biol 2008, 9: 231–241.
Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science 2016, 352: aaf2154.
Tsuchiya K, Hosojima S, Hara H, Kushiyama H, Mahib MR, Kinoshita T, et al. Gasdermin D mediates the maturation and release of IL-1α downstream of inflammasomes. Cell Rep 2021, 34: 108887.
de Vasconcelos NM, Lamkanfi M. Recent insights on inflammasomes, gasdermin pores, and pyroptosis. Cold Spring Harb Perspect Biol 2020, 12: a036392.
Fink SL, Cookson BT. Pillars Article: Caspase-1-dependent pore formation during pyroptosis leads to osmotic lysis of infected host macrophages. Cell Microbiol. 2006, 8: 1812–1825.
Zhang X, Wen X, Al-Ramahi I, Botas J, Lu B, Fu Y. Inhibition of HIPK3 by AST487 ameliorates mutant HTT-induced neurotoxicity and apoptosis via enhanced autophagy. Neurosci Bull 2022, 38: 99–103.
Jose S, Groves NJ, Roper KE, Gordon R. Mechanisms of NLRP3 activation and pathology during neurodegeneration. Int J Biochem Cell Biol 2022, 151:106273. https://doi.org/10.1016/j.biocel.2022.106273.
Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 2017, 547: 99–103.
Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 2017, 8: 14128.
Kovacs SB, Miao EA. Gasdermins: Effectors of pyroptosis. Trends Cell Biol 2017, 27: 673–684.
de Araújo FM, Cuenca-Bermejo L, Fernández-Villalba E, Costa SL, Diogenes A, Silva V, Herrero MT. Role of microgliosis and NLRP3 inflammasome in parkinson’s disease pathogenesis and therapy. Cell Mol Neurobiol 2022, 42: 1283–1300.
Holbrook JA, Jarosz-Griffiths HH, Caseley E, Lara-Reyna S, Poulter JA, Williams-Gray CH, et al. Neurodegenerative disease and the NLRP3 inflammasome. Front Pharmacol 2021, 12: 643254.
Feng YS, Tan ZX, Wu LY, Dong F, Zhang F. The involvement of NLRP3 inflammasome in the treatment of Alzheimer’s disease. Ageing Res Rev 2020, 64: 101192.
Feng YS, Tan ZX, Wu LY, Dong F, Zhang F. The involvement of NLRP3 inflammasome in the treatment of neurodegenerative diseases. Biomed Pharmacother 2021, 138: 111428.
Huang S, Chen Z, Fan B, Chen Y, Zhou L, Jiang B, et al. A selective NLRP3 inflammasome inhibitor attenuates behavioral deficits and neuroinflammation in a mouse model of Parkinson’s disease. J Neuroimmunol 2021, 354: 577543.
Zhang X, Xu A, Lv J, Zhang Q, Ran Y, Wei C, et al. Development of small molecule inhibitors targeting NLRP3 inflammasome pathway for inflammatory diseases. Eur J Med Chem 2020, 185: 111822.
Wang X, Chi J, Huang D, Ding L, Zhao X, Jiang L, et al. α-synuclein promotes progression of Parkinson’s disease by upregulating autophagy signaling pathway to activate NLRP3 inflammasome. Exp Ther Med 2020, 19: 931–938.
Fan Z, Pan YT, Zhang ZY, Yang H, Yu SY, Zheng Y, et al. Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation 2020, 17: 11.
Zhang P, Shao XY, Qi GJ, Chen Q, Bu LL, Chen LJ, et al. Cdk5-dependent activation of neuronal inflammasomes in parkinson’s disease. Mov Disord 2016, 31: 366–376.
Choong CJ, Mochizuki H. Neuropathology of α-synuclein in parkinson’s disease. Neuropathology 2022, 42: 93–103.
Tofaris GK. Initiation and progression of α-synuclein pathology in Parkinson’s disease. Cell Mol Life Sci 2022, 79: 210.
Taguchi K, Watanabe Y, Tsujimura A, Tanaka M. Brain region-dependent differential expression of alpha-synuclein. J Comp Neurol 2016, 524: 1236–1258.
Kim C, Ho DH, Suk JE, You S, Michael S, Kang J, et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat Commun 2013, 4: 1562.
Gómez-Benito M, Granado N, García-Sanz P, Michel A, Dumoulin M, Moratalla R. Modeling parkinson’s disease with the alpha-synuclein protein. Front Pharmacol 2020, 11: 356.
Le W, Dong J, Li S, Korczyn AD. Can biomarkers help the early diagnosis of parkinson’s disease? Neurosci Bull 2017, 33: 535–542.
Atik A, Stewart T, Zhang J. Alpha-synuclein as a biomarker for parkinson’s disease. Brain Pathol 2016, 26: 410–418.
Fayyad M, Salim S, Majbour N, Erskine D, Stoops E, Mollenhauer B, et al. Parkinson’s disease biomarkers based on α-synuclein. J Neurochem 2019, 150: 626–636.
Panicker N, Sarkar S, Harischandra DS, Neal M, Kam TI, Jin H, et al. Fyn kinase regulates misfolded α-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med 2019, 216: 1411–1430.
Daniele SG, Béraud D, Davenport C, Cheng K, Yin H, Maguire-Zeiss KA. Activation of MyD88-dependent TLR1/2 signaling by misfolded α-synuclein, a protein linked to neurodegenerative disorders. Sci Signal 2015, 8: ra45.
Pajares M, Rojo AI, Manda G, Boscá L, Cuadrado A. Inflammation in parkinson’s disease: Mechanisms and therapeutic implications. Cells 2020, 9: 1687.
Singh SS, Rai SN, Birla H, Zahra W, Rathore AS, Singh SP. NF-κB-mediated neuroinflammation in parkinson’s disease and potential therapeutic effect of polyphenols. Neurotox Res 2020, 37: 491–507.
Cheng X, Xu S, Zhang C, Qin K, Yan J, Shao X. The BRCC3 regulated by Cdk5 promotes the activation of neuronal NLRP3 inflammasome in Parkinson’s disease models. Biochem Biophys Res Commun 2020, 522: 647–654.
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, et al. Triggering of inflammasome by aggregated α-synuclein, an inflammatory response in synucleinopathies. PLoS One 2013, 8: e55375.
Pike AF, Varanita T, Herrebout MAC, Plug BC, Kole J, Musters RJP, et al. α-Synuclein evokes NLRP3 inflammasome-mediated IL-1β secretion from primary human microglia. Glia 2021, 69: 1413–1428.
Qin Y, Qiu J, Wang P, Liu J, Zhao Y, Jiang F, et al. Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav Immun 2021, 91: 324–338.
Cheng ZY, Xia QP, Hu YH, Wang C, He L. Dopamine D1 receptor agonist A-68930 ameliorates Aβ1-42-induced cognitive impairment and neuroinflammation in mice. Int Immunopharmacol 2020, 88: 106963.
Jiang W, Li M, He F, Bian Z, Liu J, He Q, et al. Dopamine D1 receptor agonist A-68930 inhibits NLRP3 inflammasome activation and protects rats from spinal cord injury-induced acute lung injury. Spinal Cord 2016, 54: 951–956.
Jiang W, Huang Y, He F, Liu J, Li M, Sun T, et al. Dopamine D1 receptor agonist A-68930 inhibits NLRP3 inflammasome activation, controls inflammation, and alleviates histopathology in a rat model of spinal cord injury. Spine 2016, 41: E330–E334.
Wang T, Nowrangi D, Yu L, Lu T, Tang J, Han B, et al. Activation of dopamine D1 receptor decreased NLRP3-mediated inflammation in intracerebral hemorrhage mice. J Neuroinflammation 2018, 15: 2.
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z, et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015, 160: 62–73.
Pike AF, Longhena F, Faustini G, van Eik JM, Gombert I, Herrebout MAC, et al. Dopamine signaling modulates microglial NLRP3 inflammasome activation: Implications for Parkinson’s disease. J Neuroinflammation 2022, 19: 50.
Pfanner N, Warscheid B, Wiedemann N. Mitochondrial proteins: From biogenesis to functional networks. Nat Rev Mol Cell Biol 2019, 20: 267–284.
Jornayvaz FR, Shulman GI. Regulation of mitochondrial biogenesis. Essays Biochem 2010, 47: 69–84.
Popov LD. Mitochondrial biogenesis: An update. J Cell Mol Med 2020, 24: 4892–4899.
Ploumi C, Daskalaki I, Tavernarakis N. Mitochondrial biogenesis and clearance: A balancing act. FEBS J 2017, 284: 183–195.
Frey TG, Mannella CA. The internal structure of mitochondria. Trends Biochem Sci 2000, 25: 319–324.
Nunnari J, Suomalainen A. Mitochondria: In sickness and in health. Cell 2012, 148: 1145–1159.
Sarkar S, Malovic E, Harishchandra DS, Ghaisas S, Panicker N, Charli A, et al. Mitochondrial impairment in microglia amplifies NLRP3 inflammasome proinflammatory signaling in cell culture and animal models of Parkinson’s disease. NPJ Parkinsons Dis 2017, 3: 30.
Kolmychkova KI, Zhelankin AV, Karagodin VP, Orekhov AN. Mitochondria and inflammation. Patol Fiziol Eksp Ter 2016, 60: 114–121.
Subramaniam SR, Chesselet MF. Mitochondrial dysfunction and oxidative stress in Parkinson’s disease. Prog Neurobiol 2013, 106–107: 17–32.
Banerjee R, Starkov AA, Beal MF, Thomas B. Mitochondrial dysfunction in the limelight of Parkinson’s disease pathogenesis. Biochim Biophys Acta 2009, 1792: 651–663.
Litwiniuk A, Baranowska-Bik A, Domańska A, Kalisz M, Bik W. Contribution of mitochondrial dysfunction combined with NLRP3 inflammasome activation in selected neurodegenerative diseases. Pharmaceuticals (Basel) 2021, 14: 1221.
Su CJ, Shen Z, Cui RX, Huang Y, Xu DL, Zhao FL, et al. Thioredoxin-interacting protein (TXNIP) regulates parkin/PINK1-mediated mitophagy in dopaminergic neurons under high-glucose conditions: Implications for molecular links between parkinson’s disease and diabetes. Neurosci Bull 2020, 36: 346–358.
Lee E, Hwang I, Park S, Hong S, Hwang B, Cho Y, et al. MPTP-driven NLRP3 inflammasome activation in microglia plays a central role in dopaminergic neurodegeneration. Cell Death Differ 2019, 26: 213–228.
Zhang X, Huang W, Shao Q, Yang Y, Xu Z, Chen J, et al. Drp1, a potential therapeutic target for Parkinson’s disease, is involved in olfactory bulb pathological alteration in the Rotenone-induced rat model. Toxicol Lett 2020, 325: 1–13.
Ahmed S, Panda SR, Kwatra M, Sahu BD, Naidu V. Perillyl alcohol attenuates NLRP3 inflammasome activation and rescues dopaminergic neurons in experimental in vitro and in vivo models of parkinson’s disease. ACS Chem Neurosci 2022, 13: 53–68.
Qiu J, Chen Y, Zhuo J, Zhang L, Liu J, Wang B, et al. Urolithin A promotes mitophagy and suppresses NLRP3 inflammasome activation in lipopolysaccharide-induced BV2 microglial cells and MPTP-induced Parkinson’s disease model. Neuropharmacology 2022, 207: 108963.
Li Y, Xia Y, Yin S, Wan F, Hu J, Kou L, et al. Targeting microglial α-synuclein/TLRs/NF-kappaB/NLRP3 inflammasome axis in parkinson’s disease. Front Immunol 2021, 12: 719807.
Stevenson T. Drug therapy in the management of Parkinson’s disease. Br J Nurs 1997, 6(144–148): 150.
Goetz CG, Pal G. Initial management of Parkinson’s disease. BMJ 2014, 349: g6258.
Waller S, Williams L, Morales-Briceño H, Fung VS. The initial diagnosis and management of Parkinson’s disease. Aust J Gen Pract 2021, 50: 793–800.
Qiu X, Wang Q, Hou L, Zhang C, Wang Q, Zhao X. Inhibition of NLRP3 inflammasome by glibenclamide attenuated dopaminergic neurodegeneration and motor deficits in paraquat and maneb-induced mouse Parkinson’s disease model. Toxicol Lett 2021, 349: 1–11.
Mao Z, Liu C, Ji S, Yang Q, Ye H, Han H, et al. The NLRP3 inflammasome is involved in the pathogenesis of parkinson’s disease in rats. Neurochem Res 2017, 42: 1104–1115.
Qiao C, Zhang LX, Sun XY, Ding JH, Lu M, Hu G. Caspase-1 deficiency alleviates dopaminergic neuronal death via inhibiting caspase-7/AIF pathway in MPTP/p mouse model of parkinson’s disease. Mol Neurobiol 2017, 54: 4292–4302.
Wang W, Nguyen LTT, Burlak C, Chegini F, Guo F, Chataway T, et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein α-synuclein. Proc Natl Acad Sci U S A 2016, 113: 9587–9592.
Pellegrini C, D’Antongiovanni V, Miraglia F, Rota L, Benvenuti L, Di Salvo C, et al. Enteric α-synuclein impairs intestinal epithelial barrier through caspase-1-inflammasome signaling in Parkinson’s disease before brain pathology. NPJ Parkinsons Dis 2022, 8: 9.
Vila M, Ramonet D, Perier C. Mitochondrial alterations in Parkinson’s disease: New clues. J Neurochem 2008, 107: 317–328.
Li Q, Wang Z, Xing H, Wang Y, Guo Y. Exosomes derived from miR-188-3p-modified adipose-derived mesenchymal stem cells protect Parkinson’s disease. Mol Ther Nucleic Acids 2021, 23: 1334–1344.
Cao H, Han X, Jia Y, Zhang B. Inhibition of long non-coding RNA HOXA11-AS against neuroinflammation in Parkinson’s disease model via targeting miR-124-3p mediated FSTL1/NF-κB axis. Aging 2021, 13: 11455–11469.
Zeng R, Luo DX, Li HP, Zhang QS, Lei SS, Chen JH. MicroRNA-135b alleviates MPP+-mediated Parkinson’s disease in in vitro model through suppressing FoxO1-induced NLRP3 inflammasome and pyroptosis. J Clin Neurosci 2019, 65: 125–133.
Sun Q, Wang S, Chen J, Cai H, Huang W, Zhang Y, et al. MicroRNA-190 alleviates neuronal damage and inhibits neuroinflammation via Nlrp3 in MPTP-induced Parkinson’s disease mouse model. J Cell Physiol 2019, 234: 23379–23387.
Li D, Yang H, Ma J, Luo S, Chen S, Gu Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum Cell 2018, 31: 106–115.
Wang R, Li Q, He Y, Yang Y, Ma Q, Li C. MiR-29c-3p inhibits microglial NLRP3 inflammasome activation by targeting NFAT5 in Parkinson’s disease. Genes Cells 2020, 25: 364–374.
Zhou Y, Lu M, Du RH, Qiao C, Jiang CY, Zhang KZ, et al. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol Neurodegeneration 2016, 11: 28.
Zhang Q, Huang XM, Liao JX, Dong YK, Zhu JL, He CC, et al. LncRNA HOTAIR promotes neuronal damage through facilitating NLRP3 mediated-pyroptosis activation in parkinson’s disease via regulation of miR-326/ELAVL1 axis. Cell Mol Neurobiol 2021, 41: 1773–1786.
Sun Q, Zhang Y, Wang S, Yang F, Cai H, Xing Y, et al. NEAT1 decreasing suppresses parkinson’s disease progression via acting as miR-1301-3p sponge. J Mol Neurosci 2021, 71: 369–378.
Chen L, Xue L, Zheng J, Tian X, Zhang Y, Tong Q. PPARß/δ agonist alleviates NLRP3 inflammasome-mediated neuroinflammation in the MPTP mouse model of Parkinson’s disease. Behav Brain Res 2019, 356: 483–489.
Zheng R, Ruan Y, Yan Y, Lin Z, Xue N, Yan Y, et al. Melatonin attenuates neuroinflammation by down-regulating NLRP3 inflammasome via a SIRT1-dependent pathway in MPTP-induced models of parkinson’s disease. J Inflamm Res 2021, 14: 3063–3075.
Zhang M, He Q, Chen G, Li PA. Suppression of NLRP3 inflammasome, pyroptosis, and cell death by NIM811 in rotenone-exposed cells as an in vitro model of parkinson’s disease. Neurodegener Dis 2020, 20: 73–83.
Wu AG, Zhou XG, Qiao G, Yu L, Tang Y, Yan L, et al. Targeting microglial autophagic degradation in NLRP3 inflammasome-mediated neurodegenerative diseases. Ageing Res Rev 2021, 65: 101202.
Glick D, Barth S, MacLeod KF. Autophagy: Cellular and molecular mechanisms. J Pathol 2010, 221: 3–12.
Yu L, Chen Y, Tooze SA. Autophagy pathway: Cellular and molecular mechanisms. Autophagy 2018, 14: 207–215.
Karabiyik C, Frake RA, Park SJ, Pavel M, Rubinsztein DC. Autophagy in ageing and ageing-related neurodegenerative diseases. Ageing Neurodegener Dis 2021, 1–2. https://doi.org/10.20517/and.2021.05
Coll RC, Robertson AAB, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC, et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015, 21: 248–255.
Schwaid AG, Spencer KB. Strategies for targeting the NLRP3 inflammasome in the clinical and preclinical space. J Med Chem 2021, 64: 101–122.
Klück V, Th A Jansen TL, Janssen M, Comarniceanu A, Efdé M, Tengesdal IW, et al. Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: An open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol 2020, 2: e270–e280.
Tate MD, Ong JDH, Dowling JK, McAuley JL, Robertson AB, Latz E, et al. Reassessing the role of the NLRP3 inflammasome during pathogenic influenza A virus infection via temporal inhibition. Sci Rep 2016, 6: 27912.
Witzenrath M, Pache F, Lorenz D, Koppe U, Gutbier B, Tabeling C, et al. The NLRP3 inflammasome is differentially activated by pneumolysin variants and contributes to host defense in pneumococcal pneumonia. J Immunol 2011, 187: 434–440.
Kasper L, König A, Koenig PA, Gresnigt MS, Westman J, Drummond RA, et al. The fungal peptide toxin Candidalysin activates the NLRP3 inflammasome and causes cytolysis in mononuclear phagocytes. Nat Commun 2018, 9: 4260.
Xiong B, Wang Y, Chen Y, Xing S, Liao Q, Chen Y, et al. Strategies for structural modification of small molecules to improve blood-brain barrier penetration: A recent perspective. J Med Chem 2021, 64: 13152–13173.
Tansey MG, Wallings RL, Houser MC, Herrick MK, Keating CE, Joers V. Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 2022, 22: 657–673.
Öberg M, Fabrik I, Fabrikova D, Zehetner N, Härtlova A. The role of innate immunity and inflammation in Parkinson´s disease. Scand J Immunol 2021, 93: e13022.
Li Y, Chen Y, Jiang L, Zhang J, Tong X, Chen D, et al. Intestinal inflammation and parkinson’s disease. Aging Dis 2021, 12: 2052–2068.
Quijano A, Diaz-Ruiz C, Lopez-Lopez A, Villar-Cheda B, Muñoz A, Rodriguez-Perez AI, et al. Angiotensin type-1 receptor inhibition reduces NLRP3 inflammasome upregulation induced by aging and neurodegeneration in the Substantia nigra of male rodents and primary mesencephalic cultures. Antioxidants (Basel) 2022, 11: 329.
von Herrmann KM, Salas LA, Martinez EM, Young AL, Howard JM, Feldman MS, et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. NPJ Parkinsons Dis 2018, 4: 24.
Acknowledgments
This review was supported by Shanghai Municipal Central Government funds for guiding local scientific and technological development (YDZX20213100001002).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Han, QQ., Le, W. NLRP3 Inflammasome-Mediated Neuroinflammation and Related Mitochondrial Impairment in Parkinson’s Disease. Neurosci. Bull. 39, 832–844 (2023). https://doi.org/10.1007/s12264-023-01023-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12264-023-01023-y