
Abstract
Salinity variations in restricted basins like the Baltic Sea can alter their vulnerability to hypoxia (i.e., bottom water oxygen concentrations <2 mg/l) and can affect the burial of phosphorus (P), a key nutrient for marine organisms. We combine porewater and solid-phase geochemistry, micro-analysis of sieved sediments (including XRD and synchrotron-based XAS), and foraminiferal δ18O and δ13C analyses to reconstruct the bottom water salinity, redox conditions, and P burial in the Ångermanälven estuary, Bothnian Sea. Our sediment records were retrieved during the Integrated Ocean Drilling Program (IODP) Baltic Sea Paleoenvironment Expedition 347 in 2013. We demonstrate that bottom waters in the Ångermanälven estuary became anoxic upon the intrusion of seawater in the early Holocene, like in the central Bothnian Sea. The subsequent refreshening and reoxygenation, which was caused by gradual isostatic uplift, promoted P burial in the sediment in the form of Mn-rich vivianite. Vivianite authigenesis in the surface sediments of the more isolated part of the estuary ultimately ceased, likely due to continued refreshening and an associated decline in productivity and P supply to the sediment. The observed shifts in environmental conditions also created conditions for post-depositional formation of authigenic vivianite, and possibly apatite formation, at ∼8 m composite depth. These salinity-related changes in redox conditions and P burial are highly relevant in light of current climate change. The results specifically highlight that increased freshwater input linked to global warming may enhance coastal P retention, thereby contributing to oligotrophication in both coastal and adjacent open waters.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The area of coastal waters with hypoxic bottom waters (i.e., oxygen concentrations <2 mg/l) is expanding worldwide (Diaz and Rosenberg 2008; Rabalais et al. 2014). This not only increases fish mortality but also results in increased habitat compression and changes in benthic communities (Diaz and Rosenberg 2008). Restricted basins are naturally prone to bottom water hypoxia due to a density stratified water column, which may limit the vertical mixing of oxygen-rich surface water toward deeper waters (e.g., Farmer and Freeland 1983; Officer et al. 1984; Lefort et al. 2012). Many restricted basins today suffer from bottom water hypoxia, anoxia, or even euxinia (sulfidic bottom waters). Examples include the Baltic Sea, Chesapeake Bay, and the Lower St. Lawrence Estuary (Thibodeau et al. 2006; Diaz and Rosenberg 2008; Conley et al. 2011).
Many restricted basins have been subject to salinity changes throughout the Late Pleistocene and the Holocene (Dionne 1988; Middelburg 1991; Björck 1995; Hobbs 2004) and are likely to be affected by salinity changes in the near future. For instance, the annual mean salinity in the Gotland Deep (Baltic Proper) at the end of the twenty-first century is expected to be lower than any values observed since 1850 (Meier et al. 2012b). In Chesapeake Bay, the annual mean salinity may also change in the coming century (−2 to 2; Najjar et al. 2010) due to projected changes in yearly streamflow (−40 to +30% in 2100; Najjar et al. 2010). Variations in salinity can alter the degree of water column stratification and renewal of bottom water oxygen in restricted basins and thus affect their vulnerability to hypoxia (e.g., Lefort et al. 2012; Jilbert et al. 2015). It is therefore vital to understand the close coupling between water column salinity and redox conditions in these systems.
In addition, water column salinity and water column and bottom water redox conditions can affect the burial of phosphorus (P; Ruttenberg 2003; Slomp 2011), a key nutrient for marine organisms (Tyrrell 1999). For example, a lower salinity is thought to lead to more retention of Fe-oxide-bound P in sediments because of a lower availability of sulfate (SO4 2−), and associated lower rates of SO4 2− reduction and sulfide(HS−)-induced reduction of Fe-oxides (e.g., Caraco et al. 1990; Hartzell et al. 2009). In contrast, low oxygen conditions in bottom waters may enhance SO4 2− reduction and ultimately stimulate the release of P from Fe-oxides (Rozan et al. 2002). Changes in salinity and bottom water redox conditions can also affect the formation of other P-bearing minerals such as carbonate fluorapatite (CFA) (e.g., Ruttenberg and Berner 1993; Slomp et al. 1996; Kraal et al. 2012) by influencing porewater alkalinity, dissolved calcium (Ca2+), and dissolved phosphate (predominantly present as HPO4 2− at seawater pH, simplified in the text as PO4) concentrations through a complex interplay of geochemical and microbial processes (Ruttenberg 2003; Slomp 2011). The resulting changes in burial of sediment P can play a key role in regulating P availability and, subsequently, primary productivity and oxygen consumption in coastal waters (Froelich et al. 1982; Ruttenberg 2003).
Another relevant P form that is, however, rarely considered in studies of salinity and redox-dependent P burial in coastal sediments is vivianite, a reduced iron-phosphate (Fe3(PO4)2·8H2O). The formation and burial of authigenic vivianite depends on the balance between dissolved HS−, iron (Fe2+), and PO4 in porewaters (Rothe et al. 2015). In most marine sediments, Fe is mainly captured in Fe-sulfides and no Fe2+ is left to precipitate with PO4, thereby preventing the precipitation of vivianite. In many lake sediments, in contrast, bottom water SO4 2− concentrations are low and may allow for accumulation of Fe2+ and PO4 in the porewater and subsequent authigenic vivianite formation (Nriagu and Dell 1974; Rothe et al. 2014; O’Connell et al. 2015). In sediments of brackish/marine basins, an excess supply of reactive Fe-oxides may also act as a key driver of vivianite formation (Jilbert and Slomp 2013; Dijkstra et al. 2016; Reed et al. 2016). In addition to vivianite formed near the sediment-water interface, vivianite can also form diagenetically in brackish/marine basins below the sedimentary sulfate/methane transition zone (SMTZ) when porewater Fe2+ accumulates in the absence of HS− and meets porewater PO4 (März et al. 2008; Hsu et al. 2014; Egger et al. 2015a).
Major shifts in water column conditions can also affect P diagenesis in the subsurface by creating distinct geochemical gradients. For instance, vivianite crystals can form at lake/marine transitions in sediments, where upward diffusing Fe2+ from the lacustrine deposits meets PO4 from the overlying organic-rich marine deposits, as demonstrated recently for the Baltic Sea (Dijkstra et al. 2016). Moreover, enhanced input of organic matter may result in an upward shift of the SMTZ within the sediment (Egger et al. 2015b). This, in turn, results in an upward shift of the zone below the SMTZ where vivianite formation can be favorable due to the abundant presence of porewater Fe2+ (Egger et al. 2015a). Most studies on the relationship between variations in bottom water salinity and redox conditions and P burial focus on surface sediments along a salinity and/or redox transect (e.g., Hartzell et al. 2009; Mort et al. 2010; Dijkstra et al. 2014; Oxmann and Schwendenmann 2015). As a result, post-depositional P diagenesis related to non-steady-state depositional regimes often remains unrecognized.
The Baltic Sea has been subject to variations in salinity and redox conditions throughout the Holocene (Björck 1995; Zillén et al. 2008; Andrén et al. 2011) and is thus an ideal location to study both P authigenesis upon deposition and P diagenesis at depth. Currently, one of the largest hypoxic areas is located in the Baltic Proper (Fig. 1). The Baltic Sea is experiencing widespread hypoxia and severe cyanobacteria blooms, which are linked to enhanced external inputs of nutrients into the coastal waters from agriculture and waste water (Gustafsson et al. 2012; Funkey et al. 2014; Carstensen et al. 2014). In the past, large parts of the Baltic Proper were also hypoxic after the intrusion of seawater into the freshwater Ancyclus lake and the establishment of the Littorina Sea (A/L transition; ∼ 8000 year before present (BP); Andrén et al. 2011). This interval of hypoxia occurred during the Holocene Thermal Maximum (HTM; 8000–4000 years BP) when the inflow of North Sea water and warmer sea surface temperatures together likely promoted water column stratification and the subsequent development of hypoxia in the Baltic Proper (Zillén et al. 2008).

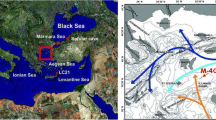
Map of the Baltic Sea with the sampling sites, bathymetry, and main basins (G.o.F. Gulf of Finland, G.o.R. Gulf of Riga). Depth is in meters below sea surface. The right panel shows a detailed map of the coring locations in the Ångermanälven estuary
A recent paleo-bathymetric model shows that the Bothnian Sea was probably well connected to the Baltic Proper between 8000 and 4000 years BP due to a deeper sill at its southern boundary. During that time, deep saline water from the Baltic Proper may have entered the Bothnian Sea (Jilbert et al. 2015), and the subsequent formation of stagnant deep waters likely promoted bottom water hypoxia in the Bothnian Sea. However, continuous isostatic shoaling of the Åland sills since deglaciation gradually restricted inflow of saline deep water into the basin, and the Bothnian Sea became oxic ∼4000 years BP (Jilbert et al. 2015). Over the past 8000 years, the coastal zones of the Bothnian Sea were also less isolated from the other parts of the Baltic Sea than at present (Lambeck et al. 1998; Berglund 2012) and may have experienced changes in salinity and redox conditions. It is unknown how these changes affected P burial in coastal areas of the Bothnian Sea.
In this study, we show that the coastal zone of the Bothnian Sea experienced anoxia after the intrusion of brackish water during the early Holocene. The later refreshening and reoxygenation affected P burial in the estuarine sediments by promoting vivianite authigenesis in the surface sediments. We further show that such shifts in environmental conditions can create settings for diagenetic vivianite formation at greater depth. Finally, we discuss the consequences of our findings within the context of increased freshwater inputs to the coastal zone linked to current climate change.
Materials and Methods
Study Area
Our study area, the Ångermanälven estuary, is one of the largest estuaries in the Bothnian Sea. It has a water volume of 3.180 km3 and is characterized by a short water residence time (5–14 days) and sediment accumulation rates of 5 to 6 mm/year (Humborg et al. 2003, and references therein). The modern estuary is oligotrophic, with mean monthly P concentrations of 0.3 ± 0.2 μM and nitrogen (N) concentrations of 18.7 ± 5.5 μM in the surface waters (0–20 m below sea surface; Humborg et al. 2003; Baltic Sea Environmental Database). The current annual discharge of the Ångermanälven River, which connects the estuary to the Bothnian Sea, is 16.7 km3 (Humborg et al. 2003). The present surface water salinity is estimated to be 2.7 in winter and 4.1 in summer (as determined with an estuarine circulation model; Stigebrandt 1981; Humborg et al. 2003). The area where the estuary is situated (Ångermanland) has experienced the highest rate of uplift of north-western Sweden, with a 250 m rise in the last 10,000 years (relative to sea level; Berglund 2012). The current rate of uplift is estimated at ∼7 mm/year (Ekman 1996; Lambeck et al. 1998).
Coring and Chronology
During the Integrated Ocean Drilling Program (IODP) Baltic Sea Paleoenvironment Expedition 347 in 2013, sediment cores were retrieved from two sites in the Ångermanälven estuary: site M0061 (62° 46.70′ N, 18° 02.95′ E) and site M0062 (62° 57.35′ N, 17° 47.70′ E). Site M0061 is located at the mouth of the Ångermanälven estuary (86 m water depth) whereas site M0062 is located further upstream (68 m water depth; Andrén et al. 2015; Fig. 1). Piston cores were collected from three holes at site M0061 (holes A–C) and four holes at site M0062 (holes A–D). The composite depth scale in Andrén et al. (2015) was used to calculate meter composite depths (mcd). All data can be found in Andrén et al. (2015) and in Online Resource 1.
We constructed an age model for site M0061 based on radiocarbon dating of seven terrestrial macrofossils that were identified in the upper ∼6 mcd (Hole C; Table 1; Fig. 2a) following the protocol summarized in Yamane et al. (2014) for ultra-small sample sizes (Yokoyama et al. 2010, 2016). Six samples are within the composite splice (Andrén et al. 2015), and one sample was obtained from a core catcher that is removed from the splice by 44 cm. While the composite depth of this sample is subject to uncertainty due to the likelihood of differential expansion/compaction between adjacent holes (e.g., Obrochta et al. 2014), the relative short length of these cores (3.3 versus 9.5 m for IODP cores obtained with the JOIDES Resolution) minimizes any potential error.

Age dating of site M0061 (a uncalibrated radiocarbon ages, 1σ uncertainty, and linear fit; b age-depth model with 2σ uncertainty, with calendar ages in years before present (1950); c sedimentation rate). Ages discussed in the text are given in years before present (y BP). Sediments that were deposited during phase B are most enriched in Mo and Corg, whereas maxima in P are observed in the phase C sediments
Age modeling was performed using six of the seven macrofossil ages, excluding one potentially reworked sample (YAUT-025815), in a routine modified from Obrochta et al. (2017). Ages were sequentially calibrated down the hole using the IntCal13 (Reimer et al. 2013) calibration curve and MatCal (Lougheed and Obrochta 2016) in a 1000-iteration Monte Carlo simulation. One probability-weighted age per iteration was selected from the 95.4% range of calibrated age probability density function (PDF). In case of an age reversal, the PDF was resampled, which is supported by the laminated nature of the sediments at site M0061. Calibrated ages are the median and 2σ range of the resulting 1000 age models (Table 2; Fig. 2b). The top of hole M0061C is assumed to be modern, though there is likely sediment missing, which is common with this type of coring apparatus.
Lithology and Biostratigraphy
The lower 20 m of the sediment record from site M0061 consists of sand, silt, or a combination of both (Fig. 3). These sediments are overlain by a 1.3 m thick section of more organic-rich laminated silty-sandy clay. The upper part of the sediment record from site M0061 consists of greenish to black laminated clay which gradually becomes lighter upward, marking a decrease in organic matter content in the younger sediments. The lower part of the sediment record from site M0062 consists of fine sands that were likely deposited in a glaciofluvial or fluvial system (holes A and B; Andrén et al. 2015). This sandy layer is overlain by a ∼10 m thick interval of very dark greenish-gray laminated silty clay. Within this layer, there is a distinct interval with greenish black intervals between 6 and 8.5 mcd. A more detailed description and interpretation of the lithology can be found in Andrén et al. (2015).

Trends of key elements with depth in the sediments from sites M0061 (holes A–C) and M0062 (holes A, C, and D). Sediment depth is in meters composite depth (mcd). A simplified lithologic column is shown on the right for each site; the detailed sediment lithology is described in Andrén et al. (2015). Note that the Mo values below background equivalent concentration (BEC) are only approximate values (precision can be >10% (1σ)). Phase A comprises the Ancyclus Lake phase. The other phases (B–D) occurred after the seawater intrusion in the Early Holocene. Sediments that were deposited during phase B are most enriched in Mo and Corg, whereas maxima in P are observed in the phase C sediments. The near-surface sediments at site M0062 (phase D) are lower in Mo, Corg, and P than the other Littorina Sea deposits
Diatom, foraminifera, and mollusk occurrences and assemblages were used to identify changes in salinity (Andrén et al. 2015; M0061: p. 15–17; M0062: p.13). At site M0061, fragments of the mollusk Mytilus edulis, which can tolerate salinities as low as 5, were found between 4.24 and 5.18 mcd. At site M0061, the first diatoms occur at ∼8 mcd. The assemblage consisted of freshwater taxa, brackish-freshwater, and more marine species (65, 15, and 20% of total assemblage, respectively). Brackish species are most abundant in the upper 6 mcd of sediments (>50%), likely representing the transition to more marine conditions. Based on the transition from freshwater to brackish diatom species at ∼7 mcd, the A/L boundary was estimated to have occurred at this depth. At site M0062, the first diatom species occurred at 8.6 mcd and mainly consisted of freshwater species. A sharp decrease in freshwater taxa occurs between 8.6 to 6.8 mcd, and the transition from a freshwater to a brackish environment was therefore set at 8.6 mcd upward. The freshwater taxa are again more abundant in the upper 3.4 mcd of sediments (>65% of total taxa), indicating a refreshening of the estuary.
Sediment samples for foraminifera were washed over a 63 μm sieve, dried, and inspected for foraminifera. Benthic foraminifera, which were observed for the first time in the Bothnian Gulf, were found between 1.7 to 6.3 mcd in the sediments at site M0061, with a maximum abundance at 3.4 mcd (Andrén et al. 2015). All foraminifera belonged to the species Elphidium albiumbilicatum or Elphidium excavatum, two species that can tolerate very low salinity (Rottgardt 1952; Lutze 1965). Foraminifera abundance was evaluated from core catchers during shipboard sampling directly after coring, while the remaining samples were taken 4–5 months later during core sampling. The observed foraminifera abundances were lower in the latter samples. This discrepancy may be caused by dissolution of foraminifera by sulfuric acid that is released by pyrite oxidation during sample storage (Kraal et al. 2009). No foraminifera were found at site M0062 (Andrén et al. 2015).
Benthic Foraminifera Stable Isotope and Trace Element Analyses
In the foraminifera-bearing interval at site M0061, foraminifera of sufficient abundance for stable oxygen and carbon isotope (δ18O and δ13C) analysis were found between 3.4 and 4.15 mcd (hole C). At the University of California Santa Cruz (UCSC), 11–18 foraminifera per sample of the species E. albiumbilicatum (size 63–250 μm) were analyzed for δ18O and δ13C using an IR-MS (MAT-256) with a Kiel auto-carbonate device. Analytical precision based on replicate measurements of the in-house carbonate standard Carrara Marble and NBS-19 included with the analytical run was ±0.03‰ for δ13CCaCO3 and ±0.06‰ for δ18OCaCO3 (1σ). One sample (M0061C, 1H–CC, 3.37 mcd) contained enough foraminifera for trace element analysis and was prepared using standard methods (Martin and Lea 2002), including reductive cleaning. Trace element analyses via ICP-MS were conducted for Mg, Mn, Fe, Al, B, U, and Ca at UCSC following techniques in Quintana Krupinski et al. (2017). Because of a lack of suitable Mg/Ca-temperature calibrations for this species and setting at this time, calculation of paleotemperature is not suitable, but the trace elemental data are provided in the Online Resources (2).
The foraminiferal δ18O data were used to estimate the maximum benthic salinity change over this interval and used in a sensitivity analysis giving a range of possible absolute salinities. However, these calculations required an assumed temperature, as a value for past bottom water temperature is not available. We assumed a range of local bottom water temperatures between 1.5 to 4 °C based on recent water temperature variations at 90-m water depth in the Bothnian Sea (Marmefelt and Omstedt 1993). We used the δ18O-temperature calibration of Epstein et al. (1953), as modified to a linear form by Bemis et al. (1998), to calculate seawater δ18O, and applied a salinity-δ18OSW mixing line which is suitable for the Baltic Sea and Skagerrak region (Frohlich et al. 1988). Trends in foraminiferal δ13C data were used to qualitatively evaluate changes in benthic water conditions (i.e., salinity, productivity, and oxygen).
Sediment Total Elements and Organic Carbon
Sediment samples were obtained during the onshore sampling at the Bremen Core Repository, stored in plastic-lined aluminum bags purged with nitrogen gas (N2) at 4 °C and transported to Utrecht University (sites M0061 and M0062). These samples were freeze-dried and ground with an agate mortar and pestle in an oxygen-free environment prior to total destruction (both sites) and additional CN analysis and sequential extractions (only site M0061).
To determine the total elemental composition, additional freeze-dried subsamples (0.125 g) were digested in a mixture of hydrofluoric acid, nitric acid, and perchloric acid at 90 °C. These acids were fumed off during the next day, and 1 M nitric acid was added to redissolve the residue. The total molybdenum (Mo), sulfur (S), Fe, aluminum (Al), manganese (Mn), and P concentrations in the final solutions were determined by inductively coupled plasma-optical emission spectrometry (ICP-OES; SPECTRO ARCOS). Note that the Mo values below background equivalent concentration (BEC) are only approximate values (precision can be ±10% (1σ)). For comparison, the Mo concentrations for a selection of the samples from site M0061 were also analyzed by ICP-MS. We observed no distinct differences between the Mo content measured by ICP-OES (including values below BEC) and by ICP-MS at site M0061 (Fig. 3). The precision for all elements, based on analyses of laboratory reference material (ISE-921), in-house standards, and sample triplicates, was generally less than 3% (1σ).
Sediment subsamples (0.2 g) from site M0061 were decalcified at Utrecht University with two washes with hydrochloric acid (1 M HCl; 4 and 12 h, respectively) and a final rinse with UHQ water (Van Santvoort et al. 2002). The decalcified samples were freeze-dried and Corg contents were determined by CN analysis (Fisons Instruments NA 1500 NCS analyzer). Precision based on duplicate samples and in-house standards was generally <5% (1σ). A set of organic carbon (Corg) analyses for site M0062 were already performed during the onshore science party). At the Bremen Core Repository, freeze-dried ground samples were decalcified using 12.5% HCl and analyzed on a LECO CS-300 carbon-sulfur analyzer. The analytical precision based on replicate sample analysis was <3% (Andrén et al. (2015) for more details).
Porewater Analysis
Porewater was collected on the ship directly after core recovery using rhizon samplers (0.2 μm pore size). Chloride (Cl−) concentrations were determined with Metrohm 882 Compact Ion Chromatography. Salinity was calculated from Cl− concentrations assuming that seawater with a salinity of 35 has a Cl− concentration of 569 mM (Dickson et al. 2007). Alkalinity was determined by single-point titration according to Grasshoff et al. (1983), and ammonium (NH4 +) was determined by conductivity following the method of Hall and Aller (1992). The total P, Fe, and Mn concentrations of filtered and acidified porewaters (0.2 μm; 10 μL of conc. HNO3 per mL) were determined by ICP-OES (Agilent Technologies 700 Series) and are assumed to mainly represent PO4, Fe2+, and Mn2+, respectively. Calibration standards for major elements and trace elements were prepared using IAPSO seawater and National Institute of Standards and Technology Certified Reference Material (NIST CRM). Measurement precision was ±3–5% (1σ). The Metrohm 882 compact ion chromatograph at the University of Bremen was used to measure SO4 2− (precision of ±1.6% (1σ)). To determine CH4 content, a 5 cm3 sediment sample was collected immediately after core recovery. The sample was extruded into a glass vial with 8 mL of 1 M NaOH solution and directly sealed and stored upside down. At Utrecht University, a volume of 250 μL from the head space of the sample vial was injected into a Thermo Finnigan Trace GC gas chromatograph to determine CH4 concentrations (precision generally less than ±10% (1 σ)). For further details on the analyses that were conducted onboard or during the sampling in the Bremen Core Repository, we refer to Andrén et al. (2015).
Sequential Phosphorus Extraction
We applied the SEDEX method (Ruttenberg 1992), as modified by Slomp et al. (1996), but including the exchangeable P step on freeze-dried and ground sediment subsamples (0.1 g) that were collected during the onshore sampling campaign (Hole M0061B). The exchangeable P fraction was targeted by an extraction with a magnesium chloride solution (1 M MgCl2 brought to pH 8 with sodium hydroxide) for 0.5 h. Afterward, Fe-bound P was targeted with a CDB solution (0.3 M trisodium citrate and 25 g/L sodium dithionite buffered to pH ∼ 7.6 in 1 M sodium bicarbonate) for 8 h, followed by a wash step with 1 M MgCl2 for 0.5 h. In addition to Fe-oxide-bound P, reduced Fe-phosphates such as vivianite also dissolve in the CDB step of the SEDEX extraction (Dijkstra et al. 2016). The authigenic Ca-P fraction was targeted with a sodium acetate solution (1 M) buffered with acetic acid (pH 4) for 6 h, and again followed by a short 1 M MgCl2 wash step (0.5 h). The subsample residue was then extracted with 1 M HCl for 24 h (detrital P). Finally, organic P was targeted with a 1 M HCl extraction for 24 h after combusting the subsample residue at 550 °C for 2 h. The P, Fe, Mn, and Ca in the CDB solutions were determined by ICP-OES (see Online Resource 3 for CDB Fe, Mn, and Ca). All other P concentrations were determined by the colorimetric molybdenum blue method (Strickland and Parsons 1972). The first step of the SEDEX was performed in a nitrogen-purged glovebox to minimize sample oxidation and associated changes in P fractionation (Kraal et al. 2009; Kraal and Slomp 2014). All other steps until the first HCl extraction were performed beneath a nitrogen gas line in order to shield the samples from the atmosphere. All extractants were purged with nitrogen for 30 min before usage. Conversion of other sedimentary P phases to Fe-oxide-bound P upon exposure to oxygen before opening and sampling of the cores is assumed to be minimal as also no major changes in the sedimentary P fractionation occurred in highly sulfidic sediments from IODP347 site M0063 during the same time of core storage (Dijkstra et al. 2016). The sum of the sequential P fractions over the whole cores was generally similar to the total P concentrations as derived from the ICP-OES (<15% difference). The precision, based on sample triplicates and in-house standards, were generally less than ±10% (1σ).
SEM-EDS of Vivianite Crystals
During the on-board sampling campaign, examination of sieved sediment with a light microscope revealed the presence of blue crystals (>63 μm). These crystals were observed at five depths in the upper 2 m of sediment at site M0061 (hole A, 0.28 and 1.19 mcd; hole B, 0.14 and 0.23 mcd; hole C, 1.55 mcd) and were collected for further analyses (SEM-EDS, XAS, XRD). In addition, blue crystals were also observed at 5.25 mcd at site M0062. The blue color is a characteristic oxidation artifact of phosphates from the vivianite group (e.g., Nriagu 1972; Yakubovich et al. 2001). This mineral group consists of minerals with the common formula M3(TO4)2·8H2O, where M represents Fe, Mg, Zn, Ni, Co, and T represents P or As. We will refer to this group of phosphates as vivianite (Fe(II)3(PO4)2, the pure Fe-phosphate form of the vivianite group (excluding arsenates). SEM-EDS was performed to estimate the abundances of major elements in these crystals. Blue crystals from site M0061 (hole C, 1.19 mcd) were mounted on an aluminum stub, coated with platinum, and analyzed by scanning electron microscope energy-dispersive spectroscopy (SEM-EDS; JCM 6000PLUS NeoScope Benchtop SEM). The SEM analysis was conducted with a 15-kV accelerating voltage, silica (Si)/Li detector, and 1 μm beam in backscatter mode. In addition, EDS analysis was performed in the 0–20-keV energy range (probe current 1 nA; acquisition time 50 s). We used SEM-EDS software to estimate the relative abundances in mol% for the major elements (P, Fe, Mn, Mg, Si, and Al). All other measured elements contributed on average less than 2% to the total molar mass.
XAS of Sieved Sediment Samples
Blue crystals from site M0061 (hole C, 1.19 mcd) were investigated by X-ray absorption spectroscopy for both X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) at the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. Prior to analysis, we concentrated the blue crystals in the sieved sediment samples by removing other large particles with a tweezer underneath a light microscope. The sieved sample was then ground with an agate mortar and pestle and mounted between Kapton® tape. The sample was investigated at the Dutch-Belgium beamline (DUBBLE, BM26) in June 2014 on the Fe and Mn energy range (7.00–7.65 and 6.50–6.90 keV, respectively). In addition, a sieved sample from 0.14 mcd was ground with an agate mortar and pestle and investigated under vacuum at the ID21 beamline in April 2015 on the P energy range (2.13–2.40 keV). After calibration of the monochromator against the maximum intensity of the first derivative of a Fe foil (7.11198 keV), Mn foil (6.53862 keV), and a tricalcium phosphate standard (2.14943 keV), the spectra were collected with an unfocused beam at room temperature in transmission (Fe) or fluorescence mode (Mn and P). Details on the layouts of the beamlines can be found in Borsboom et al. (1998), Nikitenko et al. (2008), and Salomé et al. (2013).
All individual spectra (2–8 spectra per sample) were merged to obtain average spectra. We did not collect any EXAFS spectra on the P energy range due to low signal to noise ratios. The ATHENA software package (Ravel and Newville 2005) was used for background removal and normalization. We also performed linear combination fitting with same software package (fitting range of 7100–7180 eV for XANES and 3–9 Å−1 for EXAFS). The XANES and EXAFS spectra of Fithian illite were also collected at BM26 in June 2014. The Mn-vivianite and the synthesized vivianite are described and published in Dijkstra et al. (2016) and Egger et al. (2015a). More information on the hureaulite Mn spectra can be found in Manceau et al. (2012).
XRD and Total Destruction of Sieved Sediment Sample
We investigated the minerology of the ground sample with blue crystals from site M0061 (hole C, 1.19 mcd) using a Bruker D2 diffractometer with Cobalt Kα radiation over a 5–85 2θ range (step size 0.026°; measurement time 0.4 s per step). The XRD spectrum of the vivianite standard is available in Dijkstra et al. (2016). Next, the elemental composition of the sieved sediment sample was determined after total digestion (similar method as for the freeze-dried sediment samples). We calculated the relative molar abundances of the major elements (note that Si is lost to the atmosphere during the HF digestion), excluding elements that contributed less than 2% to the total molar mass of the sample.
Results
Total Elemental Composition of the Sediment
At both sites, the sediments that were deposited during and just after the transition to more brackish conditions are enriched in Mo, Corg, and S (Fig. 3; Online Resource 1). In contrast, most surface sediments and deeper lacustrine sediments are low in those elements (<0.1 μmol Mo/g, 1 wt% Corg, and 100 μmol S/g). The organic-rich sediments are also slightly elevated in Fe/Al. At site M0062, these organic sediments were also high in Mn (>100 μmol/g). At both sites, maxima in P (>30 μmol/g) are observed in the upper part of the organic-rich sediment layer and at the transition from freshwater to brackish conditions.
Foraminiferal Stable Isotopes
Foraminiferal δ18O values decrease from −2.5‰ at 4.15 mcd to −3.1‰ at 3.40 mcd, and δ13C values decrease from −1.6‰ at 4.15 mcd to −2.3‰ at 3.40 mcd (Table 3). Both isotopes show a trend toward more negative values in younger sediments, and δ18O and δ13C are highly positively correlated (r 2 = 0.80, p < 0.05). We calculated a maximum decrease in salinity of 2.1 over the interval 4.15–3.40 mcd (Fig. 4) if we assume that all change in foraminiferal δ18O (−0.6 ‰) is due to salinity change (by assuming a constant temperature). It is unlikely that the calculated changes in salinities are overestimated due to a warming of the bottom waters during the same interval. The high magnetic inclinations in the sediments at ∼2 mcd (hole A) may correspond to the Holocene inclination features ε1 (∼2650 cal years BP) or γ (1290 cal years BP) (Snowball et al. 2007; Andrén et al. 2015). The foraminifera from 4.15 to 3.4 mcd were deposited just after the HTM (∼4200–3200 years BP; Fig. 4). At that time, the Baltic Sea experienced a general cooling trend instead of warming (e.g., Moros et al. 2004; Seppä et al. 2005). Much of the δ18O signal is therefore expected to be a response to refreshening.

Sensitivity analysis providing a range of estimated salinities and maximum salinity change, calculated from foraminiferal δ18O values from site M0061. This sensitivity analysis assumes that all change in foraminiferal δ18O in this interval is due to salinity change by using constant minimum and maximum bottom water temperature estimates (1.5 and 4 °C)
Given a range of bottom water temperatures between 1.5 and 4 °C, we can calculate a range of possible salinities during this interval. With an assumed constant temperature of 1.5 °C, the salinity would have decreased from 10.7 to 8.6 from ∼4200–3200 years BP, while for an assumed constant temperature of 4.0 °C, salinity would have decreased from 12.8 to 10.7 in the same time (Fig. 4). These salinities lie within the range of or slightly above the maximum mid-Holocene surface salinity estimates of Widerlund and Andersson (2011; 11–12 for the Bothnian Sea).
Porewater Chemistry
At both sites, salinity is highest in the lacustrine sediments (values up to 9.4) and lower in the surface sediments (∼6) (Fig. 5). Alkalinity, NH4 + and PO4 are lowest in the deeper lacustrine sediments with values below 10, 0.4, and 0.05 mmol/L, respectively. Highest values for alkalinity, NH4 + and PO4 are observed in the organic-rich deposits. Maxima in NH4 + and PO4 in the organic-rich deposits are higher at site M0061 than at site M0062. The dissolved Fe concentrations vary strongly with depth with high values in the lacustrine sediments below the transition from freshwater to brackish conditions (>0.2 mmol/L) and lowest values in the deeper part of the organic-rich sediments (<0.05 mmol/L). Concentrations of Fe2+ are highest in the youngest lacustrine deposits at site M0062. The trends in Mn2+ are generally similar to the trends in Fe2+, although Mn2+ is also very low in sediments just below the organic-rich deposits. Only the deeper sediments contain porewater SO4 2−, whereas porewaters high in CH4 are only observed in the upper sediments. An SMTZ, the transition zone where upward diffusing SO4 2− meets downward diffusing CH4, is located at ∼16 mcd at both sites.

Porewater chemistry at sites M0061 and M0062. For information on the phases A–D, see Fig. 3
Sequential Extraction of Phosphorus
Exchangable P is only a minor P burial form in the sediments at site M0061 (<2.2 μmol/g; Fig. 6). Organic P is below 1 μmol/g in the deeper lacustine sediments and highest in the upper 6 m of sediment (>2 μmol/g). Detrital P is an important P burial form throughout the sediment record (on average ∼12 μmol/g), and contents are particularly high in the lacustrine sand, clayey silt, and silty clay deposits. The upper part of these lacustrine sediments is also enriched in authigenic Ca-P. There is a distinct maximum of Fe-bound P in the upper 3 m of the sediment record with values reaching almost 60 μmol/g. In addition, there is a small peak in Fe-bound P at 7.15 mcd.

The P forms in sediments at site M0061, as determined by sequential P extraction (hole B). The fractions are exchangeable P (Ex P), organic P (Org P), detrital P (Detr P), authigenic Ca-P (Authi Ca-P), and Fe-bound P. The total extracted P pool is compared with the total P pool, as determined by total digestions and ICP-OES, in the right panel. For information on the phases A–D, see Fig. 3
Crystal Morphology, XRD, and Elemental Quantification
We observed blue crystals in the upper 2 m of the sediments at site M0061 that were all platy or needle-shaped and approximately 100 μm in size (Figs. 6 (B) and 7a). We indentified main diffraction peaks that can be assigned to vivianite in the sieved sample at 31.1, 15.3, 24.3, and 32.6° 2θ (Fig. 7c). Other significant peaks can be attributed to quartz, kaolinite, albite, or illite. Some diffraction peaks of the vivianite crystal are slightly shifted to the right in comparison to the synthesized vivianite standard (0.1–0.2° 2θ). A similar shift has been observed in other vivianite samples from natural environments (e.g., Nakano 1992; Egger et al. 2015a).

Investigation of blue crystals from 1.19 mcd at site M0061 (hole C) with light microscopy (a), SEM-EDS (b), and XRD (c). The sieved sediments that were analyzed by XRD also included other compounds than vivianite (q = quartz, k = kaolinite, a = albite, i = illite). No scale was available for the light microscope images (a)
The elemental composition of the sediment sample (Table 4) is in general accordance with the XRD analysis. The total elemental analysis of the sieved sediment indicated that it was enriched in elements associated with clay and quartz such as Al and Fe, and also contained P (11 mol%). Dissolution of salts may explain the slight enrichments of Mg, Ca, K, Na, and S in the sample (<10 mol%). With the SEM-EDS analyses, we were able to examine single blue crystals (Table 4). These crystals contained elements associated with vivianite (P, Fe, Mn, Mg). The high Si, Al, and Fe contents likely reflect clay particles that were not removed from the aggregates prior to SEM-EDS analysis.
XANES and EXAFS Spectra
The Fe-XANES and-EXAFS spectrum of the sieved sediment can be reproduced by a combination of vivianite and illite (Fig. 8a, b). The relative weights of vivianite and illite are 0.58 and 0.42 for the XANES spectrum (chi-squared = 0.09; R-factor 0.003) and 0.67 and 0.33 for the EXAFS spectrum (chi-squared = 1.31; R-factor 0.06), respectively. This suggests that vivianite is the main Fe mineral in the sieved sediment sample. The peak position of the white line of the sieved sediment is located between the peak positions of the white lines for the vivianite (7126.7 eV) and the illite standard (7132.0 eV). The post-edge oscillations of the sieved sediment and the vivianite standard are almost similar in shape and position. The Fe-EXAFS spectrum of the sieved sediment sample is also most identical to the Fe-EXAFS spectrum of the vivianite standard (Fig. 8b).

XANES and EXAFS spectra of the sieved sediment from site M0061 (1.19 mcd; hole C) and selected standards in the energy range for Fe (a, b), Mn (c, d), and P (e). Dashed lines show linear combination fit. No EXAFS spectra were collected in the P energy range due to low signal to noise ratios
The Mn XANES spectrum of the sieved sediment is almost similar to the spectra of hureaulite and Mn-rich vivianite (Fig. 8c). The peak positions of the XANES spectra are located at 6552.3, 6552.4, and 6552.6 eV, respectively. The higher amplitude of the Mn-XANES spectrum of the sediment sample compared to the spectra of hureaulite might be caused by less self-absorption in our sediment sample compared to the hureaulite standard. The EXAFS spectrum of the sieved sediment sample is almost identical to the spectra of the Mn-rich vivianite (Fig. 8d).
The position of the white line of the P-XANES of the sieved sediment sample (2153.4 eV) is close to the position of the white lines of the Mn-rich vivianite and the synthesized vivianite standard on the P energy range (2153.3 eV; Fig. 8e). The post-edge oscillations of the sieved sediment are identical but less pronounced than the post-edge oscillations of the Mn-rich vivianite and the synthesized vivianite standard.
Discussion
Holocene Reconstruction of Salinity, Redox Conditions, and P Burial
Based on lithology, biostratigraphy, chronology, geochemistry, and foraminiferal isotope data, we have reconstructed the environmental conditions in the Ångermanälven estuary during the Holocene. We have distinguished four phases that are described individually below (see Fig. 9 for a schematic overview).

Schematic overview of the Holocene history of the Ångermanälven estuary. The estuary has experienced four different phases with varying salinity, redox conditions and productivity. Due to seawater intrusion, the Ångermanälven estuary changed from an oligotrophic, freshwater, system (A) to a brackish system with productive waters (B). Afterward, isostatic uplift resulted in a gradual refreshening and subsequent reoxygenation of the estuary (C). At that time, the surface sediments were high in pore water PO4 but low in HS−, which resulted in authigenic vivianite formation (Fe(II)-P). At site M0062, the oligotrophic conditions, which lead to a decrease in organic P flux toward the seafloor, ultimately inhibited vivianite formation (D)
Phase A: the Ancyclus Lake Phase
The Ancyclus lake deposits were identified in our sediment records by their low Corg and S content and the absence of foraminifera and brackish/marine diatoms (Fig. 3; Andrén et al. 2015). The inland ice within the catchment of the Ångermanälven estuary disappeared just before the A/L transition (Cato 1985; Hyttinen et al. 2016). This likely caused a shift from glacial toward more fluvial weathering. The associated nutrient inputs may have stimulated primary productivity in the estuary, resulting in an increased organic matter flux to the seafloor. This may explain the concurrent peak in Corg and elements associated with rock and soil weathering (Al, Cr, Cu, K, and Ti; Zhou et al. 2004) in sediments just below the A/L transition (Fig. 3; Online resource 4).
Phase B: Hypoxia in the Estuary After Seawater Intrusion
Approximately 8000 y BP, seawater entered the Baltic Sea at its southwestern entrance, and the brackish waters from the Baltic proper intruded the Bothnian Sea (Jilbert et al. 2015). At that time, the Ångermanälven estuary was better connected to the Bothnian Sea than at present due to its isostatic depression (Berglund 2012), and it is possible that the seawater intrusion into the Ångermanälven estuary and open Bothnian Sea occurred nearly simultaneously. Salinities in the estuary may have been as high as 12 at site M0061 (Fig. 4), creating a suitable environment for foraminifera and brackish diatoms to establish (Andrén et al. 2015). The more inland part of the estuary (including site M0062) was likely less well connected to the Bothnian Sea and consequently less saline, as suggested earlier by Hyttinen et al. (2016). This explains the lack of foraminifera and the absence of marine diatom species at site M0062 (Andrén et al. 2015).
Due to the seawater intrusion, the estuary became more vulnerable to water column stagnation and deoxygenation of the bottom water. Hypoxia is recorded in both sediment records by the increase in sedimentary Mo to values >0.4 μmol/g (Fig. 3). Such enrichments are generally observed in sediments that are intermittently/seasonally overlain by euxinic waters (Scott and Lyons 2012). The associated abundant presence of HS− in the porewaters may have led to more sequestration of Fe as Fe-sulfides, resulting in sediments enriched in S and Fe/Al (Fig. 3). The variations in bottom water oxygen may have stimulated the transformation of Mn oxides to more stable Mn carbonates in these sediments, in particular at site M0062 where Mn content is >100 μmol/g (Fig. 3; Huckriede and Meischner 1996; Lenz et al. 2015). The high Corg content in these Mo-enriched sediments is likely the combined effect of enhanced preservation of organic matter in sediments under anoxic conditions (e.g., De Lange et al. 2008; Hartnett et al. 1998; Tsandev et al. 2012), and an increased flux of organic matter from the productive water column to the seafloor. The increased organic matter input is also reflected in maxima in porewater alkalinity, NH4 + and PO4, as part of the organic matter will degrade in the sediment (Fig. 5). The changes in salinity and productivity after the onset of the A/L transition are also clearly reflected in the sediment lithology where a shift to darker, and more greenish, clay rich in organic matter is observed at both sites (Fig. 3).
In contrast to Corg, the P content in the sediments did not increase during phase B (Fig. 3). This is in accordance with the general observation that Porg is often inefficiently retained in sediments of hypoxic basins (Algeo and Ingall 2007) due to preferential release of Porg from organic matter by C-limited microbes (Ingall et al. 1993; Steenbergh et al. 2011, 2013). Such sediments are characterized by sedimentary Corg/Porg ratios above the Redfield ratio for marine organic matter, just as we observe here (>400 versus 106 mol/mol; Online Resource 6).
Phase C: Refreshening and Vivianite Formation in the Estuary
The continuous uplift that occurred in the Baltic Sea region after the deglaciation led to gradual isolation of the estuary from the Bothnian Sea and other parts of the Baltic Sea. The subsequent refreshening likely explains the decreasing foraminifera abundance from 5 mcd upward (no foraminifera are present above 1.7 mcd; Andrén et al. 2015) and is also recorded in our foraminiferal isotopic record which indicates a decline in salinity of 2.3 between 4.15 and 3.40 m sediment depth (∼4200–3200 years BP; Table 3; Fig. 2). The strong correlation of foraminiferal δ18O and δ13C and the low regional salinity suggests that much of the δ13C signal also reflects declining salinities after the A/L transition, with impacts of bottom water oxygenation or productivity on δ13C being secondary. Refreshening of the estuary, which appears to have begun ∼5500 years BP (Fig. 2), based on decreasing foraminifera abundances from 5 mcd upward (Andrén et al. 2015), likely occurred at the same time as the basin-wide refreshening in the Baltic Sea (Gustafsson and Westman 2002).
We assume that these changes in the basin bathymetry and subsequently salinity were the main drivers for the reoxygenation of the Ångermanälven estuary that is recorded in our sediments by a sharp decrease in total Mo (Fig. 3). The thinner section of Mo-enriched sediments at Site M0062 than at Site M0061 (Fig. 3) suggests that the reoxygenation occurred first at the shallower upstream site M0062. At site M0061, bottom water hypoxia likely prevailed from ∼7000 to 4800 years BP (Figs. 2 and 3). The duration of the hypoxia in the estuary is thus less than 3800 years, the estimated interval of hypoxia in the deep part of the Bothnian Sea (Jilbert et al. 2015). At both sites in the estuary, the reoxygenation was followed by a gradual decrease in productivity in the estuary, shown by declining Corg content in the sediment (Fig. 3). Due to the refreshening and reoxygenation, the HS− concentrations in the bottom waters and sediments likely declined, resulting in less sequestration of S and Fe as Fe-sulfides in the sediments (Fig. 3).
During the refreshening and reoxygenation, Fe2+ started to accumulate in the sediments in the absence of HS− (Fig. 5). This created suitable conditions for vivianite authigenesis in the estuarine sediments, whereby the abundant Fe2+ precipitated with PO4 released from organic matter decomposition. The decline in sedimentary S/Fe ratios (Online Resource 5) is consistent with a transition to an environment that is supportive of vivianite formation (Rothe et al. 2016). Indeed, blue aggregates were observed in the upper part of the organic-rich sediments at both sites (Andrén et al. 2015). With the use of XRD and SEM-EDS, we show that these aggregates are vivianite minerals (Fig. 7; Table 4). In addition, the XAS analysis confirms that the blue crystals are vivianite and that they contain Mn (Fig. 8). Vivianite dissolves in the CDB extraction step of SEDEX (Nembrini et al. 1983; Dijkstra et al. 2016; Kraal et al. 2017), just as Fe-oxide-bound P (Ruttenberg 1992), and may thus represent the high Fe-bound P content in the upper sediments at Site M0061. We note that the Fe/P ratios of the CDB solutions are close to 1.5 mol/mol (Online Resource 3), which is the Fe/P ratio of pure vivianite. In contrast, typical Fe/P ratios for sedimentary Fe-oxides are ∼10:1 mol/mol (Slomp et al. 1996; Anschutz et al. 1998; Dellwig et al. 2010). The formation of vivianite thus likely explains the more than twofold increase in sediment P concentrations in the upper part of the organic-rich sediments at both sites (Fig. 3).
Phase D: Low Productivity in the Modern Estuary (Site M0062)
Together with the continuous isostatic uplift, the enhanced burial of P may have caused a further decline in water column productivity and lower organic matter burial at both sites. At site M0062, the Corg content is even below 1 wt% in the upper 6 mcd of sediments (Fig. 3), and low organic matter remineralization resulted in a decreased release of alkalinity, NH4 +, and PO4 to the porewaters (Fig. 5). Low PO4 concentrations in the porewaters may have ultimately inhibited vivianite authigenesis in the sediments at site M0062, resulting in sedimentary P concentrations that are similar to values below the A/L transition (Fig. 3).
Coupled Changes in Salinity and Redox Conditions in the Past and Future
The low oxygen conditions in the Ångermanälven estuary have coincided with the extensive hypoxia that occurred during the HTM throughout the Baltic Proper and in the central Bothnian Sea (8000–4000 years BP; Zillén et al. 2008; Jilbert and Slomp 2013; Jilbert et al. 2015; Fig. 2). Not only the Ångermanälven estuary but all coastal waters in the region were better connected to the open waters of the Bothnian Sea 8000 years BP (Lambeck et al. 1998; Berglund 2012). The hypoxia during the HTM thus likely covered large areas of the Baltic Sea.
In contrast, the hypoxia in the Baltic Proper during the Medieval Climate Anomaly (MCA; 2000–800 years BP) is not recorded in our coastal sediments (Fig. 3). This agrees with the study of Jilbert et al. (2015), in which the reduced exchange of deep saline water at the Åland sills during the MCA is proposed to be a primary cause of the prevalence of oxic bottom water conditions in the central part of the Bothnian Sea (Jilbert et al. 2015).
Such glacio-isostatic control on salinity and redox conditions also occurred in areas beyond the Bothnian Sea. For instance, coastal sites adjacent to the Baltic Proper were also more connected to the open waters 8000 years BP (Påsse and Andersson 2005). At Gåsfjärden, a Swedish coastal site located on the western shores of the Baltic Proper, bottom water hypoxia also occurred during the HTM whereas the area remained oxygenated during the MCA (Ning et al. 2016). The central Bothnian Bay was also likely more saline and productive in the early Holocene than today (Ingri et al. 2014). Other brackish basins have also been subject to Holocene isostatic uplift, such as the Lower St. Lawrence Estuary and Norwegian Fjords (Skei 1988; Duchesne et al. 2010). These basins may also have become more vulnerable to hypoxia since deglaciation, as proposed recently by Jilbert et al. (2015). This further illustrates that paleobathymetric changes can control salinity and past hypoxia in restricted basins.
The bottom water salinities in the Ångermanälven estuary may have decreased over the last 8000 years from ∼11 (Fig. 4) to salinities in the bottom waters that are close to current surface water salinities of 3.4 (Stigebrandt 1981; Humborg et al. 2003). This change is of similar magnitude as the decrease in bottom water and surface water salinities for other parts of the Baltic Sea during the Holocene (Gustafsson and Westman 2002). In the coming century, climate projections predict a decline in average mean salinity in the Baltic Sea of ∼2 due to increased freshwater runoff (Meier et al. 2012a). This refreshening is expected to occur at a rate that is an order of magnitude faster than during the last 8000 years and may thus impact future bottom water oxygen concentrations in the Baltic Sea.
This is supported by a coupled physical-biogeochemical modeling study of Meier et al. (2011), which predicts that increased freshwater inputs, together with wind speed changes, indeed may increase bottom water oxygen concentrations in the coastal zones of the Baltic Sea in the coming century. The Gulf of Finland (Fig. 1) is another area in which the impact of reduced stratification due to increased runoff may lead to more oxygen-rich bottom waters by the end of the twenty-first century (Meier et al. 2011). In restricted basins without severe stratification, climate-induced refreshening can thus potentially counteract the effects of warmer temperatures on water column stratification.
Vivianite and CFA Authigenesis in Semi-Enclosed Basins
The conditions that led to the formation of vivianite in the Ångermanälven estuary are schematized in Fig. 9. We demonstrate that authigenic vivianite minerals can form in near-surface sediments in restricted basins that are subject to refreshening and a recovery from hypoxia. Our results are in accordance with recent studies showing that vivianite can act as an important burial sink for P in modern Baltic Sea sediments (Jilbert and Slomp 2013; Dijkstra et al. 2014, 2016; Egger et al. 2015a). Sediments from the central coastal Bothnian Sea that were deposited after the recovery from hypoxia during the HTM were also enriched in P (>30 μmol/g; Jilbert et al. 2015). In the Archipelago Sea (Finland), weakly laminated sediments in the upper part of the Holocene deposits were enriched in Fe-bound P (>40 μmol/g; Virtasalo and Kotilainen 2008). We hypothesize that these P enrichments are the result of authigenic vivianite formation, but further analyses are needed to confirm this.
The observed coupling between water column refreshening and sedimentary vivianite burial in the Ångermanälven estuary is highly relevant in the light of current climate change as some coastal systems may experience freshening in the near future due to increased freshwater inputs (e.g., Najjar et al. 2010; Meier et al. 2012b). This decrease in water column salinity may enhance P burial (as vivianite) in the sediments, thereby contributing to oligotrophication in both coastal and adjacent open waters.
The shifts in environmental conditions in the Ångermanälven estuary not only affected the P diagenesis upon deposition, as discussed above, but also resulted in diagenetic alternations at greater depth in the sediments. We find removal of both Fe2+ and PO4 where downward diffusing PO4 from the organic-rich sediments meets the upward diffusing Fe2+ (Fig. 5). The dissolved Fe is supplied by reductive dissolution of Fe-oxides at depth in the lacustrine sediments. As these sediments are rich in CH4 but low in SO4 2− (Fig. 5), some of the Fe reduction might be coupled to anaerobic oxidation of CH4 (Beal et al. 2009), as has been suggested for other sites in the Baltic Sea (Egger et al. 2015b). In the Baltic Proper, blue aggregates were recently discovered in sediments at the A/L transition and identified as vivianite by SEM-EDS (Dijkstra et al. 2016). It is likely that the P enrichments in our sediments at the A/L transition (Fig. 3) and the peak in Fe-bound P (Fig. 6) are also the result of vivianite authigenesis at depth. Assuming a porosity of 0.8 and using sediment diffusion coefficients for Fe2+ and PO4, corrected for salinity, temperature, and tortuosity (Boudreau 1997), we calculate diffusive fluxes of Fe2+ and PO4 to the A/L transition of ∼1.7 and ∼0.6 mmol/m2/year. With an average concentration of 20 μmol/g for the Fe-P enrichment over a depth of 0.2 m, and assuming a sediment density of 2.65 g/cm3, we estimate that ∼2200 and ∼1500 mmol/m2 of Fe and P, respectively, are present as Fe-bound P. With the present-day Fe2+ and PO4 profiles, and assuming no other removal processes, it would thus take up to ∼2500 years to form the Fe-bound P enrichment at the A/L transition, which is a reasonable time frame.
Although many other restricted basins are also characterized by a clear lake-marine transition in the sediment (e.g., Björck 1995; Dionne 1988; Hobbs 2004; Middelburg 1991), this may not always lead to vivianite formation at this sedimentary boundary. In the Ångermanälven estuary, a SMTZ in the lacustrine sediments prevents SO4 2− from greater depths from diffusing up toward the lake-marine boundary and allows Fe2+ to accumulate in the methanogenic lacustrine sediments. In contrast, Black Sea lake sediments that are located just below the Holocene marine deposits are high in porewater SO4 2− and lack Fe2+. As a consequence, vivianite precipitates only in the deeper sediments far (∼2 m) below the lake-marine transition where downward diffusing SO4 2− is fully consumed by anaerobic oxidation of CH4 and organoclastic SO4 2− reduction (Egger et al. 2016). This illustrates that Fe, S, and CH4 dynamics in non-steady-state regimes act as an important control on post-depositional P diagenesis.
Some of the removal of PO4 below the A/L transition might also be the result of diagenetic formation of apatite in the lacustrine sediments of the Ångermanälven estuary. The high fraction of authigenic Ca-P below the A/L transition at Site M0061 (Fig. 6) may represent apatite minerals that form when downward diffusing PO4 meets porewaters rich in Ca2+ (>6 mM Ca2+ in the lacustrine sediments; Andrén et al. 2015). The release of PO4 from either organic matter or Fe-oxides and the formation of apatite typically occur at approximately the same depth in the sediments (Ruttenberg and Berner 1993; Slomp et al. 1996; Ruttenberg 2003). A less direct sink-switching mechanism, whereby PO4 released from brackish sediments diffuses downward and precipitates in the underlying lacustrine sediments, might thus be present in non-steady-state regimes as the Ångermanälven estuary.
Conclusion
The Ångermanälven estuary of the Bothnian Sea became hypoxic after the seawater intrusion during the Holocene, much like the central Bothnian Sea and large parts of the Baltic Proper. These low oxygen conditions were followed by a period of reoxygenation and refreshening, which resulted in massive burial of Fe-bound P in the sediments (∼50 μmol P/g). Blue crystals were observed in sediments from this interval and micro-analysis of the crystals and our sieved samples through SEM-EDS, XRD, and synchrotron-based XAS demonstrate the abundant presence of vivianite in these sediments. Vivianite authigenesis later decreased at the site in the upper part of the estuary. We also have strong indications for post-depositional vivianite authigenesis at the transition from lacustrine to brackish sediments (A/L transition), a phenomenon that might be widespread throughout the Baltic Sea. Refreshening, as may occur in some coastal systems in the near future due to increased freshwater inputs, can thus promote burial of vivianite and enhance coastal P retention.
References
Algeo, T.J., and E. Ingall. 2007. Sedimentary Corg: P ratios, paleocean ventilation, and Phanerozoic atmospheric pO2. Palaeogeography Palaeoclimatology Palaeoecology 256: 130–155. doi:10.1016/j.palaeo.2007.02.029.
Andrén, T., S. Björck, E. Andrén, and D. Conley. 2011. The development of the Baltic Sea Basin during the last 130 ka. In The Baltic Sea Basin, ed. J. Harff, S. Björck, and P. Hoth, 75–97. Berlin: Springer-Verlag.
Andrén T, Jørgensen BB, Cotterill C, Green S, Expedition 347 Scientists. 2015. Proc. IODP, 347. College Station, TX (Integrated Ocean Drilling Program).
Anschutz, P., S. Zhong, B. Sundby, A. Mucci, and C. Gobeil. 1998. Burial efficiency of phosphorus and the geochemistry of iron in continental margin sediments. Limnology and Oceanography 43: 53–64. doi:10.4319/lo.1998.43.1.0053.
Beal, E.J., C.H. House, and V.J. Orphan. 2009. Manganese- and iron-dependent marine methane oxidation. Science 325: 184–187. doi:10.1126/science.1169984.
Bemis, B.E., H.J. Spero, J. Bijma, and D.W. Lea. 1998. Reevaluation of the oxygen isotopic composition of planktonic foraminifera: Experimental results and revised paleotemperature equations. Paleoceanography 13: 150–160. doi:10.1029/98PA00070.
Berglund, M. 2012. The highest postglacial shore levels and glacio-isostatic uplift pattern in northern Sweden. Geografiska Annaler. Series A, Physical Geography 94: 321–337. doi:10.1111/j.1468-0459.2011.00443.x.
Björck, S. 1995. A review of the history of the Baltic Sea, 13.0-8.0 ka BP. Quaternary International 27: 19–40.
Borsboom, M., W. Bras, I. Cerjak, D. Detollenaere, D. Glastra van Loon, P. Goedtkindt, M. Konijnenburg, P. Lassing, Y.K. Levine, B. Munneke, M. Oversluizen, R. van Tol, and E. Vlieg. 1998. The Dutch–Belgian beamline at the ESRF. Journal of Synchrotron Radiation 5: 518–520.
Boudreau, B.P. 1997. Diagenetic models and their implementation (Vol. 505). Berlin: Springer.
Caraco, N., J. Cole, and G. Likens. 1990. A comparison of phosphorus immobilization in sediments of freshwater and coastal marine systems. Biogeochemistry 9: 277–290. doi:10.1007/BF00000602.
Carstensen, J., J.H. Andersen, B.G. Gustafsson, and D.J. Conley. 2014. Deoxygenation of the Baltic Sea during the last century. Proceedings of the National Academy of Sciences of the United States of America 111: 5628–5633. doi:10.1073/pnas.1323156111.
Cato, I. 1985. The definitive connection of the Swedish geochronological time scale with the present, and the new date of the zero year in Döviken, northern Sweden. Boreas 14: 117–122. doi:10.1111/j.1502-3885.1985.tb00901.x.
Conley, D.J., J. Carstensen, J. Aigars, P. Axe, E. Bonsdorff, T. Eremina, B.-M. Haahti, C. Humborg, P. Jonsson, J. Kotta, C. Lännegren, U. Larsson, A. Maximov, M.R. Medina, E. Lysiak-Pastuszak, N. Remeikaité-Nikiené, J. Walve, S. Wilhelms, and L. Zillén. 2011. Hypoxia is increasing in the coastal zone of the Baltic Sea. Environmental Science & Technology 45: 6777–6783. doi:10.1021/es201212r.
De Lange, G.J., J. Thomson, A. Reitz, C.P. Slomp, M. Speranza Principato, E. Erba, and C. Corselli. 2008. Synchronous basin-wide formation and redox-controlled preservation of a Mediterranean sapropel. Nature Geoscience 1: 606–610. doi:10.1038/ngeo283.
Dellwig, O., T. Leipe, C. März, M. Glockzin, F. Pollehne, B. Schnetger, E.V. Yakushev, M.E. Böttcher, and H.-J. Brumsack. 2010. A new particulate Mn–Fe–P-shuttle at the redoxcline of anoxic basins. Geochimica et Cosmochimica Acta 74: 7100–7115. doi:10.1016/j.gca.2010.09.017.
Diaz, R.J., and R. Rosenberg. 2008. Spreading dead zones and consequences for marine ecosystems. Science 321: 926–929. doi:10.1126/science.1156401.
Dickson AG, Sabine CL, Christian JR (2007) Guide to best practices for ocean CO2 measurements. PICES Spec Publ 3 3:191. doi:10.1159/000331784.
Dijkstra, N., P. Kraal, M.M.M. Kuypers, B. Schnetger, and C.P. Slomp. 2014. Are iron-phosphate minerals a sink for phosphorus in anoxic Black Sea sediments? PloS One 9: e101139. doi:10.1371/journal.pone.0101139.
Dijkstra, N., C. Slomp, T. Behrends, and Expedition 347 Scientists. 2016. Vivianite is a key sink for phosphorus in sediments of the Landsort Deep, an intermittently anoxic deep basin in the Baltic Sea. Chemical Geology 438: 58–72.
Dionne, J.C. 1988. Holocene relative sea-level fluctuations in the St. Lawrence estuary, Québec, Canada. Quaternary Research 29: 233–244. doi:10.1016/0033-5894(88)90032-4.
Duchesne, M.J., N. Pinet, K. Bédard, G. St-Onge, P. Lajeunesse, D.C. Campbell, and A. Bolduc. 2010. Role of the bedrock topography in the Quaternary filling of a giant estuarine basin: The Lower St. Lawrence Estuary, Eastern Canada. Basin Research. doi:10.1111/j.1365-2117.2009.00457.x.
Egger, M., T. Jilbert, T. Behrends, C. Rivard, and C.P. Slomp. 2015a. Vivianite is a major sink for phosphorus in methanogenic coastal surface sediments. Geochimica et Cosmochimica Acta 169: 217–235. doi:10.1016/j.gca.2015.09.012.
Egger, M., O. Rasigraf, C.J. Sapart, T. Jilbert, M.S.M. Jetten, T. Röckmann, C. Van Der Veen, N. Banda, B. Kartal, K.F. Ettwig, and C.P. Slomp. 2015b. Iron-mediated anaerobic oxidation of methane in brackish coastal sediments. Environmental Science & Technology 49: 277–283. doi:10.1021/es503663z.
Egger, M., P. Kraal, T. Jilbert, F. Sulu-Gambari, C.J. Sapart, T. Röckmann, and C.P. Slomp. 2016. Anaerobic oxidation of methane alters sediment records of sulfur, iron and phosphorus in the Black Sea. Biogeosciences 13: 5333–5355. doi:10.5194/bg-13-5333-2016.
Ekman, M. 1996. A consistent map of the postglacial uplift of Fennoscandia. Terra Nova 8: 158–165. doi:10.1111/j.1365-3121.1996.tb00739.x.
Epstein, S., R. Buchsbaum, H.A. Lowenstam, and H.C. Urey. 1953. Revised carbonate-water isotopic temperature scale. Geological Society of America Bulletin 64: 1315. doi:10.1130/0016-7606(1953)64%5B1315:RCITS%5D2.0.CO;2.
Farmer, D.M., and H.J. Freeland. 1983. The physical oceanography of Fjords. Progress in Oceanography 12: 147–219. doi:10.1016/0079-6611(83)90004-6.
Froelich, P., M. Bender, and N. Luedtke. 1982. The marine phosphorus cycle. American Journal of Science 282: 474–511.
Frohlich, K., J. Grabczak, and K. Rozanski. 1988. Deuterium and oxygen-18 in the Baltic Sea. Chem Geol Isot Geosci Sect 72: 77–83.
Funkey, C.P., D.J. Conley, N.S. Reuss, C. Humborg, T. Jilbert, and C.P. Slomp. 2014. Hypoxia sustains cyanobacteria blooms in the baltic sea. Environmental Science & Technology 48: 2598–2602. doi:10.1021/es404395a.
Grasshoff, K., Ehrhardt, M., Kremling, K. (eds) 1983. Methods of seawater analysis, Second. Weinheim/Deerfield Beach, Florida, Verlag Chemie.
Gustafsson, B.G., and P. Westman. 2002. On the causes for salinity variations in the Baltic Sea during the last 8500 years. Paleoceanography 17: 1–14. doi:10.1029/2000PA000572.
Gustafsson, B., F. Schenk, T. Blenckner, K. Eilola, H.E.M. Meier, B. Müller-Karulis, T. Neumann, T. Ruoho-Airola, O.P. Savchuk, and E. Zorita. 2012. Reconstructing the development of Baltic Sea eutrophication 1850–2006. Ambio 41: 534–548.
Hall, P.O.J., and R.C. Aller. 1992. Rapid, small-volume, flow injection analysis for SCO2, and NH4+ in marine and freshwaters. Limnology and Oceanography 37: 1113–1119. doi:10.4319/lo.1992.37.5.1113.
Hartnett, H.E., R.G. Keil, J.I. Hedges, and A.H. Devol. 1998. Influence of oxygen exposure time on organic carbon preservation in continental margin sediments. Nature 391: 572–575. doi:10.1038/35351.
Hartzell, J.L., T.E. Jordan, and J.C. Cornwell. 2009. Phosphorus burial in sediments along the salinity gradient of the Patuxent River, a subestuary of the Chesapeake Bay (USA). Estuaries and Coasts 33: 92–106. doi:10.1007/s12237-009-9232-2.
Hobbs, C. 2004. Geological history of Chesapeake Bay, USA. Quaternary Science Reviews 23: 641–661.
Hsu, T., W. Jiang, and Y. Wang. 2014. Authigenesis of vivianite as influenced by methane-induced sulfidization in cold-seep sediments off southwestern Taiwan. Journal of Asian Earth Sciences 89: 88–97.
Huckriede, H., and D. Meischner. 1996. Origin and environment of manganese-rich sediments within black-shale basins. Geochimica et Cosmochimica Acta 60: 1399–1413. doi:10.1016/0016-7037(96)00008-7.
Humborg, C., Å. Danielsson, B. Sjöberg, and M. Green. 2003. Nutrient land–sea fluxes in oligothrophic and pristine estuaries of the Gulf of Bothnia, Baltic Sea. Estuarine, Coastal and Shelf Science 56: 781–793. doi:10.1016/S0272-7714(02)00290-1.
Hyttinen, O., Kotilainen, A.T., Virtasalo, J.J., Kekäläinen, P., Snowball, I., Obrochta, S., Andrén, T. 2016. Holocene stratigraphy of the Ångermanälven River estuary, Bothnian Sea. Geo-Marine Letters 1–16. doi:10.1007/s00367-016-0490-2.
Ingall, E.D., R.M. Bustin, and P. Van Cappellen. 1993. Influence of water column anoxia on the burial and preservation of carbon and phosphorus in marine shales. Geochimica et Cosmochimica Acta 57: 303–316. doi:10.1016/0016-7037(93)90433-W.
Ingri, J., A. Widerlund, T. Suteerasak, S. Bauer, and S.-Å. Elming. 2014. Changes in trace metal sedimentation during freshening of a coastal basin. Marine Chemistry 167: 2–12. doi:10.1016/j.marchem.2014.06.010.
Jilbert, T., and C.P. Slomp. 2013. Iron and manganese shuttles control the formation of authigenic phosphorus minerals in the euxinic basins of the Baltic Sea. Geochimica et Cosmochimica Acta 107: 155–169. doi:10.1016/j.gca.2013.01.005.
Jilbert, T., D.J. Conley, B.G. Gustafsson, C.P. Funkey, and C.P. Slomp. 2015. Glacio-isostatic control on hypoxia in a high-latitude shelf basin. Geology 43: 427–430. doi:10.1130/G36454.1.
Kraal, P., and C.P. Slomp. 2014. Rapid and extensive alteration of phosphorus speciation during Oxic storage of wet sediment samples. PloS One 9: e96859. doi:10.1371/journal.pone.0096859.
Kraal, P., C.P. Slomp, A. Forster, M.M.M. Kuypers, and A. Sluijs. 2009. Pyrite oxidation during sample storage determines phosphorus fractionation in carbonate-poor anoxic sediments. Geochimica et Cosmochimica Acta 73: 3277–3290. doi:10.1016/j.gca.2009.02.026.
Kraal, P., C.P. Slomp, D.C. Reed, G.-J. Reichart, and S.W. Poulton. 2012. Sedimentary phosphorus and iron cycling in and below the oxygen minimum zone of the northern Arabian Sea. Biogeosciences 9: 2603–2624. doi:10.5194/bg-9-2603-2012.
Kraal, P., N. Dijkstra, T. Behrends, and C.P. Slomp. 2017. Phosphorus burial in sediments of the sulfidic deep Black Sea: Key roles for adsorption by calcium carbonate and apatite authigenesis. Geochimica et Cosmochimica Acta 204: 140–158.
Lambeck, K., C. Smither, and M. Ekman. 1998. Tests of glacial rebound models for Fennoscandinavia based on instrumented sea- and lake-level records. Geophysical Journal International 135(2): 375–387.
Lefort, S., Y. Gratton, A. Mucci, I. Dadou, and D. Gilbert. 2012. Hypoxia in the Lower St. Lawrence estuary: how physics controls spatial patterns. Journal of Geophysical Research 117: 1–14. doi:10.1029/2011JC007751.
Lenz, C., T. Jilbert, D.J. Conley, and C.P. Slomp. 2015. Hypoxia-driven variations in iron and manganese shuttling in the Baltic Sea over the past 8 kyr. Geochemistry, Geophysics, Geosystems 16: 3754–3766. doi:10.1002/2015GC005960.
Lougheed, B.C., and S.P. Obrochta. 2016. MatCal: open source Bayesian 14C age calibration in Matlab. Journal of Open Research Software. doi:10.5334/jors.130.
Lutze, G. 1965. Zur foraminiferen-Fauna der Ostsee. Meyniana 15: 75–142.
Manceau, A., M.A. Marcus, and S. Grangeon. 2012. Determination of Mn valence states in mixed-valent manganates by XANES spectroscopy. American Mineralogist 97: 816–827. doi:10.2138/am.2012.3903.
Marmefelt, E., and A. Omstedt. 1993. Deep water properties in the Gulf of Bothnia. Continental Shelf Research 13: 169–187. doi:10.1016/0278-4343(93)90104-6.
Martin, P.A., and D.W. Lea. 2002. A simple evaluation of cleaning procedures on fossil benthic foraminiferal Mg/Ca. Geochemistry, Geophysics, Geosystems 3: 1–8.
März, C., J. Hoffmann, U. Bleil, G.J. de Lange, and S. Kasten. 2008. Diagenetic changes of magnetic and geochemical signals by anaerobic methane oxidation in sediments of the Zambezi deep-sea fan (SW Indian Ocean). Marine Geology 255: 118–130. doi:10.1016/j.margeo.2008.05.013.
Meier, H.E.M., H.C. Andersson, K. Eilola, B.G. Gustafsson, I. Kuznetsov, B. Müller-Karulis, T. Neumann, and O.P. Savchuk. 2011. Hypoxia in future climates: A model ensemble study for the Baltic Sea. Geophysical Research Letters 38: 1–6. doi:10.1029/2011GL049929.
Meier, H., R. Hordoir, H. Andersson, and C. Dieterich. 2012a. Modeling the combined impact of changing climate and changing nutrient loads on the Baltic Sea environment in an ensemble of transient simulations for 1961–2099. Climate Dynamics 39: 2412–2441.
Meier, H.E.M., H.C. Andersson, B. Arheimer, T. Blenckner, B. Chubarenko, C. Donnelly, K. Eilola, B.G. Gustafsson, A. Hansson, J. Havenhand, A. Höglund, I. Kuznetsov, B.R. MacKenzie, B. Müller-Karulis, T. Neumann, S. Niiranen, J. Piwowarczyk, U. Raudsepp, M. Reckermann, T. Ruoho-Airola, O.P. Savchuk, F. Schenk, S. Schimanke, G. Väli, J.-M. Weslawski, and E. Zorita. 2012b. Comparing reconstructed past variations and future projections of the Baltic Sea ecosystem—first results from multi-model ensemble simulations. Environmental Research Letters 7: 34005. doi:10.1088/1748-9326/7/3/034005.
Middelburg, J.J. 1991. Organic carbon, sulphur, and iron in recent semi-euxinic sediments of Kau Bay, Indonesia. Geochimica et Cosmochimica Acta 55: 815–828. doi:10.1016/0016-7037(91)90344-5.
Moros, M., K. Emeis, B. Risebrobakken, I. Snowball, A. Kuijpers, J. McManus, and E. Jansen. 2004. Sea surface temperatures and ice rafting in the Holocene North Atlantic: climate influences on northern Europe and Greenland. Quaternary Science Reviews 23: 2113–2126.
Mort, H.P., C.P. Slomp, B.G. Gustafsson, and T.J. Andersen. 2010. Phosphorus recycling and burial in Baltic Sea sediments with contrasting redox conditions. Geochimica et Cosmochimica Acta 74: 1350–1362. doi:10.1016/j.gca.2009.11.016.
Najjar, R.G., C.R. Pyke, M.B. Adams, D. Breitburg, C. Hershner, M. Kemp, R. Howarth, M.R. Mulholland, M. Paolisso, D. Secor, K. Sellner, D. Wardrop, and R. Wood. 2010. Potential climate-change impacts on the Chesapeake Bay. Estuarine, Coastal and Shelf Science 86: 1–20. doi:10.1016/j.ecss.2009.09.026.
Nakano, S. 1992. Manganoan vivianite in the bottom sediments of Lake Biwa, Japan. Mineralogical Journal 16: 96–107. doi:10.2465/minerj.16.96.
Nembrini, G.P., J.A. Capobianco, M. Viel, and A.F. Williams. 1983. A Mössbauer and chemical study of the formation of vivianite in sediments of Lago Maggiore (Italy). Geochimica et Cosmochimica Acta 47: 1459–1464. doi:10.1016/0016-7037(83)90304-6.
Nikitenko, S., A. Beale, A.M.J. van der Eerden, S.D.M. Jacques, O. Leynaud, M.G. O’Brien, D. Detollenaere, R. Kaptein, B.M. Weckhuysen, and W. Bras. 2008. Implementation of a combined SAXS/WAXS/QEXAFS set-up for time-resolved in situ experiments. Journal of Synchrotron Radiation 15: 632–640.
Ning, W., A. Ghosh, T. Jilbert, C.P. Slomp, M. Khan, J. Nyberg, D.J. Conley, and H.L. Filipsson. 2016. Evolving coastal character of a Baltic Sea inlet during the Holocene shoreline regression: impact on coastal zone hypoxia. Journal of Paleolimnology 55: 319–338. doi:10.1007/s10933-016-9882-6.
Nriagu, J.O. 1972. Stability of vivianite and ion-pair formation in the system fe3(PO4)2-H3PO4H3PO4-H2o. Geochimica et Cosmochimica Acta 36: 459–470. doi:10.1016/0016-7037(72)90035-X.
Nriagu, J., and C. Dell. 1974. Diagenetic formation of iron phosphates in recent lake sediments. American Mineralogist 59: 934–946.
O’Connell, D.W., M. Mark Jensen, R. Jakobsen, B. Thamdrup, T. Joest Andersen, A. Kovacs, and H.C. Bruun Hansen. 2015. Vivianite formation and its role in phosphorus retention in Lake Ørn, Denmark. Chemical Geology 409: 42–53. doi:10.1016/j.chemgeo.2015.05.002.
Obrochta, S.P., T.J. Crowley, J.E.T. Channell, D.A. Hodell, P.A. Baker, A. Seki, and Y. Yokoyama. 2014. Climate variability and ice-sheet dynamics during the last three glaciations. Earth and Planetary Science Letters 406: 198–212. doi:10.1016/j.epsl.2014.09.004.
Obrochta, S.P., Andrén, T., Fazeka, S.Z., Lougheed, B.C.. Snowball, I., Yokoyama, Y., Miyairi, Y., Kondo, R., Kotilainen, A.T. Hyttinene, O., Fehr, A. 2017. The undatables: Quantifying uncertainty in a highly expanded Late Glacial-Holocene sediment sequence recovered from the deepest Baltic Sea basin - IODP Site M0063.
Officer, C., Biggs, R., Taft, J. 1984. Chesapeake Bay anoxia: origin, development, and significance.
Oxmann, J., and L. Schwendenmann. 2015. Authigenic apatite and octacalcium phosphate formation due to adsorption–precipitation switching across estuarine salinity gradients. Biogeosciences (BG) 12: 723–738.
Påsse, T., and L. Andersson. 2005. Shore-level displacement in Fennoscandia calculated from empirical data. GFF 127: 253–268. doi:10.1080/11035890501274253.
Quintana Krupinski, N.B., A.D. Russell, D.K. Pak, and A. Paytan. 2017. Core-top calibration of B/Ca in Pacific Ocean Neogloboquadrina incompta and Globigerina bulloides as a surface water carbonate system proxy. Earth and Planetary Science Letters 466: 139–151.
Rabalais, N., W. Cai, J. Carstensen, D. Conley, B. Fry, X. Hu, Z. Quinones-Rivera, R. Rosenberg, C.P. Slomp, and R. Turner. 2014. Eutrophication-driven deoxygenation in the coastal ocean. Oceanography 27: 172–183.
Ravel, B., and M. Newville. 2005. ATHENA, ARTEMIS, HEPHAESTUS: data analysis for X-ray absorption spectroscopy using IFEFFIT. Journal of Synchrotron Radiation 12: 537–541. doi:10.1107/S0909049505012719.
Reed, D.C., B.G. Gustafsson, and C.P. Slomp. 2016. Shelf-to-basin iron shuttling enhances vivianite formation in deep Baltic Sea sediments. Earth and Planetary Science Letters 434: 241–251. doi:10.1016/j.epsl.2015.11.033.
Reimer, P.J., E. Bard, A. Bayliss, J.W. Beck, P.G. Blackwell, C. Bronk Ramsey, C.E. Buck, H. Cheng, R.L. Edwards, M. Friedrich, P.M. Grootes, T.P. Guilderson, H. Haflidason, I. Hajdas, C. Hatté, T.J. Heaton, D.L. Hoffmann, A.G. Hogg, K.A. Hughen, K.F. Kaiser, B. Kromer, S.W. Manning, M. Niu, R.W. Reimer, D.A. Richards, E.M. Scott, J.R. Southon, R.A. Staff, C.S.M. Turney, and J. van der Plicht. 2013. IntCal13 and Marine13 radiocarbon age calibration curves 0–50,000 years cal BP. Radiocarbon 55: 1869–1887. doi:10.2458/azu_js_rc.55.16947.
Rothe, M., T. Frederichs, M. Eder, A. Kleeberg, and M. Hupfer. 2014. Evidence for vivianite formation and its contribution to long-term phosphorus retention in a recent lake sediment: a novel analytical approach. Biogeosciences 11: 5169–5180.
Rothe, M., A. Kleeberg, B. Grüneberg, K. Friese, M. Pérez-Mayo, and M. Hupfer. 2015. Sedimentary sulphur:iron ratio indicates vivianite occurrence: a study from two contrasting freshwater systems. PloS One 10: e0143737. doi:10.1371/journal.pone.0143737.
Rothe, M., A. Kleeberg, and M. Hupfer. 2016. The occurrence, identification and environmental relevance of vivianite in waterlogged soils and aquatic sediments. Earth-Science Reviews 158: 51–64. doi:10.1016/j.earscirev.2016.04.008.
Rottgardt, D. 1952. Mikropaläontologisch wichtige Bestandteile recenter brackischer Sedimente an den Küsten Schleswig-Holsteins.
Rozan, T., M. Taillefert, R. Trouwborst, B.T. Glazer, S. Ma, J. Herszage, L.M. Valdes, K.S. Price, and G.W. Luther. 2002. Iron-sulfur-phosphorus cycling in the sediments of a shallow coastal bay: Implications for sediment nutrient release and benthic macroalgal blooms. Limnology and Oceanography 47: 1346–1354.
Ruttenberg, K.C. 1992. Development of a sequential extraction method for different forms of phosphorus in marine sediments. Limnology and Oceanography 37: 1460–1482.
Ruttenberg, K.C. 2003. The global phosphorus cycle. In Treatise on geochemistry 8, 585–643.
Ruttenberg, K.C., and R.A. Berner. 1993. Authigenic apatite formation and burial in sediments from non-upwelling, continental margin environments. Geochimica et Cosmochimica Acta 57: 991–1007. doi:10.1016/0016-7037(93)90035-U.
Salomé, M., M. Cotte, R. Baker, R. Barrett, N. Benseny-Cases, G. Berruyer, D. Bugnazet, H. Castillo-Michel, C. Cornu, B. Fayard, E. Gagliardini, R. Hino, J. Morse, E. Papillon, E. Pouyet, C. Rivard, V.A. Solé, J. Susini, and G. Veronesi. 2013. The ID21 scanning X-ray microscope at ESRF. Journal of Physics Conference Series 425: 182004. doi:10.1088/1742-6596/425/18/182004.
Scott, C., and T. Lyons. 2012. Contrasting molybdenum cycling and isotopic properties in euxinic versus non-euxinic sediments and sedimentary rocks: refining the paleoproxies. Chemical Geology 324–325: 19–27.
Seppä, H., D. Hammarlund, and K. Antonsson. 2005. Low-frequency and high-frequency changes in temperature and effective humidity during the Holocene in south-central Sweden: Implications for atmospheric and oceanic forcings of climate. Climate Dynamics 25: 285–297.
Skei, J.M. 1988. Framvaren—environmental setting. Marine Chemistry 23: 209–218. doi:10.1016/0304-4203(88)90093-X.
Slomp, C.P. 2011. Phosphorus cycling in the estuarine and coastal zones: sources, sinks, and transformations. In Treatise on estuarine and coastal science, ed. E. Wolanski and D.S. McLusky, 201–229. Waltham: Academic Press.
Slomp, C.P., E.H.G. Epping, W. Helder, and W. Van Raaphorst. 1996. A key role for iron-bound phosphorus in authigenic apatite formation in North Atlantic continental platform sediments. Journal of Marine Research 54: 1179–1205. doi:10.1357/0022240963213745.
Snowball, I., L. Zillén, A. Ojala, T. Saarinen, and P. Sandgren. 2007. FENNOSTACK and FENNORPIS: Varve dated Holocene palaeomagnetic secular variation and relative palaeointensity stacks for Fennoscandia. Earth Planet 225: 106–116.
Steenbergh, A.K., P.L.E. Bodelier, H.L. Hoogveld, C.P. Slomp, and H.J. Laanbroek. 2011. Phosphatases relieve carbon limitation of microbial activity in Baltic Sea sediments along a redox-gradient. Limnology and Oceanography 56: 2018–2026. doi:10.4319/lo.2011.56.6.2018.
Steenbergh, A.K., P.L.E. Bodelier, M. Heldal, C.P. Slomp, and H.J. Laanbroek. 2013. Does microbial stoichiometry modulate eutrophication of aquatic ecosystems? Environmental Microbiology 15: 1572–1579. doi:10.1111/1462-2920.12042.
Stigebrandt, A. 1981. A mechanism governing the estuarine circulation in deep, strongly stratified fjords. Estuarine, Coastal and Shelf Science 13: 197–211. doi:10.1016/S0302-3524(81)80076-X.
Strickland, J.D., and T.R. Parsons. 1972. A practical handbook of seawater analysis. Ottawa: Fisheries Research Board of Canada.
Thibodeau, B., A. de Vernal, and A. Mucci. 2006. Recent eutrophication and consequent hypoxia in the bottom waters of the Lower St. Lawrence estuary: micropaleontological and geochemical evidence. Marine Geology 231: 37–50.
Tsandev, I., D.C. Reed, and C.P. Slomp. 2012. Phosphorus diagenesis in deep-sea sediments: sensitivity to water column conditions and global scale implications. Chemical Geology 330–331: 127–139. doi:10.1016/j.chemgeo.2012.08.012.
Tyrrell, T. 1999. The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400: 525–531. doi:10.1038/22941.
Van Santvoort, P.J.M., G.J. De Lange, J. Thomson, S. Colley, F.J.R. Meysman, and C.P. Slomp. 2002. Oxidation and origin of organic matter in surficial eastern Mediterranean Hemipelagic sediments. Aquatic Geochemistry 8: 153–175. doi:10.1023/A:1024271706896.
Virtasalo, J.J., and A.T. Kotilainen. 2008. Phosphorus forms and reactive iron in lateglacial, postglacial and brackish-water sediments of the Archipelago Sea, northern Baltic Sea. Marine Geology 252: 1–12. doi:10.1016/j.margeo.2008.03.008.
Widerlund, A., and P.S. Andersson. 2011. Late Holocene freshening of the Baltic Sea derived from high-resolution strontium isotope analyses of mollusk shells. Geology 39: 187–190. doi:10.1130/G31524.1.
Yakubovich, O.V., W. Massa, R.P. Liferovich, and C.A. McCammon. 2001. The crystal structure of baricite (Mg1.70Fe1.30)(PO4)2.8H2O, the magnesium-dominant member of the vivianite group. The Canadian Mineralogist 39: 1317–1324. doi:10.2113/gscanmin.39.5.1317.
Yamane, M., Y. Yokoyama, Y. Miyairi, H. Suga, H. Matsuzaki, R.B. Dunbar, and N. Ohkouchi. 2014. Compound-specific 14C dating of IODP Expedition 318 Core U1357A obtained off the Wilkes land coast, Antarctica. Radiocarbon 56: 1009–1017. doi:10.2458/56.17773.
Yokoyama, Y., M. Koizumi, H. Matsuzaki, Y. Miyairi, and N. Ohkouchi. 2010. Developing ultra small-scale radiocarbon sample measurement at the University of Tokyo. Radiocarbon 52: 310–318.
Yokoyama, Y., J.B. Anderson, M. Yamane, L.M. Simkins, Y. Miyairi, T. Yamazaki, M. Koizumi, H. Suga, K. Kusahara, L. Prothro, H. Hasumi, J.R. Southon, and N. Ohkouchi. 2016. Widespread collapse of the Ross Ice Shelf during the late Holocene. Proceedings of the National Academy of Sciences 113: 2354–2359. doi:10.1073/pnas.1516908113.
Zhou, H., X. Peng, and J. Pan. 2004. Distribution, source and enrichment of some chemical elements in sediments of the Pearl River estuary, China. Continental Shelf Research 24: 1857–1875. doi:10.1016/j.csr.2004.06.012.
Zillén, L., D.J. Conley, T. Andrén, E. Andrén, and S. Björck. 2008. Past occurrences of hypoxia in the Baltic Sea and the role of climate variability, environmental change and human impact. Earth-Science Reviews 91: 77–92. doi:10.1016/j.earscirev.2008.10.001.
Acknowledgements
This research was funded by the European Research Council under the European Community’s Seventh Framework Programme (FP7/2007-2013)/ERC Starting Grant 278364 and the Netherlands Organisation for Scientific Research (NWO Vici Grant 865.13.005). This research used samples and/or data provided by the Integrated Ocean Drilling Program (IODP). We thank the captain, crew and scientists on board on the Greatship Manisha from 12 September until and 1 November 2013 and the scientists that were part of the onshore science party in Bremen from 22 January to 20 February 2014. Funding for beam time at the DUBBLE beamline was granted by the Netherlands Organisation for Scientific Research (NWO experiment 26-01-1002) and for the ID21 beamline via the European Synchrotron Radiation Facility (experiment numbers #ES45 and #ES171). We further acknowledge C. Rivard and D. Banerjee for their assistance at the beamlines and thank E. Smedberg for providing us with the Baltic Sea bathymetric map. Thilo Behrends is thanked for his guidance at the beamlines and our helpful discussions. We thank Wilrieke Boterblom for her help with the laboratory analyses.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by David Reide Corbett
Electronic supplementary material
Online Resource 1
(XLS 142 kb)
Online Resource 2
(XLS 32 kb)
Online Resource 3
(DOCX 75 kb)
Online Resource 4
(DOCX 134 kb)
Online Resource 5
(DOCX 75 kb)
Online Resource 6
(DOCX 66 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Dijkstra, N., Quintana Krupinski, N.B., Yamane, M. et al. Holocene Refreshening and Reoxygenation of a Bothnian Sea Estuary Led to Enhanced Phosphorus Burial. Estuaries and Coasts 41, 139–157 (2018). https://doi.org/10.1007/s12237-017-0262-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12237-017-0262-x