
Abstract
Objective
To study whether furosemide infusion in early-onset acute kidney injury (AKI) in critically ill children would be associated with a reduced proportion of patients progressing to the higher stage (Injury or Failure) as compared to placebo.
Method
A double-blind, placebo-controlled, randomized pilot trial was conducted. The authors enrolled children aged 1-mo (corrected) to 12-y, who were diagnosed with AKI (“risk” stage) using pediatric-Risk, Injury, Failure, Loss, End stage kidney disease (p-RIFLE) criteria, and achieved immediate resuscitation goals within 24 h of admission. Participants received either furosemide (0.05 to 0.4 mg/kg/h) or placebo (5%-dextrose) infusion. The primary outcome was the proportion of patients progressing to a higher stage (injury or failure). Secondary outcomes were (i) need for renal replacement therapy, (ii) the effect on neutrophil gelatinase-associated lipocalin (urine and blood), (iii) fluid balance, (iv) adverse effects, (v) time to achieve renal recovery, (vi) duration of hospital stay and mechanical ventilation, and (vii) all-cause 28-d mortality.
Results
The trial was stopped for futility, and data were analyzed on an intention-to-treat basis (furosemide-group: n = 38; placebo-group: n = 37). No significant difference was noted in the progression of AKI to a higher stage between furosemide and placebo groups (10.5% vs. 21.6%; relative risk = 0.49, 95% CI 0.16 to 1.48) (p = 0.22). There were no differences in the secondary outcomes between the study groups. All-cause 28-d mortality was similar between the groups (10.5% vs. 10.8%). No trial-related severe adverse events occurred.
Conclusions
Furosemide infusion in early-onset AKI did not reduce the progression to a higher stage of AKI. A future trial with large sample size is warranted.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Acute kidney injury (AKI) is a common problem in the pediatric intensive care unit (PICU) with an incidence of up to 51.2% [1, 2]; however, 82% of AKI occurred within the first 7 d of admission [3]. Patients with AKI have higher morbidity-mortality and health resource use than those without AKI [4]. The management of AKI is a predominantly conservative approach. There are few if any, interventions that proved to have an impact on the clinical course and outcome of AKI [5]. Hence the question—what is the role of furosemide in the management of AKI?
Furosemide is the most commonly used diuretic in critically ill children [6]. It acts on the thick ascending limb of the loop of Henle and inhibits the Na-K-Cl pump on the luminal surface of the tubular epithelium, and can theoretically reduce renal tubular oxygen demand [7]. Low-dose furosemide may reduce the ischemia/reperfusion-induced apoptosis and associated gene transcription in AKI [8]. Furosemide infusion had varied effects on daily urine output in critically ill adult and pediatric patients; however, there were no benefits in renal function and mortality. Systematic reviews recommend the need for controlled trials to fill up this knowledge gap [9, 10]. Pediatric clinical management reviews did not arrive at a meaningful conclusion for use of furosemide in AKI due to the limited number of controlled studies [11, 12]. It was hypothesized that furosemide infusion in early-onset AKI [by pediatric-Risk, Injury, Failure, Loss, End stage kidney disease (p-RIFLE) criteria] in critically ill children would be associated with a reduced proportion of patients progressing to the higher stage (injury or failure) as compared to placebo.
Material and Methods
This double-blind, placebo-controlled, randomized pilot trial was conducted in the PICU of a tertiary-care institution from 1st October 2016 to 31st December 2018. The institutional ethics committee approved the study and, a written informed consent was obtained from parents/legal guardians. Children aged 1-mo (corrected) to 12-y, who were diagnosed with early-onset AKI (risk stage), and achieved immediate resuscitation goals were enrolled within 24 h of admission. AKI was defined by p-RIFLE criteria (either urine output or serum creatinine criterion or both) [3]. The immediate resuscitation goals were defined as directed by the treating physician, which included one or more of the following: fluid resuscitation and/or vasoactive therapy to achieve (i) capillary refill of ≤ 2 s, (ii) > 5th percentile mean arterial blood pressure (MABP), (iii) normal pulse volume with no differential peripheral and central pulse, (iv) central venous pressure (CVP) ≥ 8 cm H2O (if measured), (v) central venous oxygen saturation (ScvO2) ≥ 70% (if measured), (vi) cardiac index between 3.3 to 6.0 L/min/m2 (if measured). Children with any of the following conditions were excluded: (i) stage-4 or more chronic kidney disease, end-stage kidney disease on renal replacement therapy (RRT), kidney transplantation or already received RRT in PICU, (ii) acute pulmonary edema requiring urgent furosemide use or RRT, (iii) patient already receiving furosemide (infusion or bolus) irrespective of dose and duration, (iv) known or suspected allergy to furosemide, and (v) known or suspected obstructive etiology for AKI.
A web-based, computer-generated, unstratified block randomization with variable block sizes of four, six, and eight were used. The random number allocation was performed by a person not involved in the study. Individual assignments were placed in sequentially numbered, opaque sealed envelopes (SNOSE). The envelope contained an instruction slip showing the dilution and preparation of the trial drug. Nursing personnel, who was not part of the study and working in the pediatric emergency, opened the envelope and prepared the infusion. Injection furosemide [FRUSEMIDE, 2 mL/20 mg, MODERN Laboratories, Indore (M.P.), India] was used. Four milliliters of furosemide (40 mg) was diluted in 36 mL of 5%-dextrose [= 1 mg/mL].
Five percent dextrose solution was used as a placebo. Both the drugs were identical in appearance. The Institute’s central pharmacy supplied the trial drugs. The infusion syringe was labeled with random numbers, three alphanumerical codes, and drug dose (0.05 mL/kg/h). The volume of the infusion was also included in the calculation of the daily fluid balance. The person who prepared the trial drug was blinded to the patient’s identity. The participants, nurses administering the drugs, treating doctors, and the investigators and research personnel who collected the data and study statistician, were unaware of the treatment assignments. The treatment allocation was disclosed only after the finalization of the first draft of the results.
The patients were managed by stabilizing the airway, breathing, and circulation as per standard protocol. Maintenance fluid was calculated using the Holiday–Segar formula, and 80% of the calculated fluid was started, as per the study unit protocol [13]. After that, every six-hourly (more frequently, if needed) fluid charting was done based on fluid balance, clinical, and laboratory parameters. The trial drug titration concept was adapted from the SPARK study [14]. The intravenous (peripheral/central) infusion of the trial drug was commenced at 0.05 mL/kg/h (= 0.05 mg/kg/h of furosemide) using a dedicated infusion pump and titrated at the rate of 0.05 mL/kg/h up to a maximum infusion of 0.4 mL/kg/h. The trial drug infusion was titrated to achieve the minimum urine output of 1 to 2 mL/kg/h or 1 to 2 mL/kg/h from baseline. If the target urine output was achieved, the same rate of infusion was continued. If the urine output was more than the targeted range for more than two consecutive hours, the infusion rate was reduced by 0.05 mL/kg/h every hour. The infusion was discontinued if the urine output remained more than the target range with the infusion at the lowest rate (0.05 mL/kg/h). It was started again at the lowest rate when the urine output fell below 1 mL/kg/h (or 1 mL/kg/h from baseline). The infusion was stopped, if any of the following occurred: (i) MABP below 5th percentile and/or addition of and/or increase in vasoactive therapy > 20% from baseline to achieve the target MABP or (ii) CVP < 8 cmH2O (if measured) or (iii) ScvO2 < 60% (if measured) or (iv) cardiac index < 3.3 L/min/m2 (if measured). The trial drug infusion was also discontinued in the event of adverse effects related to the intervention, as described by Naranjo et al. [15]. By protocol and in order to minimize the potential bias of clinician discretion on when to initiate RRT, at least one of the following criteria must be fulfilled before initiation of RRT: (i) refractory oliguria (urine output < 0.5 mL/kg/h in preceding 6 h, despite fluid resuscitation and/or vasoactive therapy or maximum dose of the trial drug), (ii) refractory extravascular fluid overload and/or hypoxemia and/or pulmonary edema [Fraction of inspired oxygen (FiO2) ≥ 60%, receiving mechanical ventilation, Partial pressure of oxygen (PaO2)/FiO2 ratio ≤ 200], (iii) symptomatic azotemia (i.e., encephalopathy, pericarditis), and (iv) metabolic acidosis (pH < 7.2 or HCO3 < 15); hyperkalemia [K+ ≥ 6.0 mEq/dL or electrocardiogram changes, and/or administration of at least one dose of potassium binder and/or intravenous insulin and/or intravenous bicarbonate].
The trial drug infusion was continued for a minimum of 24 h, and discontinued if any one of the following events occurred: (i) patient was initiated on RRT, or as per the decision of the treating team, (ii) patient was discharged from the PICU, (iii) attained operational definition of renal recovery, or (iv) patient died. The operational definition of renal recovery was defined by the return of serum creatinine to 25% of baseline levels and spontaneous urine output ≥ 1.0 mL/kg/h for a minimum of 24 h independent of RRT [16]. Serum creatinine was estimated by the modified Jaffe method using an autoanalyzer (Olympus® AU 680, Beckman Coulter, California, USA). Creatinine clearance was calculated using the modified Schwartz formula (= Length in cm × 0.413 ÷ Serum creatinine in mg/dL). If a child had documented creatinine values in the past three months, the lowest value was taken as baseline; otherwise, 100 mL/min/1.73 m2 was taken as the baseline creatinine clearance [3]. Urine output was measured with a Foley catheter or by diaper weighing or by the noninvasive collection of urine in a collection bag.
Serum electrolytes and renal function tests (urea and creatinine) were done at admission; six-hourly on day 1 and then once a day or as and when required. Fluid balance [percentage of fluid overload (%FO)] was calculated daily {%FO = [(Fluid input in liter − Output in liter) ÷ Admission weight in kg] × 100} [17]. The acid-base disturbance was defined using standard criteria [18]. Sepsis and multiorgan dysfunction were defined as per the International Pediatric Sepsis Consensus Conference definition [19]. For neutrophil gelatinase-associated lipocalin (NGAL) assay, 1 mL of plasma and 1 mL of urine were collected at admission, 6, 12, and 24 h and daily. All the blood investigations data were collected up to 7-d, and patients were followed up till 28-d. The NGAL samples were stored at −20 °C for batched analysis by enzyme-linked immunosorbent assay method (ELISA; Human NGAL-ELISA kit-E1719Hu by Bioassay Technology Laboratory, China).
The primary outcome was the proportion of patients with the “risk” stage progressing to injury or failure. Secondary outcomes were the proportion of patients requiring RRT, the effect on NGAL (plasma and urine), fluid balance, adverse effects of furosemide, time to achieve the operational definition of renal recovery, and length of hospital stay (including PICU), and mechanical ventilation, and all-cause hospital and 28-d mortality.
Statistical analysis
The sample size was calculated based on the assumption that furosemide infusion reduces the progression to a higher stage from 35% to 15% (author center unpublished data, January 2016 to March 2016, [3]). Assuming an attrition rate of 10%, it was estimated that 110 patients would need to be enrolled in each group with an alpha level of 5% and 90% power and an allocation ratio of 1:1 (nQuery advisor 4.0). The trial progress was reviewed yearly by the institute’s ethics and data and safety monitoring committees, including an independent statistician who was also a physician.
Data were analyzed on an intention-to-treat basis. The normality of data was checked with Kolmogorov–Smirnov z test. Continuous variables were compared by Student’s t test if normally distributed or Mann–Whitney U test, if data were non-normally distributed. The proportion was compared by Chi-square test (Fisher exact test when cell frequencies were < 5). Kaplan–Meier and log-rank test followed by Cox proportional model adjusted for age, gender, and severity, were used for time to event data. Relative risk/hazard ratio with 95% confidence interval was calculated wherever appropriate. All tests were two-tailed, and a p value of < 0.05 was considered statistically significant. Data analysis was performed using IBM-SPSS, version 20.0 (SPSS Inc. Chicago, Illinois) and Epi Info ™ 7 (7.0.9.7, CDC).
Results
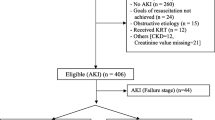
The trial was stopped after the planned interim analysis (at two years of study review) contended that it was futile to study further and was decided to present as a pilot trial. The trial flow is depicted in Fig. 1. Seventy-five patients were enrolled (furosemide-group, n = 38, and placebo-group, n = 37). All patients met the ‘risk’ stage according to the creatinine criteria, and 11/38 (29%) patients in the furosemide-group and 8/37 (21%) in the placebo-group met both creatinine and urine output criteria. The baseline serum creatinine was available in 12 (16%) patients. There was no protocol violation noted. No mechanical problem was encountered during the infusion of trial drugs. No patient required urgent administration of furosemide after enrollment. Baseline characteristics were comparable, between the two groups, except hyperchloremia and blood urea (Table 1). The proportion of patients requiring maximum trial drug infusion was similar in both groups (21.1% vs. 22.2%). The mean (SD) duration of trial drug infusion was similar in both groups (29.7 ± 17 vs. 25.5 ± 14.5 h; p = 0.25). The proportion of patients who required suspension of the trial drug at least a single point of time was similar in both groups (47.4%, n = 18/38 vs. 37.8%, n = 14/37; p = 0.40). The mean (SD) duration of suspension of the trial drug was similar in both groups (3.3 ± 1.1 vs. 4.0 ± 1.3 h; p = 0.12].

Trial flow. AKI Acute kidney injury; CKD Chronic kidney disease; GFR Glomerular filtration rate; RRT Renal replacement therapy
No significant difference was noted in the progression of AKI from the “risk” to “injury” or “failure” stage in furosemide and placebo groups (10.5% vs. 21.6%; relative risk = 0.49, 95% CI 0.16 to 1.48; p = 0.22) (Table 2). No significant difference was noted on the hazard of progression to a higher stage in both groups (adjusted hazard ratio = 0.32, 95% CI 0.10 to 1.47; p = 0.14) (Fig. 2). Two patients in the placebo group received continuous renal replacement therapy (CRRT) (for fluid overload due to persistent oliguria and associated metabolic abnormalities) and none in the Furosemide group. No significant difference was noted in other secondary outcomes (Table 2). No serious trial-related adverse effects were noted.

Kaplan–Meier curve showing the progression of AKI from the early-stage to the higher stage in the study groups. AKI Acute kidney injury
Discussion
In this controlled pilot study, no significant difference was noted in the progression of AKI to a higher stage in furosemide (10.5%) and placebo groups (21.6%). There were no differences in the other secondary outcomes. Pediatric AKI is different from adults in many ways. Ongoing growth, with greater renal reserve and regeneration capacity in children compared to adults, might affect pediatric AKI outcomes [20]. Hence, the response to furosemide can also be expected to be different in pediatric AKI. In adults with AKI, furosemide bolus followed by infusion was associated with increased urine output in oliguric postoperative patients. Nevertheless, it did not reduce the progression of AKI, the need for RRT, and renal recovery [21,22,23]. The method, settings, and study results were similar to the present study except that in the present study only furosemide infusion was used to avoid bolus-associated complications.
Furosemide had varied responses on urine output depending upon the method of delivery. A meta-analysis found that continuous infusion of furosemide was associated with a significant increase in daily urine output, a net decrease in body weight, and lower adverse effects than the bolus group. However, this effect was not seen in pediatric studies [10]. A recent systematic review also confirmed the greater diuretic effect associated with continuous infusion of furosemide than bolus dose. Nevertheless, it was associated with an extended hospital stay, and there was no difference in mortality or change in renal function [9]. However, analysis was limited by high heterogeneity among the included studies [9].
Fluid overload is a significant problem in PICU. A controlled study on furosemide vs. peritoneal dialysis in infants with postcardiac surgery found no difference in negative fluid balance on the first postoperative day, hospital stay, and mortality. Furosemide group had a longer duration of inotropic use, higher electrolyte abnormality score, prolonged ventilator use, and more likely to have 10% fluid overload than peritoneal dialysis [24]. In the present study, no significant difference were found in urine output, changes of creatinine, electrolyte abnormality, renal recovery and PICU and hospital stay, duration of ventilation, and mortality between study groups. The adaptation of restrictive fluid strategy in the present study, which was evident at the time of enrollment, and fluid responsive renal injury, could be the potential reasons. Avoiding fluid overload in AKI patients was associated with favorable outcomes [25, 26].
Plasma and urine NGAL levels for early diagnosis of AKI were also compared. Moreover, NGAL can also predict AKI progression. There was no significant change in urine and plasma NGAL in this study. This is in contrast to the previous studies, which showed elevation of NGAL in pediatric AKI [27, 28]. However, the study population predominantly constituted by the post-cardiac surgery and inborn errors of metabolism groups, compared to the present study where the infection or sepsis was predominantly observed. Similar to the present study, Hamishehkar et al. reported that NGAL was not found to reflect any effects of furosemide in adults with AKI [29].
The present study is the first placebo-controlled pilot study with a robust methodology involving critically ill children with AKI. The authors also followed the children up to 28-d and studied the NGAL in AKI of various etiologies, in contrast to previously published studies [22, 27, 28]. There are few limitations. It is a single-center study. The majority (84%) of the patient’s baseline serum creatinine was assumed as 100 mL/min/1.73 m2. Measuring acute renal function change is challenging in infants, requiring interpretation of serum creatinine increments referenced to low baseline levels. The ototoxicity was also not assessed. A multicentric study with larger sample size is warranted in the future.
Conclusion
The study concludes that furosemide infusion did not reduce the progression of AKI to a higher stage. Further, furosemide infusion was not associated with the difference in the need for RRT, fluid balance, length of stay in hospital and ventilation, biomarker (NGAL), electrolyte disturbances, and mortality.
References
Sutherland SM, Byrnes JJ, Kothari M, et al. AKI in hospitalized children: comparing the pRIFLE, AKIN, and KDIGO definitions. Clin J Am Soc Nephrol. 2015;10:554–61.
Rameshkumar R, Krishnamurthy S, Ganesh RN, et al. Histopathological changes in septic acute kidney injury in critically ill children: a cohort of post-mortem renal biopsies. Clin Exp Nephrol. 2017;21:1075–82.
Akcan-Arikan A, Zappitelli M, Loftis LL, Washburn KK, Jefferson LS, Goldstein SL. Modified RIFLE criteria in critically ill children with acute kidney injury. Kidney Int. 2007;71:1028–35.
Fortenberry JD, Paden ML, Goldstein SL. Acute kidney injury in children: an update on diagnosis and treatment. Pediatr Clin N Am. 2013;60:669–88.
Basu RK, Wheeler DS. Approaches to the management of acute kidney injury in children. Recent Pat Biomark. 2011;1:49–59.
Mehta RL, Pascual MT, Soroko S, Chertow GM, Group PS. Diuretics, mortality, and nonrecovery of renal function in acute renal failure. JAMA. 2002;288:2547–53.
Kramer HJ, Schuurmann J, Wassermann C, Dusing R. Prostaglandin-independent protection by furosemide from oliguric ischemic renal failure in conscious rats. Kidney Int. 1980;17:455–64.
Aravindan N, Aravindan S, Riedel BJ, Weng HR, Shaw AD. Furosemide prevents apoptosis and associated gene expression in a rat model of surgical ischemic acute renal failure. Ren Fail. 2007;29:399–407.
Ng KT, Velayit A, Khoo DKY, Mohd Ismail A, Mansor M. Continuous infusion versus intermittent bolus injection of furosemide in critically ill patients: a systematic review and meta-analysis. J Cardiothorac Vasc Anesth. 2018;32:2303–10.
Alqahtani F, Koulouridis I, Susantitaphong P, Dahal K, Jaber BL. A meta-analysis of continuous vs intermittent infusion of loop diuretics in hospitalized patients. J Crit Care. 2014;29:10–7.
Lethaby D, Cyriac J, Bockenhauer D. Question 1: is the use of furosemide beneficial in the treatment of acute kidney injury in the paediatric population including neonates? Arch Dis Child. 2015;100:713–5.
Sparrow HG, Swan JT, Moore LW, Gaber AO, Suki WN. Disparate outcomes observed within kidney disease: improving global outcomes (KDIGO) acute kidney injury stage 1. Kidney Int. 2019;95:905–13.
Holliday MA, Segar WE. The maintenance need for water in parenteral fluid therapy. Pediatrics. 1957;19:823–32.
Bagshaw SM, Gibney RT, McAlister FA, Bellomo R. The SPARK study: a phase II randomized blinded controlled trial of the effect of furosemide in critically ill patients with early acute kidney injury. Trials. 2010;11:50.
Naranjo CA, Busto U, Sellers EM, et al. A method for estimating the probability of adverse drug reactions. Clin Pharmacol Ther. 1981;30:239–45.
Soler YA, Nieves-Plaza M, Prieto M, Garcia-De Jesus R, Suarez-Rivera M. Pediatric risk, injury, failure, loss, end-stage renal disease score identifies acute kidney injury and predicts mortality in critically ill children: a prospective study. Pediatr Crit Care Med. 2013;14:e189–95.
Goldstein SL, Currier H, Graf C, Cosio CC, Brewer ED, Sachdeva R. Outcome in children receiving continuous venovenous hemofiltration. Pediatrics. 2001;107:1309–12.
Greenbaum L. Electrolyte and acid-base disorders. In: SB KR, St Geme J, Schor N, editors. Nelson textbook of pediatrics. I. Philadelphia: Elsevier - Health Sciences Division; 2016. p. 369–83.
Goldstein B, Giroir B, Randolph A, International Consensus Conference on Pediatric S. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005;6:2–8.
Greenberg JH, Parikh CR. Biomarkers for diagnosis and prognosis of AKI in children: one size does not fit all. Clin J Am Soc Nephrol. 2017;12:1551–7.
Bagshaw SM, Gibney RTN, Kruger P, Hassan I, McAlister FA, Bellomo R. The effect of low-dose furosemide in critically ill patients with early acute kidney injury: a pilot randomized blinded controlled trial (the SPARK study). J Crit Care. 2017;42:138–46.
Mahesh B, Yim B, Robson D, Pillai R, Ratnatunga C, Pigott D. Does furosemide prevent renal dysfunction in high-risk cardiac surgical patients? Results of a double-blinded prospective randomised trial. Eur J Cardiothorac Surg. 2008;33:370–6.
Shilliday IR, Quinn KJ, Allison ME. Loop diuretics in the management of acute renal failure: a prospective, double-blind, placebo-controlled, randomized study. Nephrol Dial Transplant. 1997;12:2592–6.
Kwiatkowski DM, Goldstein SL, Cooper DS, Nelson DP, Morales DL, Krawczeski CD. Peritoneal dialysis vs furosemide for prevention of fluid overload in infants after cardiac surgery: a randomized clinical trial. JAMA Pediatr. 2017;171:357–64.
McGuire MD, Heung M. Fluid as a drug: balancing resuscitation and fluid overload in the intensive care setting. Adv Chronic Kidney Dis. 2016;23:152–9.
Zhang L, Chen Z, Diao Y, Yang Y, Fu P. Associations of fluid overload with mortality and kidney recovery in patients with acute kidney injury: a systematic review and meta-analysis. J Crit Care. 2015;30:860 e7–13.
Wheeler DS, Devarajan P, Ma Q, et al. Serum neutrophil gelatinase-associated lipocalin (NGAL) as a marker of acute kidney injury in critically ill children with septic shock. Crit Care Med. 2008;36:1297–303.
Polat M, Fidan K, Derinoz O, Gonen S, Soylemezoglu O. Neutrophil gelatinase-associated lipocalin as a follow-up marker in critically ill pediatric patients with established acute kidney injury. Ren Fail. 2013;35:352–6.
Hamishehkar H, Sanaie S, Fattahi V, Mesgari M, Mahmoodpoor A. The effect of furosemide on the level of neutrophil gelatinase-associated lipocalin in critically hospitalized patients with acute kidney injury. Indian J Crit Care Med. 2017;21:442–7.
Acknowledgments
The authors acknowledge the contribution of Mrs. S. Raja Deepa B.Com, MCA (JIPMER Campus, Puducherry, India) for data handling, review and editing of the manuscript; Mr. Rakesh Mohindra (Punjab University, Chandigarh, India) and Mrs. Thenmozhi M. (MSc, PhD, Senior Demonstrator, CMC, Vellore, India) for helping with statistical analysis and Mrs. Harpreet Kaur (Punjab University, Chandigarh, India), and Mrs. Neelima Chadha (Tulsi Das Library, PGIMER, Chandigarh, India) for helping with the medical literature search. The authors acknowledge the member of the data and safety monitoring committee (Dr Ebor Jacob, Professor and senior consultant of PICU, CMC Vellore, India; Dr Sriram Krishnamurthy, Additional Professor of Pediatrics, JIPMER, Puducherry, India; Dr J Venkatachalam, Associate Professor of Social and Preventive Medicine, JIPMER, Puducherry, India; Mrs. Lakshmi Kumaran, Nursing Sister, JIPMER, Puducherry, India).
Author information
Authors and Affiliations
Contributions
RR: Conceptualization of the study, review of literature, and critical review of the manuscript. SA, SS, RB, MC, AS, KM: Data collection, review of literature, and manuscript writing. RS: Protocol writing, supervision of biological sample analysis, and manuscript writing. SM: Protocol development, review of literature and manuscript writing. All authors were involved in the management of patients and approved the final version of the manuscript. RR is the guarantor of the paper.
Corresponding author
Ethics declarations
Conflict of Interest
None.
Source of Funding
JIPMER Intramural Research Grant [JIP/RES/INTRA-DM-MCH/PHS1/01/2016–17 Dt 9/9/2016 to RR].
Protocol accessible
Clinical Trial Registry of India (CTRI) www.ctri.nic.in. (Trial Registered Prospectively and Registration No. CTRI/2016/09/007321) (Title: “Effect of furosemide on progression of early onset of acute kidney injury in critically ill children: A randomized double blinded, placebo controlled trial.”).
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Abraham, S., Rameshkumar, R., Chidambaram, M. et al. Trial of Furosemide to Prevent Acute Kidney Injury in Critically Ill Children: A Double-Blind, Randomized, Controlled Trial. Indian J Pediatr 88, 1099–1106 (2021). https://doi.org/10.1007/s12098-021-03727-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12098-021-03727-3