
Abstract
Recycling of phosphorus (P) from sediments contributes to the development of bottom-water hypoxia in many coastal systems. Here, we present results of a year-long assessment of P dynamics in sediments of a seasonally hypoxic coastal marine basin (Lake Grevelingen, the Netherlands) in 2012. Sequential phosphorus extractions (SEDEX) and X-ray absorption spectroscopy (XAS) indicate that P was adsorbed to Fe-(III)-(oxyhydr)oxides when cable bacteria were active in the surface sediments in spring. With the onset of summer hypoxia, sulphide-induced dissolution of the Fe-(III)-(oxyhydr)oxides led to P release to the pore water and overlying water. The similarity in authigenic Ca-P concentrations in the sediment and suspended matter suggest that Ca-P is not formed in situ. The P burial efficiency was ≤ 32%. Hypoxia-driven sedimentary P recycling had a major impact on the water-column chemistry in the basin in 2012. Water-column monitoring data indicate up to ninefold higher surface water concentrations of phosphate in the basin in the late 1970s and a stronger hypoxia-driven seasonal P release from the sediment. The amplified release of P from the sediment in the past is attributed to the presence of a larger pool of Fe-bound P in the basin prior to the first onset of hypoxia. Given that P is not limiting, primary production in the basin has not been affected by the decadal changes in P availability and recycling over the past 40 years. The changes in P dynamics on decadal time scales were not recorded in sediment profiles of total P or organic C/total P.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Phosphorus (P) is an essential nutrient for marine organisms (Arrigo 2005). Human activities on land, such as the widespread use of fertiliser and the release of waste water to streams and rivers have led to increased inputs of nutrients to many coastal environments since the 1950s (Beusen et al. 2016; Diaz and Rosenberg 2008; Rabalais et al. 2010; Ruttenberg 2003). Enhanced P availability in coastal systems can stimulate algal blooms and the development of bottom-water hypoxia (i.e. oxygen concentrations < 63 μM; (Carstensen et al. 2014; Middelburg and Levin 2009). The number of coastal environments suffering from hypoxia is still increasing worldwide (Diaz and Rosenberg 2008; Rabalais et al. 2014).
Sedimentary P cycling is very sensitive to changes in bottom-water oxygen. In sediments overlain by oxic bottom waters, phosphate released to the pore water from degrading sediment organic matter can be bound to iron (Fe)(oxyhydr)oxides (Anschutz et al. 1998; Mort et al. 2010; Mortimer 1941; Slomp et al. 1996; Sundby et al. 1992) or can be taken up by aerobic micro-organisms in the form of polyphosphates (Gächter and Meyer 1993; Ingall et al. 1993), thereby preventing the escape of P to the overlying water. With the onset of bottom-water hypoxia, P bound to Fe-(oxyhydr)oxides and in polyphosphates is generally remobilised and released to the overlying water, where it can support primary productivity and contribute to the further development of hypoxia (Ingall et al. 1993; Ruttenberg 2003). Some of the phosphate released to the pore water can also precipitate in the form of authigenic P minerals such as the calcium (Ca)-P mineral carbonate fluorapatite (Ruttenberg and Berner 1993; Slomp et al. 1996) and vivianite, an Fe(II)-P mineral (Egger et al. 2015; Jilbert and Slomp 2013; Martens et al. 1978; März et al. 2008). The factors controlling the formation of such authigenic minerals, which can act as a key sink for P in sediments of seasonally hypoxic basins, are still incompletely understood. This hampers accurate model projections of the recovery of eutrophic systems from hypoxia (Gustafsson et al. 2012; Meier et al. 2011; Soetaert and Middelburg 2009).
Large filamentous sulphur-oxidising bacteria such as Beggiatoa and Thiomargarita have been suggested to play a critical role in the formation of authigenic Ca-P in hypoxic marine systems (Dale et al. 2013; Goldhammer et al. 2010; Schulz and Schulz 2005). These bacteria release phosphate from internally stored polyphosphates upon exposure to anoxia and sulphide, as demonstrated in laboratory experiments for Beggiatoa (Brock and Schulz-Vogt 2011). The bacterial release of phosphate from polyphosphates in Thiomargarita namibiensis has been shown to lead to the episodic formation of phosphorus-rich minerals in sediments off the coast of Namibia (Schulz and Schulz 2005). This effect of sulphur-oxidising bacteria on authigenic Ca-P formation is likely not ubiquitous in marine sediments, however. For example, no evidence for authigenic Ca-P formation was found in hypoxic sediments of the Baltic Sea, despite the known presence of Beggiatoa (Mort et al. 2010). Furthermore, in a recent study of P cycling at a site in a seasonally hypoxic coastal marine basin (Lake Grevelingen, the Netherlands), Sulu-Gambari et al. (2016a) demonstrated a lack of authigenic Ca-P formation at a site where polyphosphate-bearing Beggiatoaceae were abundant in autumn. In the latter setting, cable bacteria, a group of sulphur-oxidising bacteria from the Desulfobulbaceae family (Burdorf et al. 2016; Nielsen et al. 2010; Pfeffer et al. 2012; Risgaard-Petersen et al. 2012; Trojan et al. 2016) that can also form polyphosphates in their cells were active in spring (Sulu-Gambari et al. 2016a).
Cable bacteria can directly link the oxidation of sulphide to the reduction of oxygen over distances of several centimetres (Nielsen et al. 2010; Pfeffer et al. 2012). This typically leads to a distinct pH fingerprint with a surficial pH peak due to proton consumption via the reduction of oxygen and a pH minimum at depth due to the oxidation of sulphide (H2S) deeper in the sediment (Meysman et al. 2015). Both laboratory (Rao et al. 2016; Risgaard-Petersen et al. 2012) and field studies (Seitaj et al. 2015; Sulu-Gambari et al. 2016a) have demonstrated that the acidification of the pore water at depth leads to the dissolution of iron monosulphide (FeS) and calcium carbonate (CaCO3) and subsequent formation of Fe-(oxyhydr)oxides upon oxidation of upward diffusing Fe2+, with oxygen (Rao et al. 2016; Risgaard-Petersen et al. 2012; van de Velde et al. 2016) or with Mn-oxides (Sulu-Gambari et al. 2016a; b).
In sediments where cable bacteria are active, upward-diffusing dissolved phosphate is retained in the surface sediment and does not escape to the overlying water (Rao et al. 2016; Sulu-Gambari et al. 2016a). This has been attributed to binding of P to Fe-(oxyhydr)oxides, which is supported by the presence of a large stock of Fe-bound P in the surface sediment (Sulu-Gambari et al. 2016a). In this latter study, however, the stock of Fe-bound P was determined with an operational SEDEX extraction (Ruttenberg 1992) that can also extract vivianite (Dijkstra et al. 2014; Dijkstra et al. 2016; Egger et al. 2015). Thus, a role for vivianite besides P bound to Fe-(oxyhydr)oxides cannot, at present, be excluded.
In this study, we build on our prior work on Fe and P dynamics in sediments subject to seasonal bottom-water hypoxia (Sulu-Gambari et al. 2016a; b). We present data for the same study area, Lake Grevelingen (the Netherlands), but specifically focus on determining the types of P phases present in the surface sediment using X-ray absorption spectroscopy (XAS). We also assess hypoxia-driven changes in recycling of P from the sediment in the basin on decadal time scales using water-column data for the period from 1973 to 2012 combined with geochemical data from the sediment record for the same period. Our results demonstrate that the formation of poorly crystalline Fe-(oxyhydr)oxides induced by cable bacteria plays a key role in retaining P in the surface sediment. They also highlight a strong, hypoxia-driven recycling of P from the sediments in the basin. Trends in phosphate concentrations in surface waters of the basin since 1973 indicate an even greater role for P recycling from the sediment in earlier decades. These decadal changes in P recycling are, however, not recorded in bulk geochemical depth profiles.
Materials and Methods
Study Area
Lake Grevelingen is a former estuary along the North Sea coast of the Netherlands that was formed by the construction of a landward and a seaward dam in 1964 and 1971, respectively. The lake has an average water depth of 5 m, but is intersected by deep channels, with a maximum water depth of 48 m (Hagens et al. 2015; Sulu-Gambari et al. 2016a; b). After a period of isolation from the North Sea that led to freshening of the lake (characterised by a drop in salinity from ~ 29 in 1971 to ~ 22 in 1978; (Bannink et al. 1984), in 1978, a connection with the North Sea was re-established through an underwater seaward sluice that was open year-round in 1979 (Nolte et al. 2008; Wetsteyn 2011). From 1980 onward, water was exchanged with the North Sea 7 months per year (October to April), resulting in a stable lake salinity at present of 29 to 32 (Paulij et al. 1990). Daily exchange was subsequently introduced in 1999 (Wetsteyn 2011). The lake is subject to temperature-dependent stratification of the water column in late spring and summer, which restricts the transport of oxygen to deeper parts (Hagens et al. 2015). Since 1971, bottom-water hypoxia typically develops in summer in the main channel (Wetsteyn 2011).
Sediment Sampling and Site Characteristics
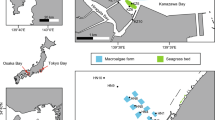
Sediment was sampled each month in 2012 at three sites in the main channel of Lake Grevelingen on board RV Luctor (Den Osse basin) (Fig. 1). Short sediment cores (~ 40 cm) with ~ 20 cm of overlying water were collected at the three sites with a gravity corer (UWITEC, Austria) using Plexiglas® core liners (6 cm inner diameter; 60 cm length). Further details on sediment sampling are given by Seitaj et al. (2015) and Sulu-Gambari et al. (2016b). The sites are characterised by sediment accumulation rates ranging from ~ 0.4 to 2 cm year−1 (Fig. 1). In addition to the monthly sampling in 2012, a short sediment core was collected at site 2 in August 2011 and a long core capturing the upper 90 cm of the sediment was obtained at site 1 in May 2012. With an average sediment accumulation rate of 2 cm year−1 at this site, and duration of the lake phase of ~ 41 years, the estuarine-lacustrine transition in this core is expected near a depth of 82 cm.

Location of Den Osse basin in Lake Grevelingen. Sediment samples were collected each month from three sites (S1–S3) along a water depth gradient from January to December 2012 (inset). The water-column oxygen concentration as a function of water depth for site 1 is plotted for the most anoxic period of the sampling year, August 2012. For the sediment accumulation rate at site 1, we refer to Malkin et al. (2014). For sites 2 and 3, sediment accumulation rates were estimated from the total thickness of the fine-grained sediment deposit and the total duration of sediment deposition in the lake after closure of the seaward dam
The bacterial succession in the sediment in Lake Grevelingen is described in detail elsewhere and is only briefly repeated here. Cable bacteria were identified in the surface sediment at sites 1 and 3 in March and May 2012, using fluorescence in situ hybridisation (FISH) and based on typical pH, O2 and H2S profiles, but were not detected at site 2 (Seitaj et al. 2015; Sulu-Gambari et al. 2016a, b). Beggiatoaceae were present at all three sites in autumn (Seitaj et al. 2015; Sulu-Gambari et al. 2016a, b). The clear signals of activity in the bulk pore-water geochemistry (e.g. G. maxima in calcium ion (Ca2+) and sulphate (SO4 2−) concentrations) (Sulu-Gambari et al. 2016b) suggested that cable bacteria were likely also present at site 2 in spring but were not detected with FISH because of patchiness of the sediment or because the bacteria were present at densities that fall below the detection limit of the method.
We note that part of the data for 2012 presented in this paper have been published previously in studies of P, Fe, Mn and Mo dynamics in the lake (Sulu-Gambari et al. 2016a, b, c). This refers to the bottom-water O2 at the three sites in 2012 and at site 1 between 1978 and 2012, the pore water profiles of PO4, ammonium (NH4 +) and Fe2+ and solid-phase depth profiles of Corg and Ptot over 10 cm at all sites and the solid phase depth profile of Corg over 90 cm at site 1 in May, and the measured and calculated PO4 fluxes. These data are repeated here to provide context for the current study and to allow additional interpretation and discussion of the P data.
Water-Column Measurements
Water-column measurements for oxygen were taken each month in 2012 at 1-m depth resolution with a CTD device equipped with an optical oxygen sensor (YSI6600 V2 sensor). At site 1, water samples were additionally collected from 8 depths (1, 3, 6, 10, 15, 20, 25 and 32 m), using a 12-l Niskin bottle. Oxygen concentrations were determined using an automated Winkler titration (Mettler Toledo DL50 titrator and a platinum redox electrode) and were used to calibrate the CTD sensor. Suspended particulate matter (SPM) was collected by filtration at each depth using 0.7-μm Whatman® glass microfiber GF/F filters as described in Sulu-Gambari et al. (2016c). SPM samples were freeze-dried and fractionated for P phases according to a modified SEDEX procedure (Ruttenberg 1992) as detailed in the ‘Sediment and Suspended Matter Analyses’ section below. Additional water-column samples were filtered and analysed for phosphate (PO4) on a nutrient auto-analyser (Seal QuAAtro).
Bottom-water oxygen concentrations for each month in 1978 to 2011 were made available for site 1 by the Ministry of Infrastructure and the Environment (RWS; (Wetsteyn 2011) Oxygen concentrations were assigned to specific time intervals, where each time interval was classified as ‘anoxic’ (< 1 μM oxygen), ‘hypoxic’ (1–62.5 μM oxygen), ‘oxic’ (> 62.5 μM oxygen) or ‘not determined’, depending on the oxygen concentration and whether or not sampling was conducted (Sulu-Gambari et al. 2016c). Surface water concentrations of dissolved PO4, NH4 +, NOx (i.e. NO3 − + NO2 −) and chlorophyll-a values were available from monthly monitoring for the lake and for the adjacent North Sea between 1973 and 2014 from a public database (Water database).
Sediment Slicing, Bottom- and Pore-Water Analyses, Benthic Fluxes
The long core and the upper 10 cm of the sediment in each short core was sliced at a resolution of 0.5 cm in a N2-purged glove bag, after taking a bottom-water sample from the overlying water. Pore-water samples were collected through centrifugation from the short cores (15 min at 4500g). Supernatant liquid was filtered (0.45 μm) and subsampled for PO4, NH4 + and total dissolved iron (assumed to be present as Fe2+) in a N2-purged glove bag. Samples for PO4 and Fe2+ were acidified with concentrated HCl (37%, 10 μl per ml), stored at 4 °C and analysed by Inductively Coupled Plasma Emission Spectroscopy (ICP-OES; Perkin Elmer Optima 3000). Sub-samples for ammonium were frozen (−20 °C) until colourimetric analysis with a nutrient auto-analyser (Seal QuAAtro) (Middelburg and Nieuwenhuize 2000). Sediment water exchange fluxes of PO4 from the sediment into the overlying water were measured on board each month using intact cores (triplicates) in closed-chamber incubations and calculated from pore-water profiles and bottom-water concentrations of PO4 as described in (Sulu-Gambari et al. 2016a).
PHREEQC calculations were conducted for the pore water from selected depths at site 1 in January, March, June and November 2012, to assess the potential for formation of various pure end-member P minerals such as fluorapatite (Ca5(PO4)3F), hydroxylapatite (Ca5(PO4)3(OH)), carbonated hydroxyapatite (CHAP, Ca10(PO4)6(CO3)), strengite (FePO4.2H2O), vivianite, (Fe(II)3(PO4)2·8H2O), chlorapatite (Ca10(PO4)6Cl2), carbonate fluorapatite (Ca5(PO4,CO3)3F), and an apatite precursor, octacalcium phosphate (OCP, Ca8H2(PO4)6:5H2O) (Supplementary information file) (Tables S1-S5).
Sediment and Suspended Matter Analyses
Centrifuged sediment samples from January, March, May, August and November were freeze-dried and ground in a N2-purged glove box. The samples from the long core taken at site 1 were treated in the same way as the short cores. Total organic carbon was determined using an elemental analyser (Fison Instruments, model NA 1500 NCS), after removing carbonate from the sediment with 1 M HCl (Van Santvoort et al. 2002). Total P was determined by ICP-OES, following acid destruction of ground samples in a closed Teflon bomb at 90 °C (12 h) using a mixture of 2.5 ml HF (40%) and 2.5 ml HClO4/HNO3, evaporation of the acids at 190 °C and dissolution of the resulting gel in 1 M HNO3.
Sediment and suspended matter at site 1 from 2012, and sediment from August 2011 at site 2, were fractionated into exchangeable P, Fe-bound P, authigenic Ca-P, detrital P and organic P using the SEDEX extraction procedure (Ruttenberg 1992), as modified by Slomp et al. (1996) but with the inclusion of the first 1 M MgCl2 wash. For sediments, the first two steps of the procedure were performed in a N2-purged glove box to prevent oxidation artefacts (Kraal et al. 2009).
A small sub-core capturing the upper 7 cm of the sediment was taken from a sediment core at site 1 in January 2012 as described in (Jilbert and Slomp 2013). The sediment was dehydrated with acetone and then set in an epoxy resin, such that it could be sliced into sections for high-resolution elemental analysis. Resin-embedded sediment sections were subsequently analysed with X-ray fluorescence (XRF) (Sulu-Gambari et al. 2016b) and, at selected locations based on Fe and/or P enrichments detected by XRF, with phosphorus X-ray absorption near edge structure (XANES) analysis at beamline I18 at the Diamond Light Source in Oxfordshire. A description of the microfocus spectroscopy beamline I18 can be found at (Mosselmans et al. 2009).
The experimental beam size was approximately 7 × 9 μm. Sediment sections were mapped for Al, Si, P, S, K, Ca, Si, Mn and Fe by μ-XRF at a beam energy of 2.8 keV, with a horizontal and vertical step size of 10 μm and a dwell time of 0.2 s per point. During mapping, the Ni/Rh-coated mirrors for harmonic rejection were not used. The intensities of the higher harmonics of the beam were high enough to measure the spatial distribution of elements with a K-edge above 2.8 keV, by using their K emission lines in the fluorescence spectra.
After mapping, spots observed to be Fe-rich with an elevated P signal were selected for analysis by X-ray absorption spectroscopy at the P-K edge. Spectra were collected in the 2.10–2.35-keV energy range. Energy calibration was performed by aligning XANES spectra of reference materials, measured at I18 at the Diamond Light Source, to those previously collected at ID21 at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. At ID21, energy calibration was conducted by positioning the maximum of the hydroxyapatite white-line at 2152.7 eV. At least two XANES spectra were collected per spot and merged to improve the signal to noise ratio. Merging and normalisation of spectra was performed with the Athena data analysis package (Ravel and Newville 2005). For comparison, reference spectra for a suite of P minerals were obtained from Dijkstra et al. (2016) and Egger et al. (2015) and supplemented with adsorbed-P spectra, taken from the ID21beamline P-XANES spectra database at the ESRF (ID21 P-XANES database).
Burial Efficiency of P
The burial efficiency of total P, defined here as the percentage of the total P input at the sediment-water interface that is buried below 10-cm depth; (Jensen et al. 1995) and of reactive P (‘biologically available’ P; (Ruttenberg 2003) was calculated from the burial rate of P at a depth of 10 cm and the measured phosphate efflux from the sediment following:
Reactive P is defined as the sum of exchangeable P, Fe-bound P, organic P and authigenic Ca-P thus omitting detrital P that is assumed to be inert in marine environments (Ruttenberg 2003). Since authigenic-P could also be largely of detrital origin (Ruttenberg 2003), we also calculated the burial efficiency of the sum of exchangeable P, Fe-bound P and organic P. For site 3, the burial flux of reactive P was estimated from the total P flux assuming a similar P speciation as at site 2.
Results
Bottom-Water Oxygen, Pore-Water Geochemistry and Benthic Fluxes of Phosphate
Bottom waters at sites 1 (34 m) and 2 (23 m) in the Den Osse basin were oxic (O2 ≥ 63 μM) from September to April, became hypoxic in late spring (O2 < 63 μM) and briefly became anoxic (O2 < 1 μM) in August (Figs. 1 and 2). At site 3 (17 m), oxygen concentrations remained above 100 μM in summer (Fig. 2).

Monthly bottom-water oxygen concentrations (in μM) at all three sites from January to December 2012
Pore-water PO4 concentrations generally increase with depth at all three sites, with concentrations generally being highest at depth in the sediment at site 1 and lowest at site 3 (Fig. 3). There is a distinct seasonality in the trends in concentrations with depth at all three sites, with generally low concentrations in the surface sediment from January to May, maxima near the sediment-water interface in June and/or July and site-dependent concave or convex downward trends from August onward. Pore-water NH4 + also increases with depth at all three sites and similar differences between sites are observed as for PO4 (Fig. 4). While the increase is nearly linear in spring, concave depth profiles are observed from August onward. In spring, release of Fe2+ to the pore water is observed in the upper 4 cm of the sediment in several of the profiles (Fig. 5). The maxima in Fe2+ are observed more frequently at site 1 than at sites 2 and 3 and, with the exception of June, these peaks in Fe2+ do not coincide with depths at which maxima are observed in dissolved PO4 depth profiles. From August to October, practically no Fe2+ is detected in the pore water. In November and December, some Fe2+ is observed in the pore water near the sediment-water interface.

Monthly depth profiles of pore-water phosphate for January to December 2012 for the sediment at all three sites

Monthly depth profiles of pore-water ammonium for January to December 2012 for the sediment at all three sites

Monthly depth profiles of pore-water iron for January to December 2012 for the sediment at all three sites
PHREEQC calculations indicate that at site 1, the pore water in the upper 10 cm was supersaturated with respect to fluorapatite and carbonate fluorapatite but undersaturated with respect to other apatite forms as well as vivianite (Fe(II)3(PO4)2·8H2O) in January, March, June and November 2012; Supplementary Information; Table S5).
Fluxes of PO4 determined from closed-core incubations show a marked seasonality at site 1, with low effluxes in winter and early spring (< 0.5 mmol m−2 day−1) and significantly higher effluxes in late spring and summer (1.5–3.4 mmol m−2 day−1) (Fig. 6). The release of PO4 from the sediment is highest at site 1 and decreases with decreasing water depth (site 1 > site 2 > site 3). Calculated sediment-water PO4 effluxes (Fig. S1) show the same seasonal trend but the differences between the sites are less clear and sometimes trends are even opposite to those observed in the closed-core incubations (e.g. July 2012).

Geochemistry of the Sediment and Suspended Matter
The sediments at all three sites are rich in organic carbon (Corg), with concentrations ranging from 3 to 5 wt% in the upper 10 cm of the sediment (Fig. 7). Total P concentrations range between 20 and 60 μmol g−1 and show a strong seasonality at all sites, with a distinct maximum near the sediment surface in January, March and May and only a minor enrichment in August and November. Molar ratios of organic C (Corg) to total P (Ptot) are generally close to Redfield (106:1) in the sediment at all sites, with the exception of the surface sediments in January, March and May, when Corg/Ptot ratios are relatively low due to the increase in total P, and those in August at site 1, when Corg/Ptot ratios are relatively low due to an exceptionally low Corg content.

Depth profiles of sediment organic C (Corg), total phosphorus (Ptot) and molar ratios of Corg/Ptot for all three sites in January, March, May, August and November 2012. The Redfield ratio (C:P = 106:1) is indicated by the dashed arrows. Data are provided in Table S8
Most sedimentary P at sites 1 (5 months) and 2 (August) is present in the form of Fe-bound P, authigenic Ca-P and organic P (Fig. 8). Exchangeable and detrital P concentrations are low. The profiles for site 1 show that there is a distinct seasonality in Fe-bound P with very strong enrichments in the surface sediment from January to May. Both authigenic Ca-P and organic P concentrations are relatively constant with depth. The profiles at site 2 in August also indicate a negligible role for exchangeable and detrital P. At this site, Fe-bound P, authigenic Ca-P and organic P are also the major P phases and their concentrations decrease below ca. 4-cm depth.

Depth profiles of sediment phosphorus forms at site 1 in January, March, May, August and November 2012 and at site 2 in August 2011. Ex-P: exchangeable P, Fe-P: Fe-bound P, Auth-P: authigenic Ca-P, Det-P: detrital P, Org-P: organic P. Data are provided in Table S9
The suspended matter (SPM) at site 1 is enriched in P (Fig. 9) relative to the underlying sediment (Fig. 8). Exchangeable (SPM: ca. 20 μmol g−1 and sediment: < 10 μmol g−1), Fe-bound (SPM: 20–100 μmol g−1 and sediment: ca. 25 μmol g−1) and organic P (SPM: ca. 25 μmol g−1 and sediment: < 20 μmol g−1) are the most abundant P phases. Fe-bound P and organic P show strong changes in concentration between sampling times, with values being highest in March and November 2012. Authigenic Ca-P concentrations in the suspended matter are near constant with depth and are generally similar to concentrations in the surface sediment.

P XANES
Normalised P-XANES spectra were collected at various locations on the resin embedded core sections from January 2012 (Fig. S2). All spectra are virtually identical and are characterised by a peak maximum (white line) at 2153.3 eV followed by an elongated hump with a maximum at 2169.5 eV (Fig. 10). The XANES spectra of the samples differ from those of Ca, Mn(II) and Fe(II) phosphates, condensed phosphates and those of other phosphate minerals reported in literature (e.g. (Hesterberg 2010; Ingall et al. 2011); Fig. 10) with regard to the shape of the spectrum and the position and intensity of the white line. Characteristic features of the reference mineral spectra, such as a shoulder on the low (Al) and high (Ca) energy side of the white line or other continuum resonances (Hesterberg 2010), are absent. The lack of characteristic oscillations on the high-energy side of the white line indicates that the XANES spectra cannot be attributed to a single crystalline P phase.

Normalised P-XANES spectra collected at various spots on a resin embedded core sample (P3, P5 and P7) from site 1 for January 2012 (See Fig. S2 for the locations on the resin-block) and spectra of P-bearing reference materials for comparison
Instead, the XANES spectra bear a similarity to those of adsorbed P or P associated with minerals in a dispersed form, which are characterised by relatively featureless spectra (Fig. 10). The position of the white line of the samples lies in between those measured for P adsorbed onto Fe, Mn and Al oxides (2153.2–2153.4 eV). The spectra of the sediment samples also show a small pre-edge feature at about 4 eV in front of the white line (Fig. S3). This pre-edge feature is an indicator of P bound to transition elements, a feature which can be attributed to the excitation of 1 s electrons into antibonding molecular orbitals of the transition metal in question and oxygen (Hesterberg 2010). Pre-edge features are very pronounced in spectra from P adsorbed onto Fe-(oxyhydr)oxides and Mn-oxides. No pre-edge features are observed when P is adsorbed onto Ca carbonates, clay minerals or aluminium oxides. Taking the intensity of the pre-edge as an indicator, about one quarter (25%) of the P at the analysed spots is associated with Fe(III), suggesting that it corresponds to the relative enrichment in Fe-P, at the core top in January. Presumably, the remaining P at the analysed spots is adsorbed onto Fe(II) solids, clay minerals, or partly incorporated as detrital P in silicates.
Burial Efficiency of P
The burial efficiencies of total P at sites 1, 2 and 3 were calculated as 27, 32 and 26%, respectively (Fig. 11; Table 1). The input of total P, calculated from the measured efflux and burial rate, increased with water depth (Fig. 11) and rate of sedimentation (Fig. 1). Burial efficiencies of reactive P and the sum of exchangeable P, Fe-bound P and organic P are significantly lower at 16–23% and 10–18%, respectively.

Phosphorus budget for the year 2012 at the three sites and corresponding burial efficiencies of total P. The P efflux is the integrated yearly P flux based on the measured fluxes (Fig. 6). The burial efficiency is calculated as the percentage of the total input that is buried below a depth of 10 cm (e.g. Jensen et al. 1995) as described in the text
Historical Records of Oxygen, Nutrients, Chlorophyll-a and Carbon in Lake Grevelingen
Monitoring data confirm that bottom-water anoxia occurred regularly in summer from 1979 to 1997, while hypoxia was more prevalent from 1998 to 2010 (Fig. 12a). In 2011 and 2012, the summer was characterised by a period of anoxia and hypoxia, respectively (Fig. 12a and Fig. 2). Water-column monitoring data for surface water PO4 in the lake and the adjacent North Sea (Fig. 12b) reveal a strong seasonal cycle between 1973 and 2012, with low concentrations in spring and high concentrations in summer and autumn. Both the amplitude of the seasonal change and the maximum PO4 concentrations were significantly higher prior to 1998, with a distinct maximum in 1978, prior to the re-establishment of the connection with the North Sea. Concentrations of PO4 are generally higher in the lake than in the North Sea, but since the changes in flushing regime in 1978 and 1999, the differences in PO4 concentration have gradually declined. Trends in water-column NH4 +, chlorophyll-a and NOx (Fig. 12c–e) also show a distinct seasonality, but apart from somewhat higher concentrations in NH4 + prior to 1978, there is no distinct trend with time. While NH4 + concentrations in the lake are higher than in the North Sea, the opposite is the case for chlorophyll-a and NOx.

a Periods of oxic, hypoxic and anoxic bottom water conditions from 1999 to 2012 at site 1 (data between 1999 and 2011 from Ministry of Infrastructure and Environment (Wetsteyn 2011)). Figure adapted from Sulu-Gambari et al. (2016c). Evolution of surface water b PO4, c NH4 +, d chlorophyll-a and e NOx (i.e. NO2 + NO3) at site 1 and in the North Sea adjacent to the lake from 1973 to 2012
Organic carbon and total P concentrations in the sediments deposited since the closure of the estuary, generally decline gradually with depth, with the exception of a maximum at the sediment-water interface and a number of smaller maxima in the upper 30 cm of the sediment (Fig. 13). Ratios of Corg/Ptot are mostly close to Redfield (106:1), with somewhat higher values above 30-cm depth than below.

Sediment depth profile of Corg, Ptot and Corg/Ptot for the long core (90 cm) at site 1 for May 2012. The estuarine-lacustrine transition is indicated by a dotted black line and the Redfield ratio (C:P = 106:1) is indicated by the grey arrow. Data are provided in Table S11
Discussion
Impact of Sulphide-Oxidising Bacteria on P Mineral Formation
Low oxygen concentrations have occurred annually in summer in the bottom waters of Lake Grevelingen since the closure of the former estuary in 1971 (Wetsteyn 2011; Fig. 12). A seasonal succession in the dominant type of sulphur-oxidising bacteria in the sediments was observed from 2011 to 2015 with cable bacteria dominating in spring and Beggiatoaceae in autumn (Seitaj et al. 2015). The seasonal changes in bottom-water oxygen and the bacterial community have major implications for the release of P from the sediment, as shown previously for site 1 by Sulu-Gambari et al. (2016a). In spring, when cable bacteria are active, an Fe-(III)-(oxyhydr)oxide-rich surface layer is formed from upward diffusing Fe2+ released through FeS dissolution (Fig. 5). Upward-diffusing PO4 is removed to an Fe-associated phase in this surface layer (Fig. 3). In early summer, when the cable bacteria population has declined, reductive dissolution of the Fe-(oxyhydr)oxides by sulphides (Seitaj et al. 2015), leads to a release of associated PO4 (Sulu-Gambari et al. 2016a). The production of Fe2+ and PO4 in the pore water of the surface sediment is then coupled. At depth, most PO4 is thought to be released from organic matter degradation, together with NH4 + (Figs. 3 and 4). Results of Fe XANES showed that the Fe (oxy)hydroxides formed in the surface sediment in spring are mostly poorly crystalline (Sulu-Gambari et al. 2016b). Our P XANES results indicate that at least part of the P is adsorbed to the surface of these Fe(III)-(oxyhydr)oxides (Figs. 10 and S3). Pore waters in the surface sediments are undersaturated with respect to vivianite and there is no evidence for the presence of this mineral from P XANES (Fig. 10). Below the surface sediments, no vivianite is expected since it dissolves when sulphide concentrations in the pore water are high (Egger et al. 2016). Our results thus indicate that Fe-(oxyhydro)xides are likely the primary temporary sink for P in Grevelingen surface sediments. The results also highlight that a decoupling of the release of pore-water Fe2+ and PO4 directly below the oxic zone may be an indicator for the metabolic activity of cable bacteria.
Sulphur-oxidising bacteria are also thought to play a critical role in the formation of authigenic Ca-P minerals by releasing PO4 from polyphosphates in their cells (Brock and Schulz-Vogt 2011; Schulz and Schulz 2005) and driving pore waters to supersaturation with respect to authigenic Ca-P minerals. Our results show, however, that there is no clear difference between authigenic Ca-P concentrations in suspended matter (Fig. 9) and in the surface sediment (Fig. 8). This suggests that there is no authigenic Ca-P formation in Grevelingen sediments, despite the presence of polyphosphates in cable bacteria and Beggiatoa (Sulu-Gambari et al. 2016a) and despite the supersaturation of the pore water with respect to carbonate fluorapatite in the surface sediment (Table S1), which is typical for marine sediments (Ruttenberg 2003). Possible explanations for the lack of authigenic Ca-P formation may include inhibition by Mg2+, where these ions take up potential binding sites for Ca2+ on the apatite precursor surface (Gunnars et al. 2004; Martens et al. 1978) and undersaturation with respect to octacalcium phosphate, a precursor for carbonate fluorapatite (Oxmann and Schwendenmann 2015); Table S1). Furthermore, we speculate that authigenic Ca-P formed in surface sediments may be re-dissolved together with Ca- and Mn-carbonates linked to the activity of cable bacteria and the associated acidity of the pore waters (e.g. Risgaard-Petersen et al. 2012). Further work is required to assess whether the activity of cable bacteria indeed explains a lack of authigenic Ca-P formation in the sediments of our study area.
Burial Efficiency of P
The burial efficiency of P provides key information on the role sediments play as a temporary source and permanent sink for P, with high values pointing towards effective sequestration of P and low values emphasising strong recycling of P (e.g. Jensen et al. 1995). The burial efficiency of a sediment for P is thought to be influenced by a wide range of factors, including, for example, the rate of sediment accumulation, the bottom-water redox conditions, the input of Fe-(oxyhydr)oxides to a sediment and whether or not sink-switching to authigenic Ca-P occurs (e.g. Ruttenberg 2003). High rates of sediment accumulation are typically thought to promote retention of P, as observed for example in Cape Lookout Bight where sedimentation rates are > 10 cm year−1 and 85% of the incoming P is buried (Klump and Martens 1987); Table 2). Oxic bottom waters are also beneficial for P retention because of sequestration of P in Fe-(oxyhydr)oxides, thereby promoting the potential for sink-switching to authigenic Ca-P (Ruttenberg and Berner 1993). Bottom-water hypoxia and anoxia, in contrast, generally promote recycling of P from sediments (Ingall and Jahnke 1994; Ingall and Jahnke 1997), as also suggested by observations of burial efficiencies for P below 35% in Aarhus Bay (Jensen et al. 1995); Table 2) and below 12% in the Baltic Sea (Viktorsson et al. 2013). The burial efficiencies for total P at our study sites range from 26 to 32% and thus are rather similar to those for the seasonally hypoxic Aarhus Bay.
We note that in our study area, there is a distinct gradient in the input of P to the sediment with water depth, with the highest input of total P at the deepest site and the lowest at the shallowest site (Fig. 11). This is also in line with the gradient in sediment accumulation with the highest rate at the deepest site (Fig. 1) and a major role for lateral input and focussing of fine-grained material in the basin (e.g. Hagens et al. 2015; Seitaj et al. 2016). Concentrations of total P in the suspended matter are on average a factor 3 higher than in the sediment, which is in line with calculated burial efficiencies suggesting that nearly one third of the incoming P is buried (Table 1). Analyses of the suspended matter also indicate that exchangeable P, Fe-bound P and organic P dominate the inputs to the basin (Fig. 9). Besides these reactive P forms, non-reactive P in clays and detrital apatite can contribute to total P burial in the basin, making it useful to consider the burial efficiency of total P versus that of reactive P. Assuming that reactive P consists of exchangeable P, Fe-bound P, organic P and authigenic Ca-P, we calculate a burial efficiency of 16 to 23% (Table 1). If we assume that the P extracted with acetate buffer that is labelled authigenic Ca-P is not truly authigenic and consists of P in clays (Ruttenberg 1992) and/or consists of authigenic Ca-P formed elsewhere, the burial efficiency becomes even lower at 10 to 18%. This emphasises the strong potential for recycling of P in hypoxic systems. This also highlights the need for accurate insight in what proportion of P is truly reactive (Ruttenberg 2003). Regardless, at values ≤ 32% the burial efficiency of P at site 1 (Table 1) is consistently below that of organic C, which is estimated at ~ 50% (Seitaj 2016).
Sediment Corg/Ptot ratios, a commonly used indicator for bottom water hypoxia and anoxia (Algeo and Ingall 2007), in the sediments in the lake are close to Redfield (106:1) in the short cores and show subtle changes with depth that mostly follow the changes in organic C, suggesting highest organic Corg/Ptot ratios during summer periods of hypoxia (Fig. 13). This is in accordance with enhanced recycling of P from sediments when oxygen in bottom waters is low (Algeo and Ingall 2007). In the long core, the depth resolution is insufficient to track seasonal changes. Surprisingly, Corg/Ptot ratios are lowest in the sediments deposited below depths of 30 cm (Fig. 13) corresponding to the period before 1998 when the system was more frequently anoxic (Fig. 12). This unexpected result may be explained by changes in the composition of the incoming particulate material from 1998 onward, in particular the increased input of organic matter from the North Sea with elevated C:P ratios (e.g. Phaeocystis globosa Schoemann et al. 2005). This highlights that, in systems where hypoxia-driven changes in Corg/Ptot ratios are relatively subtle, long-term trends in Corg/Ptot due to changes in bottom-water redox conditions may be overprinted by changes in the C:P ratio of the incoming organic matter.
Historical Trends in P Recycling and its System-Scale Impact
The hypoxia-driven recycling of P from the sediment had a major impact on the water-column geochemistry in Lake Grevelingen in 2012 (Sulu-Gambari et al. 2016a) and has contributed to a strong seasonality in PO4 concentrations in the surface waters since 1971 (Fig. 12; Bannink et al. 1984; Kelderman 1980; Wetsteyn 2011). Importantly, both the bottom-water hypoxia and the seasonal trend in surface-water PO4 were absent in the area prior to the closure of the estuary (Bannink et al. 1984; Kelderman 1980). Concentrations of PO4 in surface waters of the former estuary ranged from 1.1 to 2.5 μM (Kelderman 1980) but increased up to a maximum of ~ 24 μM (or 120 mmol m−2 given the average water depth of 5 m) in the years directly following closure of the estuary (Fig. 12). Mass balance calculations that included estimates of inputs from land suggested that a significant proportion (45%) of the increase of PO4 in the system was the result of mobilisation of P from the sediment (Bannink et al. 1984; Kelderman 1980). In these earlier studies, the responsible mechanisms were not identified.
Our results for 2012 demonstrate that Fe-P dynamics likely played a central role in controlling P dynamics in the lake upon closure, with the seasonality possibly being enhanced by the presence of cable bacteria. This would also imply that a large pool of Fe-bound P was initially present in the estuarine sediments, as is the case at present-day depositional sites in the nearby Western Scheldt estuary (e.g. Hyacinthe and Van Cappellen 2004). At the Waarde site in the latter study, for example, ~ 500 mmol P m−2 in the sediment was Fe-bound P (based on a porosity of 0.95 and average Fe-P content of 20 μmol g−1 for the upper 20 cm of the sediment). We suggest that an increasing proportion of the Fe-bound P pool was mobilised upon the onset of hypoxia after closure of the estuary, contributing to the dramatic rise in PO4 concentrations between 1973 and 1978.
The increased flushing of the lake with North Sea water from 1978 and 1999 onward removed PO4 from the system (e.g. period 1978–1980, ~ 8 μmol l−1 or ~ 40 mmol m−2; Fig. 12) and contributed to the improved bottom-water oxygen concentrations from the late 1990s onward (Fig. 12; (Wetsteyn 2011). Budget calculations for P in the lake water column for the period 2000–2005 indicate that there now is a minor import of P from the North Sea of 1 mmol P m−2 year−1 (Meijers and Groot, 2007). In these budget calculations, the removal of P to the sediment on a lake-wide scale is estimated at 10 to 12 mmol P m−2 year−1, with the difference between North Sea input and burial being explained by discharge of P from land (Meijers and Groot 2007). We note that the burial rates of P at our three sites (24–162 mmol P m−2 year−1) are higher than average in the lake (Table 1), in accordance with the preferential accumulation of fine-grained material in the former channels (Fig. 1; Egger et al. 2016).
Strikingly, the changes in availability of PO4 between 1973 and the present day, have not lead to major changes in the burial of P in the sediment (Figs. 7 and 13).There are two explanations for the lack of a change in P burial: first, P is not limiting for primary productivity in this system (Bannink et al. 1984; Spiteri and Nolte 2010). Thus, the changes in P availability have had no impact on productivity (and chlorophyll-a concentrations) in the lake (Fig. 12). Instead, nitrogen is limiting (Spiteri and Nolte 2010), and because NH4 + and NOx concentrations did not change greatly after the closure, the productivity in the basin and the burial of organic C and P also remained relatively constant (Hagens et al. 2015; Nienhuis and Huis in ‘t Veld 1984). Second, our results suggest there was no authigenic mineral P formation to sequester part of the additional P that was recycled from organic matter and Fe in the surface sediments of the lake, as is typical in other coastal systems (e.g. Ruttenberg and Berner 1993).
These results illustrate that strong changes in P recycling will only have a major impact on coastal marine ecosystems when P is limiting during at least part of the year. It also highlights that hypoxia-driven enhanced recycling of P is not necessarily registered in sediment records of Ptot or Corg/Ptot, even upon order-of-magnitude changes in P availability in coastal systems. This is because the resulting large pool of Fe-(oxyhydr)oxide P is only a temporary sink for P in the sediment and the major permanent sinks for P, organic P and authigenic Ca-P, are not affected by the changes in P availability in this system. These findings are relevant when using sediment records of P and organic C to reconstruct P dynamics in coastal systems (e.g. Jilbert et al. 2011; Mort et al. 2010; Tsandev and Slomp 2009).
Cable bacteria were found to play a key role in the recycling of P from sediments in Lake Grevelingen in 2012 (Sulu-Gambari et al., 2016a). Given the strong decadal scale changes in Fe-driven recycling of P in the lake, the question arises what role cable bacteria played in these past changes. Our work thus emphasises the need for tools to determine the past activity of cable bacteria in sediments and the need for a better understanding of the influence of cable bacteria on P recycling in coastal systems, including their potential impact on authigenic Ca-P formation.
References
Algeo, T.J., and E. Ingall. 2007. Sedimentary Corg:P ratios, paleocean ventilation, and Phanerozoic atmospheric pO2. Palaeogeography, Palaeoclimatology, Palaeoecology 256 (3–4): 130–155.
Anschutz, P., S. Zhong, B. Sundby, A. Mucci, and C. Gobeil. 1998. Burial efficiency of phosphorus and the geochemistry of iron in continental margin sediments. Limnology and Oceanography 43 (1): 53–64.
Arrigo, K.R. 2005. Marine microorganisms and global nutrient cycles. Nature 437 (7057): 349–355.
Bannink, B.A., J.H.M. Van der Meulen, and P.H. Nienhuis. 1984. Lake Grevelingen: From an estuary to a saline lake: An introduction. Netherlands Journal of Sea Research 18 (3–4): 179–190.
Beusen, A.H.W., A.F. Bouwman, L.P.H. Van Beek, J.M. Mogollón, and J.J. Middelburg. 2016. Global riverine N and P transport to ocean increased during the 20th century despite increased retention along the aquatic continuum. Biogeosciences 13 (8): 2441–2451.
Brock, J., and H.N. Schulz-Vogt. 2011. Sulfide induces phosphate release from polyphosphate in cultures of a marine Beggiatoa strain. The ISME Journal 5 (3): 497–506.
Burdorf, L.D.W., et al. 2016. Long-distance electron transport occurs globally in marine sediments. Biogeosciences Discussions 2016: 1–35.
Carstensen, J., et al. 2014. Hypoxia in the Baltic Sea: Biogeochemical cycles, benthic Fauna, and management. Ambio 43 (1): 26–36.
Dale, A.W., V.J. Bertics, T. Treude, S. Sommer, and K. Wallmann. 2013. Modeling benthic–pelagic nutrient exchange processes and porewater distributions in a seasonally hypoxic sediment: Evidence for massive phosphate release by Beggiatoa? Biogeosciences 10 (2): 629–651.
Diaz, R.J., and R. Rosenberg. 2008. Spreading dead zones and consequences for marine ecosystems. Science 321 (5891): 926–929.
Dijkstra, N., P. Kraal, M.M.M. Kuypers, B. Schnetger, and C.P. Slomp. 2014. Are iron-phosphate minerals a sink for phosphorus in anoxic Black Sea sediments? PloS One 9 (7): e101139.
Dijkstra, N., C.P. Slomp, and T. Behrends. 2016. Vivianite is a key sink for phosphorus in sediments of the Landsort deep, an intermittently anoxic deep basin in the Baltic Sea. Chemical Geology 438: 58–72.
Egger, M., T. Jilbert, T. Behrends, C. Rivard, and C.P. Slomp. 2015. Vivianite is a major sink for phosphorus in methanogenic coastal surface sediments. Geochimica et Cosmochimica Acta 169: 217–235.
Egger, M., et al. 2016. Rapid sediment accumulation results in high methane effluxes from coastal sediments. PloS One 11 (8): e0161609.
Gächter, R., and J.S. Meyer. 1993. The role of microorganisms in mobilization and fixation of phosphorus in sediments. In Proceedings of the third international workshop on phosphorus in sediments, ed. P.C.M. Boers, T.E. Cappenberg, and W. van Raaphorst, 103–121. Dordrecht: Springer Netherlands.
Goldhammer, T., V. Bruchert, T.G. Ferdelman, and M. Zabel. 2010. Microbial sequestration of phosphorus in anoxic upwelling sediments. Nature Geoscience 3 (8): 557–561.
Gunnars, A., S. Blomqvist, and C. Martinsson. 2004. Inorganic formation of apatite in brackish seawater from the Baltic Sea: An experimental approach. Marine Chemistry 91 (1–4): 15–26.
Gustafsson, B.G., et al. 2012. Reconstructing the development of Baltic Sea eutrophication 1850–2006. Ambio 41 (6): 534–548.
Hagens, M., et al. 2015. Biogeochemical processes and buffering capacity concurrently affect acidification in a seasonally hypoxic coastal marine basin. Biogeosciences 12 (5): 1561–1583.
Hesterberg, D. 2010. Chapter 11 - macroscale chemical properties and X-ray absorption spectroscopy of soil phosphorus. In Developments in Soil Science, ed. S. Balwant and G. Markus, 313–356. Amsterdam: Elsevier.
Hyacinthe, C., and P. Van Cappellen. 2004. An authigenic iron phosphate phase in estuarine sediments: Composition, formation and chemical reactivity. Marine Chemistry 91 (1–4): 227–251.
Ingall, E., and R. Jahnke. 1994. Evidence for enhanced phosphorus regeneration from marine sediments overlain by oxygen depleted waters. Geochimica et Cosmochimica Acta 58 (11): 2571–2575.
Ingall, E., and R. Jahnke. 1997. Influence of water-column anoxia on the elemental fractionation of carbon and phosphorus during sediment diagenesis. Marine Geology 139 (1–4): 219–229.
Ingall, E.D., et al. 2011. Phosphorus K-edge XANES spectroscopy of mineral standards. Journal of Synchrotron Radiation 18 (2): 189–197.
Ingall, E.D., R.M. Bustin, and P. Van Cappellen. 1993. Influence of water column anoxia on the burial and preservation of carbon and phosphorus in marine shales. Geochimica et Cosmochimica Acta 57: 303–316.
Jensen, H.S., P.B. Mortensen, F.Ø. Andersen, E. Rasmussen, and A. Jensen. 1995. Phosphorus cycling in a coastal marine sediment, Aarhus Bay, Denmark. Limnology and Oceanography 40 (5): 908–917.
Jilbert, T., C. Slomp, B.G. Gustafsson, and W. Boer. 2011. Beyond the Fe-P-redox connection: Preferential regeneration of phosphorus from organic matter as a key control on Baltic Sea nutrient cycles. Biogeosciences 8: 1699–1720.
Jilbert, T., and C.P. Slomp. 2013. Iron and manganese shuttles control the formation of authigenic phosphorus minerals in the euxinic basins of the Baltic Sea. Geochimica et Cosmochimica Acta 107 (0): 155–169.
Kelderman, P. 1980. Phosphate budget and sediment-water exchange in lake grevelingen (SW Netherlands). Netherlands Journal of Sea Research 14 (3–4): 229–236.
Klump, J.V., and C.S. Martens. 1987. Biogeochemical cycling in an organic-rich coastal marine basin. 5. Sedimentary nitrogen and phosphorus budgets based upon kinetic models, mass balances, and the stoichiometry of nutrient regeneration. Geochimica et Cosmochimica Acta 51 (5): 1161–1173.
Kraal, P., C.P. Slomp, A. Forster, M.M.M. Kuypers, and A. Sluijs. 2009. Pyrite oxidation during sample storage determines phosphorus fractionation in carbonate-poor anoxic sediments. Geochimica et Cosmochimica Acta 73 (11): 3277–3290.
Malkin, S.Y. et al. 2014. Natural occurrence of microbial sulphur oxidation by long-range electron transport in the seafloor. The ISME Journal
Martens, C.S., R.A. Berner, and J.K. Rosenfeld. 1978. Interstitial water chemistry of anoxic Long Island sound sediments. 2. Nutrient regeneration and phosphate removal 1. Limnology and Oceanography 23 (4): 605–617.
März, C., J. Hoffmann, U. Bleil, G.J. de Lange, and S. Kasten. 2008. Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean). Marine Geology 255 (3–4): 118–130.
Meier, H.E.M. et al. 2011. Hypoxia in future climates: A model ensemble study for the Baltic Sea. Geophysical Research Letters 38(24): n/a-n/a.
Meijers, E., Groot, S., 2007. Deltamodel-hulpmiddel ter ondersteuning van het beheer en beleid van de zuidwestelijke Delta, Deltares (WL).
Meysman, F.J.R., N. Risgaard-Petersen, S.Y. Malkin, and L.P. Nielsen. 2015. The geochemical fingerprint of microbial long-distance electron transport in the seafloor. Geochimica et Cosmochimica Acta 152: 122–142.
Middelburg, J.J., and L.A. Levin. 2009. Coastal hypoxia and sediment biogeochemistry. Biogeosciences 6 (7): 1273–1293.
Middelburg, J.J., and J. Nieuwenhuize. 2000. Uptake of dissolved inorganic nitrogen in turbid, tidal estuaries. Marine Ecology Progress Series 192: 79–88.
Mort, H., C. Slomp, B.G. Gustafsson, and T.J. Andersen. 2010. Phosphorus recycling and burial in Baltic Sea sediments with contrasting redox conditions. Geochimica & Cosmochimica Acta 74: 1350–1362.
Mortimer, C.H. 1941. The exchange of dissolved substances between mud and water in lakes. Journal of Ecology 30: 280–329.
Mosselmans, J.F.W., et al. 2009. I18 - the microfocus spectroscopy beamline at the diamond light source. Journal of Synchrotron Radiation 16 (6): 818–824.
Nielsen, L.P., N. Risgaard-Petersen, H. Fossing, P.B. Christensen, and M. Sayama. 2010. Electric currents couple spatially separated biogeochemical processes in marine sediment. Nature 463 (7284): 1071–1074.
Nienhuis, P.H., and J.C. Huis in’t Veld. 1984. Grevelingen: From an estuary to a saline Lake. Water Science and Technology 16 (1–2): 27–50.
Nolte, A., Troost, T., de Boer, G., Spiteri, C., van Wesenbeeck, B., 2008. Verkenning oplossingsrichtingen voor een betere waterkwaliteit en ecologische toestand van het Grevelingenmeer. Delft, Netherlands.
Oxmann, J.F., and L. Schwendenmann. 2015. Authigenic apatite and octacalcium phosphate formation due to adsorption–precipitation switching across estuarine salinity gradients. Biogeosciences 12 (3): 723–738.
Paulij, W.P., R.H. Bogaards, and J.M. Denucé. 1990. Influence of salinity on embryonic development and the distribution of Sepia Officinalis in the Delta area (south western part of The Netherlands). Marine Biology 107 (1): 17–23.
Pfeffer, C., et al. 2012. Filamentous bacteria transport electrons over centimetre distances. Nature 491 (7423): 218–221.
Rabalais, N.N. et al., 2014. Eutrophication-Driven Deoxygenation in the Coastal Ocean Oceanography, 27.
Rabalais, N.N., et al. 2010. Dynamics and distribution of natural and human-caused hypoxia. Biogeosciences 7 (2): 585–619.
Rao, A.M.F., S.Y. Malkin, S. Hidalgo-Martinez, and F.J.R. Meysman. 2016. The impact of electrogenic sulfide oxidation on elemental cycling and solute fluxes in coastal sediment. Geochimica et Cosmochimica Acta 172: 265–286.
Ravel, B., and M. Newville. 2005. ATHENA, ARTEMIS, HEPHAESTUS: Data analysis for X-ray absorption spectroscopy using IFEFFIT. Journal of Synchrotron Radiation 12 (4): 537–541.
Risgaard-Petersen, N., A. Revil, P. Meister, and L.P. Nielsen. 2012. Sulfur, iron-, and calcium cycling associated with natural electric currents running through marine sediment. Geochimica et Cosmochimica Acta 92 (0): 1–13.
Ruttenberg, K.C. 1992. Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnology and Oceanography 37 (7): 1460–1482.
Ruttenberg, K.C., 2003. 8.13 - The Global Phosphorus Cycle. In: Editors-in-Chief: Heinrich, D.H., Karl, K.T. (Eds.), Treatise on Geochemistry. Pergamon, Oxford, pp. 585–643.
Ruttenberg, K.C., and R.A. Berner. 1993. Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochimica et Cosmochimica Acta 57 (5): 991–1007.
Schoemann, V., S. Becquevort, J. Stefels, V. Rousseau, and C. Lancelot. 2005. Phaeocystis blooms in the global ocean and their controlling mechanisms: A review. Journal of Sea Research 53 (1–2): 43–66.
Schulz, H.N., and H.D. Schulz. 2005. Large sulfur bacteria and the formation of Phosphorite. Science 307 (5708): 416–418.
Seitaj, D. 2016. Impact of cable bacteria on the biogeochemical cycling in a seasonally Hypoxic Basin. Brussels: Vrije Universiteit Brussel 217 pp.
Seitaj, D., et al. 2015. Cable bacteria generate a firewall against euxinia in seasonally hypoxic basins. Proceedings of the National Academy of Sciences 112 (43): 13278–13283.
Seitaj, D. et al., 2016. Sedimentary oxygen dynamics in a seasonally hypoxic basin. Limnology and Oceanography.
Slomp, C., E. Epping, H. Willem, and W. Van Raaphorst. 1996. A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments. Journal of Marine Research 54: 1179–1205.
Soetaert, K., and J.J. Middelburg. 2009. Modeling eutrophication and oligotrophication of shallow-water marine systems: The importance of sediments under stratified and well-mixed conditions. Hydrobiologia 629 (1): 239–254.
Spiteri, C., Nolte, A., 2010. Validatie van het 3D model van het Grevelingenmeer voor hydrodynamica, waterkwaliteit en primaire productie, Deltares.
Sulu-Gambari, F. et al., 2016c. Molybdenum dynamics in sediments of a seasonally-hypoxic coastal marine basin. Biogeochemistry, (submitted).
Sulu-Gambari, F. et al., 2016b. Impact of Cable Bacteria on Sedimentary Iron and Manganese Dynamics in a Seasonally-Hypoxic Basin. Geochimica et Cosmochimica Acta.
Sulu-Gambari, F., et al. 2016a. Cable bacteria control iron–phosphorus dynamics in sediments of a coastal Hypoxic Basin. Environmental Science & Technology 50 (3): 1227–1233.
Sundby, B., C. Gobeil, N. Silverberg, and M. Alfonso. 1992. The phosphorus cycle in coastal marine sediments. Limnology and Oceanography 37 (6): 1129–1145.
Trojan, D., et al. 2016. A taxonomic framework for cable bacteria and proposal of the candidate genera Electrothrix and Electronema. Systematic and Applied Microbiology 39 (5): 297–306.
Tsandev, I., and C.P. Slomp. 2009. Modeling phosphorus cycling and carbon burial during cretaceous oceanic anoxic events. Earth and Planetary Science Letters 286 (1–2): 71–79.
van de Velde, S., et al. 2016. The impact of electrogenic sulfur oxidation on the biogeochemistry of coastal sediments: A field study. Geochimica et Cosmochimica Acta 194: 211–232.
Van Santvoort, P.J.M., et al. 2002. Oxidation and origin of organic matter in surficial eastern Mediterranean Hemipelagic sediments. Aquatic Geochemistry 8 (3): 153–175.
Viktorsson, L., N. Ekeroth, M. Nilsson, M. Kononets, and P.O.J. Hall. 2013. Phosphorus recycling in sediments of the central Baltic Sea. Biogeosciences 10 (6): 3901–3916.
Wetsteyn, L.P.M.J. 2011. Grevenlingenmeer: meer kwetsbaar? Lelystad: RWS Waterdienst.
Acknowledgements
We thank P van Rijswijk, S Hidalgo, A Zuidgeest, T Boerman, the crew of the R/V Luctor (P Coomans and M Kristalijn) and various members of the NIOZ and UU teams for their support during the sampling campaigns. We are also grateful to J Sinke, A Tramper, T Zalm, A van Dijk and D van de Meent for analytical support, M Wolthers for consultation with PHREEQC, as well as to P Mason and R D Schuiling for providing mineral samples for XANES analysis. We also extend our gratitude to F Mosselmans and the staff of beamline ID18 at the Diamond Light Source for their support, and to the Diamond Light Source for allocating beam time and providing financial support for performing the analyses. Finally, we thank C Rivard and D Stier for setting up the phosphorus XANES spectra database at ID21 and at the ESRF. This research was financially supported by the Darwin Centre for Biogeosciences, the European Research Council, under the European Community’s Seventh Framework Programme (ERC Starting Grants 278364 to CPS and 306933 to FJRM), the Netherlands Organisation for Scientific Research (NWO Vici 865.13.005 to CPS), the National Ocean and Coastal Research Programme (Grant 83910502 to JM) and the Netherlands Earth System Science Center (NESSC).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Margaret R. Mulholland
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Sulu-Gambari, F., Hagens, M., Behrends, T. et al. Phosphorus Cycling and Burial in Sediments of a Seasonally Hypoxic Marine Basin. Estuaries and Coasts 41, 921–939 (2018). https://doi.org/10.1007/s12237-017-0324-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12237-017-0324-0