
Summary
Allogeneic stem cell transplantation (alloSCT) is the only curative treatment option for patients with high-risk myelofibrosis (MF). However, it is important to bear in mind that alloSCT in MF is associated with a nonrelapse mortality that should not be underestimated. Therefore, both exact disease risk categorization and thorough evaluation of the individual transplant-related risk are mandatory to identify those patients to whom alloSCT should be offered. This short review is intended to provide a concise overview on relevant aspects to be considered for patient selection, planning, and performing alloSCT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Myelofibrosis (MF), including the subentities primary myelofibrosis (PMF), prefibrotic myelofibrosis (prePMF), postessential thrombocythemia myelofibrosis (postET MF), and postpolycythemia vera myelofibrosis (postPV MF), in general is a high-risk disease, mainly due to the risk of leukemic transformation in about 25% of patients within 10 years [1]. Despite recent progress of drug therapy (especially janus kinase (JAK) inhibition), allogeneic stem cell transplantation (alloSCT) is still the only curative treatment with improvement in overall survival (OS) during the last 20 years, reaching 5‑year OS rates > 50% [2,3,4]. The challenge is to identify the right candidates and the best timepoint for alloSCT and to define optimized strategies of how to perform the procedure. This article provides an overview of these key issues in the treatment of patients with MF.
Methods
This review is based on Onkopedia guidelines by the Deutsche/Österreichische/Schweizerische Gesellschaften für Hämatologie und Onkologie, the European Society for Blood and Marrow Transplantation (EBMT), and the European Leukemia Net (ELN), including a screening of PubMed with the keywords myelofibrosis and allogeneic SCT.
Patient selection for alloSCT
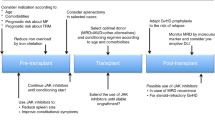
After diagnosis of MF, classification of the imminent disease risk is crucial. The risk scores established for the respective MF subentity should be used for this purpose. For PMF and prePMF, these are the Dynamic International Prognostic Scoring System (DIPSS(-plus)) score or Mutation-enhanced International Prognostic Scoring System Version2 (MIPSS-v2) [5,6,7]. The latter should be preferred, as beyond cytogenetics prognostically important molecular genetic parameters are also included. Therefore, in addition to the karyogram and the classical driver mutations, the well-known molecular high-risk mutations should be determined (Table 1). With this information, MF patients can be categorized into different risk groups. This should not only be done at initial diagnosis, but also repeatedly during follow-up to detect possible progression. For patients with postET/PV MF, the MYelofibrosis SECondary to PV and ET (MYSEC score) has been established [8].
Based on these scores, the disease can then be classified into four risk categories (low/intermediate-1/intermediate-2/high-risk) according to DIPSS-plus or the 5‑level classification according to MIPPS-v2 (very low/low/intermediate/high/very high risk). Ten-year overall survival (OS) according to MIPPS-v2 ranges from 86 to 3% [7]. In general, the lower risk groups are initially just monitored (watch and wait strategy) or treated by drug therapy, in particular JAK inhibitors. In contrast, higher risk patients in principle have an indication for alloSCT (Fig. 1). This separation is mainly based on a large retrospective study published before the JAK inhibitor era, including 443 patients from several registries, 188 treated by alloSCT and 255 by conventional therapy [3]. The study showed that patients with DIPSS intermediate‑2 risk or high risk benefit from an alloSCT in terms of OS, while patients with low risk had a worse outcome after transplantation. For patients with intermediate‑1 risk, OS benefit was evident after 15–20 years only. Hence, current guidelines recommend alloSCT in intermediate‑1 risk if additional individual risk factors, such as TP53 mutations, are identified [9, 10].

Algorithm of action after diagnosis of myelofibrosis (MF)
Independently from current risk status, each newly diagnosed MF patient that might be eligible for alloSCT based on age and comorbidities should be referred to a transplant center for counseling. In patients without a clear transplant indication at that timepoint, we find it reasonable to perform HLA typing and screening of the core family for a potential donor to be prepared for the case of a later disease progression, which may then define the indication for alloSCT. These patients must be carefully monitored in order not to miss the timepoint for another referral to the transplant center once disease progression occurs.
In patients with indication for alloSCT on the basis of their individual risk score, an immediate search for both related and unrelated donors is indicated. Furthermore, the particular probability of survival after alloSCT must be calculated using the Myelofibrosis Transplantation Scoring System (MTSS) [11]. In addition to clinical and molecular parameters (age, KPS, leucocytes, platelets, CARL/MPL/ASXL1), the score considers the degree of HLA match between patient and a potential donor. This results in a 4-level score for the 5‑year OS probability, ranging from 34 to 90%. Hence, patients with a disease risk suggesting an indication for alloSCT are finally regarded as good transplant candidates if they achieve low and intermediate transplant-related risk according to MTSS, while the decision in patients with MTSS high risk or very high risk must be individualized (Fig. 1; [10, 11]). Since the MTSS does not include comorbidities, it may be useful to additionally use the general risk score for alloSCT (HCT-CI), especially since it has recently been shown to be a valid tool for MF as well [12]. Once indicated, alloSCT should be planned and performed as soon as possible. As outlined below, specific treatment in particular using ruxolitinib might be indicated before proceeding to alloSCT.
Performing alloSCT
Prospective clinical trials in MF are scarce in the context of alloSCT. Hence, recommendations are essentially based on retrospective analyses, indirect comparisons, and expert opinions. In addition to the general proceedings of alloSCT, three main questions are of particular interest when alloSCT is performed for MF: (1) treatment prior to planned alloSCT, (2) donor type, and (3) the conditioning regimen.
Treatment prior to alloSCT
Constitutional symptoms and symptomatic splenomegaly are relevant clinical problems in MF in the context of alloSCT, as they are associated with a higher rate of graft failure. To address this problem, pretreatment with the JAK inhibitor ruxolitinib before alloSCT has become an established standard. Two prospective single-arm phase 2 studies showed that ruxolitinib can reduce spleen size and constitutional symptoms before alloSCT with good engraftment and posttransplant outcome [13, 14]. In a large EBMT study, engraftment was superior in patients who responded to ruxolitinib prior to alloSCT compared to nonresponders or patients not receiving ruxolitinib. Two-year event-free survival was superior after ruxolitinib pretreatment, while OS was similar [15]. The use of second-generation JAK inhibitors (e.g., fedratinib or momelotinib) before alloSCT is not yet supported by large-scale data, but may represent an option in case of ruxolitinib failure [16]. Patients with persisting splenomegaly after treatment with JAK inhibitors may benefit from splenectomy or splenic irradiation with respect to reduction of graft failure and relapse risk. However, the relevance of these procedures for final outcome has been debated. Especially splenectomy is associated with risk of the procedure itself, whereas splenic irradiation might be associated with hematotoxicity and limited efficacy. Therefore, both procedures should be evaluated and weighted due to local experience and individual patient conditions [16, 17].
The treatment of patients with blast phase/sAML prior to alloSCT is a challenging condition which is discussed elsewhere [10].
Donor selection
Even more than in other diseases, the availability of an HLA-matched sibling or unrelated donor plays a major role in MF, as HLA-mismatched transplantation has been associated with inferior outcome. This had been described earlier, but was confirmed recently in the context of the establishment of the MTSS [11, 18]. Umbilical cord blood transplantation was associated with a high rate of graft failure and is not routinely recommended [19]. The increasingly widespread use of haploidentical (haplo) SCT has also reached MF. Improving results in terms of OS suggested that haploSCT could be an option in lack of a matched donor [20, 21].
Conditioning
According to EBMT definitions, reduced intensity conditioning (RIC) regimen can be distinguished from standard, myeloablative (MAC) protocols [22]. In MF, two large registry studies have shown comparable OS following RIC and MAC transplants (5-year OS 51% versus 53%, and 54 versus 49%, respectively) [23, 24]. In a recent analysis, MAC was not even beneficial in patients with genetically defined high-risk disease [25]. Nevertheless, MAC could be an option for younger and fit patients, as GvHD-free, relapse-free survival was significantly superior for MAC versus RIC and KPS > 80% or age < 50 years showed to be associated with superior OS and NRM in the retrospective EBMT study [23]. The two most frequently used RIC regimen comprise fludarabine/busulfan and fludarabine/melphalan. OS was not different between these two protocols (7-year OS 59% versus 52%), while relapse incidence was higher and GvHD was lower with fludarabine/busulfan in a retrospective EBMT analysis [26]. In a further study, fludarabine/busulfan led to significant superior survival compared to fludarabine/melphalan [27]. Recently, other approaches such as the use of fludarabine/treosulfan or the addition of low-dose total body irradiation or thiotepa to fludarabine/busulfan have been published showing further improvement in alloSCT for MF [28,29,30].
Follow up after alloSCT
Patient care after alloSCT for MF follows the standard principles. Molecular monitoring in particular JAK 2 is a useful tool for early identification of incipient relapse.
Conclusion
Despite improved drug therapy, stem cell transplantation (alloSCT) is still the only curative treatment for myelofibrosis (MF) and should be offered to higher risk patients, with an acceptable estimated probability of survival after alloSCT. Validated scores are available to calculate both disease and transplant risk at diagnosis and during the course of the disease. To identify the best timepoint for alloSCT is challenging. Therefore, patients should be offered to be presented to an experienced transplant center after diagnosis. Patients initially not selected for alloSCT due to low-risk status should be closely monitored for progression. Patients selected for alloSCT should receive ruxolitinib to reduce constitutional symptoms and spleen volume before alloSCT. For the implementation of alloSCT, an HLA-matched donor and a reduced intensity conditioning (RIC) regimen (exception: young and fit patients) are preferred.
Take home message
AlloSCT is the only curative treatment option. Therefore, a risk assessment should be done at diagnosis and during the disease course to offer alloSCT to eligible patients at the optimal timepoint.
References
Barbui T, Thiele J, Gisslinger H, et al. The 2016 WHO classification and diagnostic criteria for myeloproliferative neoplasms: document summary and in-depth discussion. Blood Cancer J. 2018;8:15.
Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT‑I and -II pooled analyses. J Hematol Oncol. 2017;10:156.
Kroger N, Giorgino T, Scott BL, et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood. 2015;125:3347–50.
CIBMTR. summary slides. 2019. http://cibmtr.org.
Gangat N, Caramazza D, Vaidya R, et al. DIPSS plus: a refined dynamic international prognostic scoring system for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. 2011;29:392–7.
Guglielmelli P, Lasho TL, Rotunno G, et al. MIPSS70: mutation-enhanced international prognostic score system for transplantation-age patients with primary myelofibrosis. J Clin Oncol. 2018;36:310–8.
Tefferi A, Guglielmelli P, Lasho TL, et al. MIPSS70+ version 2.0: mutation and karyotype-enhanced international prognostic scoring system for primary myelofibrosis. J Clin Oncol. 2018; 1769–70.
Passamonti F, Giorgino T, Mora B, et al. A clinical-molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia. 2017;31:2726–31.
Gagelmann N, Badbaran A, Salit RB, et al. Impact of TP53 on outcome of patients with myelofibrosis undergoing hematopoietic stem cell transplantation. Blood. 2023;141:2901–11.
Kröger N, Bacigalupo A, Barbui T, et al. Indication and management of allogeneic haematopoietic stem-cell transplantation in myelofibrosis: updated recommendations by the EBMT/ELN International Working Group. Lancet Haematol. 2024;11:e62–e74.
Gagelmann N, Ditschkowski M, Bogdanov R, et al. Comprehensive clinical-molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood. 2019;133:2233–42.
Polverelli N, Bonneville EF, de Wreede LC, et al. Impact of comorbidities and body mass index on the outcomes of allogeneic hematopoietic cell transplantation in myelofibrosis: a study on behalf of the chronic malignancies working party of EBMT. Am J Hematol. 2024;99:993–6.
Salit RB, Scott BL, Stevens EA, et al. Pre-hematopoietic cell transplant ruxolitinib in patients with primary and secondary myelofibrosis. Bone Marrow Transplant. 2020;55:70–6.
Gupta V, Kosiorek HE, Mead A, et al. Ruxolitinib therapy followed by reduced-intensity conditioning for hematopoietic cell transplantation for myelofibrosis: myeloproliferative disorders research consortium 114 study. Biol Blood Marrow Transplant. 2019;25:256–64.
Kroger N, Sbianchi G, Sirait T, et al. Impact of prior JAK-inhibitor therapy with ruxolitinib on outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis: a study of the CMWP of EBMT. Leukemia. 2021;35:3551–60.
Polverelli N, Hernández-Boluda JC, Czerw T, et al. Splenomegaly in patients with primary or secondary myelofibrosis who are candidates for allogeneic hematopoietic cell transplantation: a position paper on behalf of the chronic malignancies working party of the EBMT. Lancet Haematol. 2023;10:e59–e70.
Gagelmann N, Hobbs GS, Campodonico E, et al. Splenic irradiation for myelofibrosis prior to hematopoietic cell transplantation: a global collaborative analysis. Am J Hematol. 2024;99:844–53.
Kröger N, Holler E, Kobbe G, et al. Allogeneic stem cell transplantation after reduced-intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the chronic leukemia working party of the European group for blood and marrow transplantation. Blood. 2009;114:5264–70.
Robin M, Giannotti F, Deconinck E, et al. Unrelated cord blood transplantation for patients with primary or secondary myelofibrosis. Biol Blood Marrow Transplant. 2014;20:1841–6.
Raj K, Eikema DJ, McLornan DP, et al. Family mismatched allogeneic stem cell transplantation for myelofibrosis: report from the chronic malignancies working party of European Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2019;25:522–8.
Kunte S, Rybicki L, Viswabandya A, et al. Allogeneic blood or marrow transplantation with haploidentical donor and post-transplantation cyclophosphamide in patients with myelofibrosis: a multicenter study. Leukemia. 2022;36:856–64.
Shimoni ARV, Nagler A. Conditioning. The EBMT handbook. 2024. pp. 125–134.
McLornan D, Szydlo R, Koster L, et al. Myeloablative and reduced-intensity conditioned allogeneic hematopoietic stem cell transplantation in myelofibrosis: a retrospective study by the chronic malignancies working party of the European Society for Blood and Marrow Transplantation. Biol Blood Marrow Transplant. 2019;25:2167–71.
Kim DH, Seo J, Shin DY, et al. Reduced-intensity conditioning versus myeloablative conditioning allogeneic stem cell transplantation for patients with myelofibrosis. Blood Res. 2022;57:264–71.
Gagelmann N, Salit RB, Schroeder T, et al. High molecular and Cytogenetic risk in Mmelofibrosis does not benefit from higher intensity conditioning before Hematopoietic cell transplantation: an international collaborative analysis. Hemasphere. 2022;6:e784.
Robin M, Porcher R, Wolschke C, et al. Outcome after transplantation according to reduced-intensity conditioning regimen in patients undergoing transplantation for myelofibrosis. Biol Blood Marrow Transplant. 2016;22:1206–11.
Murthy GSG, Kim S, Estrada-Merly N, et al. Association between the choice of the conditioning regimen and outcomes of allogeneic hematopoietic cell transplantation for myelofibrosis. Haematologica. 2023;108:1900–8.
Robin M, Iacobelli S, Koster L, et al. Treosulfan compared to busulfan in allogeneic haematopoietic stem cell transplantation for myelofibrosis: a registry-based study from the chronic malignancies working party of the EBMT. Bone Marrow Transplant. 2024. e-pub ahead of print 2024/03/16 21:43
Freyer CW, Babushok DV, Frey NV, et al. Low-dose total body irradiation added to fludarabine and busulfan reduced-intensity conditioning reduces graft failure in patients with myelofibrosis. Transplant Cell Ther. 2022;28:590–6.
Battipaglia G, Mauff K, Wendel L, et al. Thiotepa-busulfan-fludarabine (TBF) conditioning regimen in patients undergoing allogeneic hematopoietic cell transplantation for myelofibrosis: an outcome analysis from the chronic malignancies working party of the EBMT. Bone Marrow Transplant. 2021;56:1593–602.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
K. Hirschbühl and C. Schmid declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hirschbühl, K., Schmid, C. Importance of allogeneic stem cell transplantation in myelofibrosis. memo (2024). https://doi.org/10.1007/s12254-024-00987-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12254-024-00987-5