
Abstract
Pheochromocytomas and paragangliomas (PPGLs) are rare neuroendocrine tumors that arise from chromaffin cells of the adrenal medulla and the sympathetic/parasympathetic neural ganglia, respectively. The heterogeneity in its etiology makes PPGL diagnosis and treatment very complex. The aim of this article was to provide practical clinical guidelines for the diagnosis and treatment of PPGLs from a multidisciplinary perspective, with the involvement of the Spanish Societies of Endocrinology and Nutrition (SEEN), Medical Oncology (SEOM), Medical Radiology (SERAM), Nuclear Medicine and Molecular Imaging (SEMNIM), Otorhinolaryngology (SEORL), Pathology (SEAP), Radiation Oncology (SEOR), Surgery (AEC) and the Spanish National Cancer Research Center (CNIO). We will review the following topics: epidemiology; anatomy, pathology and molecular pathways; clinical presentation; hereditary predisposition syndromes and genetic counseling and testing; diagnostic procedures, including biochemical testing and imaging studies; treatment including catecholamine blockade, surgery, radiotherapy and radiometabolic therapy, systemic therapy, local ablative therapy and supportive care. Finally, we will provide follow-up recommendations.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pheochromocytomas (PCCs) and paragangliomas (PGLs)—hereinafter PPGL to include both entities—are rare neuroendocrine tumors (NETs) that arise from chromaffin cells of the adrenal medulla and the sympathetic/parasympathetic neural ganglia, respectively. PPGLs often secrete catecholamines (CMNs) that can mimic a wide range of medical disorders and may be lethal if misdiagnosed or improperly handled. PPGLs are also characterized by a very heterogeneous natural history and a low to moderate but unpredictable ability to metastasize. Surgical resection may be challenging, including adequate timing and perioperative medical management, and optimal treatment of advanced disease is controversial. Over one-third of PPGLs are inherited, and adequate genetic counseling is key to implementing screening strategies and tailoring therapy. All these factors make PPGL diagnosis and treatment very complex, and clinical experience is difficult to achieve due to their low incidence. In this context, the aim of this article was to provide practical clinical guidelines for the diagnosis and treatment of PPGLs from a multidisciplinary perspective. Experts from the national societies of the different disciplines involved participated in the elaboration of these guidelines, including clinical specialists from the Spanish Societies of Endocrinology and Nutrition (SEEN), Medical Oncology (SEOM), Medical Radiology (SERAM), Nuclear Medicine and Molecular Imaging (SEMNIM), Otorhinolaryngology (SEORL), Pathology (SEAP), Radiation Oncology (SEOR), and Surgery (AEC), and geneticists from the Spanish National Cancer Research Center (CNIO).
Epidemiology
The joint annual incidence of PPGL is estimated to be 2–8 cases per million inhabitants. A recent study from Canada disclosed an annual incidence of 6.6 cases per million inhabitants, half of them corresponding to pheochromocytomas and 37% of them to head and neck paragangliomas [1], most of them known to be parasympathetic. Head and neck paragangliomas have geographic variations as a function of altitude [2]. In another study from Holland [3], the annual incidence of PCCs and of sympathetic PGLs were 4.6 and 1.1 cases per million inhabitants, respectively, which had increased compared to previous years likely due to improved diagnostic techniques and clinical awareness.
The distribution by gender does not show significant differences, although a greater incidence in females has been observed for vagal and jugulotympanic PGLs [4], and in high-altitude PGLs [2]. PPGLs are commonly diagnosed within the 4th and 6th decades, although these neoplasms can occur over a wide age range. They appear at a younger age when they occur as part of a hereditary syndrome [5]. At pediatric ages, extra-adrenal PGLs account for more than two-third of the cases, and four of five cases are associated with a hereditary form of the disease [6, 7].
Bilateral or multiple forms are mostly associated with hereditary syndromes, but an advanced age at diagnosis or the absence of family history does not exclude the possibility of carrying a germline mutation. In fact, between 14 and 24% of clinically sporadic tumors may also be due to a germline mutation [8, 9].
The rate of metastatic disease (mPPGL) ranges from less than 1% to 79%, depending upon tumor site and size, age at diagnosis and genotype [10, 11]. Although some features included size greater than 5 cm, extra-adrenal primary tumor site, or high levels of plasma 3-metoxitiramine (3-MT) [12] provide useful information to assess the risk of metastasis, the presence of mutations in the succinate dehydrogenase complex iron sulfur subunit B (SDHBMut) is the only universally accepted criterion associated with a high risk of distant disease, both at diagnosis or during follow-up, ranging from 20 to 70% in different patient cohorts [11,12,13,14]. Recent data also suggest a higher metastatic risk in patients with mutations in other genes involved in the Krebs cycle [15].
Overall, the prognosis of PPGL is heterogeneous. Goffredo et al. analyzed 508 PPGL patients from 18 US registries (time frame 1988–2009) and reported a 5-year overall survival (OS) rate of 58% for metastatic PCCs and 80% for metastatic PGLs [16]. More recently, a retrospective study of 169 patients from 18 European centers (time frame 1998–2010) by Hescot et al. [14] reported a global 5-year OS rate of 62% and a median OS of 6.7 years for mPPGL.
Anatomy, pathology and molecular pathways
Anatomy
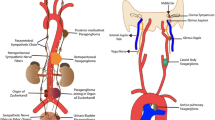
PCC (adrenal) and PGL (extra-adrenal) are neoplasms that originate from chromaffin cells of the autonomic nervous system, derived from the neural crest, and can be classified as either sympathetic or parasympathetic. The sympathetic system is distributed through the paraganglia of the prevertebral and paravertebral axes, reaching the abdominal organs and innervating the urogenital system, and it includes the adrenal medulla, which is considered the greatest paraganglion. The parasympathetic system is distributed through the head and neck and mediastinum, following the territories of the glossopharyngeal (carotid and tympanic paraganglia) and vagus (jugular, upper and lower laryngeal, subclavian and aortopulmonary) nerves [17]. These neoplasms are also grouped into cervicocephalic, thoracic and abdominal PPGLs [18].
Histopathological features
The characteristic microscopic feature of PPGLs is the proliferation of polygonal chromaffin cells distributed in nests (zellballen, in German), surrounded by a fine capillary network and sustentacular cells, easily identifiable through immunohistochemical (IHC) detection of S-100. This pattern is more evident in parasympathetic PGLs. Chromogranin A (CgA) and synaptophysin, but not cytokeratins, are expressed in tumor cells. Tyrosine hydroxylase is also expressed in PCCs and sympathetic PGLs but not in parasympathetic PGLs. IHC studies with Ki67 (Mib-1) are also recommended to estimate the cellular proliferation index [5].
Stratification of risk and staging
All PPGLs are potentially metastatic; therefore, benign vs malignant discrimination was eliminated in the last World Health Organization (WHO) classification for endocrine tumors [5]. Several scoring systems have been established to estimate the metastatic risk of these tumors. The Pheochromocytoma of the Adrenal Gland Scaled Score (PASS) [19], applicable only to PCCs, is based solely on morphological criteria, some of which may be too subjective to evaluate (Table 1). The Grading of Adrenal Pheochromocytoma and Paraganglioma (GAPP) scale [20] is applicable to both PCCs and PGLs and combines morphological, IHC and analytical findings (Table 1).
IHC studies of SDH components are indicated and are especially useful in the study of subunit B of SDH (SDHB), as the loss of expression indicates the presence of an SDH germline mutation and an increased risk of aggressive behavior [21, 22]. In fact, SDHB loss of expression is included in the most recent risk stratification score, COPPS (Composite Pheochromocytoma/paraganglioma Prognostic Score) (Table 1) [23]. The eighth edition of the American Joint Committee on Cancer (AJCC) staging system [24] includes a new chapter to address PCC and sympathetic PGL (Table 2), but not parasympathetic PGL, given its low risk of malignant behavior (~ 5%) [25, 26].
Molecular basis of PPGL
A total of 30–50% of PPGLs occur in the context of a hereditary syndrome [27,28,29,30]. The most common hereditary syndromes are those derived from germline mutations in genes encoding the different subunits of SDH (type 1: SDHD; type 2: SDHAF2; type 3: SDHC; type 4: SDHB; type 5: SDHA; including Carney-Stratakis syndrome) (15–20%), Von Hippel-Lindau (VHL) syndrome due to mutations in the VHL gene (9%), multiple endocrine neoplasia-2 (MEN2) syndrome due to mutations in the RET proto-oncogene (5%) and neurofibromatosis type 1 (NF-1) syndrome due to mutations in the NF1 gene (2%). Less frequent familial forms (< 1–2%) are caused by mutations in the transmembrane protein 127 (TMEM127), MYC-associated factor X (MAX), fumarate hydratase (FH), multiple endocrine neoplasia type 1 (MEN1), egg-laying-defective nine (egl-9) family hypoxia-inducible factor 1 gene (EGLN1), egl-9 family hypoxia-inducible factor 2 (EGLN2), malate dehydrogenase 2 (MDH2), kinesin family member 1B (KIF1B) genes [31,32,33,34], solute carrier family 25 Member 11 (SLC25A11) and dihydrolipoamide S-succinyltransferase (DLST) [35, 36]. Susceptibility genes and familial PPGL syndromes have been comprehensively reviewed recently [37], and their main features are summarized in Table 3.
Molecular classification of PPGL
The genomic characteristics of PCCs and PGLs allow us to distinguish among three groups or main clusters. Cluster 1 groups tumors with germinal or somatic mutations in genes related to the Krebs cycle (SDHA, SDHAF2, SDHB, SDHC, SDHD, FH, MDH2, GOT2, IDH1, SLC23A11, etc.), in EPAS1 and in VHL. The presence of mutations in these genes leads directly or indirectly to HIF1a and HIF2a stabilization and, therefore, to a situation of pseudohypoxia, which causes an increase in angiogenesis and cell proliferation. PPGLs associated with mutations in genes of the Krebs cycle show a characteristic pattern of higher overall methylation, known as the CpG island methylator phenotype (CIMP). The level of CIMP is higher among SDHBMut tumors. This phenotype leads to expression deregulation of genes involved in neuroendocrine differentiation or in the epithelial-mesenchymal transition process, findings that could explain the increased risk of metastasis among patients with SDHBMut tumors. In a recent study higher DNA methylation levels were found in metastatic SDHB-PPGLs as compared to SDHB-PPGLs without metastasis, and this included de novo methylation of protocadherins (PCDH). Furthermore, in vitro assays suggested PCDHGC3 as a putative suppressor gene and a potential biomarker to identify patients with SDHB-mutated cancer at high risk of metastasis [38]. Cluster 2 groups tumors with mutations in NF1, RET, HRAS and TMEM127 genes, which activate the MAP kinase signaling pathway [39]. The Cancer Genome Atlas (TCGA) project has identified a third group or cluster related to alterations in the Wnt pathway [34], which also seems to be associated with an increased risk of developing metastatic disease (Table 3).
Clinical presentation
The clinical presentation of PPGLs is extremely variable and depends on the anatomical location, tumor size and extent of locoregional or distant involvement; the secretion or not of catecholamines (CMNs), including type, amount and pattern of secretion (adrenaline/epinephrine: A/E; noradrenaline/norepinephrine: NA/NE, and dopamine: DA); the hereditary or sporadic nature; the malignancy potential; and the time elapsed from initiation of symptoms to diagnosis.
PPGLs arising from sympathetic paraganglia are characterized by adrenergic and noradrenergic symptoms, such as the classic triad of palpitations, headache and diaphoresis or tremor, facial pallor and dyspnea. However, the predominant symptom remains severe, variable hypertension (65%), with target tissue damage such as hypertrophic or dilated cardiomyopathy (including Tako Tsubo idiopathic cardiomyopathy-like forms), potentially leading to fatal cardiac events, arrhythmias, myocardial infarction, congestive heart failure and chronic lung disease. Hemodynamic instability due to alterations in sympathetic vascular tone and orthostatic hypotension has also been described [12, 40,41,42,43,44,45,46,47]. Other symptoms of CMN excess include increased basal metabolism, weight loss, sweating, heat intolerance, altered glucose homeostasis resulting in type 2 diabetes mellitus, polyuria, polydipsia, constipation, ischemic colitis, altered vision, increased erythrocyte sedimentation rate and leukocytosis, psychiatric symptoms, and, rarely, hypercalcemia and polycythemia.
Symptoms are variable in duration and frequency and can be spontaneous or induced by various stimuli, such as food with high tyramine content (chocolate, coffee, smoked meat, cheese, red wine), sustained physical exercise (sometimes during urination in bladder PGLs), delivery, trauma, the induction of anesthesia, invasive diagnostic or therapeutic procedures, surgery, tumor biopsy or fine-needle aspiration and some medications (DA-2 antagonists, β-adrenergic blockers, sympathomimetics, opioids, tricyclic antidepressants, serotonin reuptake inhibitors, monoamine oxidase (MAO) inhibitors, corticosteroids, peptides or neuromuscular blocking agents) [12, 47]. Patients with A/NA PPGLs may be asymptomatic due to early diagnosis in the setting of abdominal imaging performed for other reasons (5% of adrenal incidentalomas) or screening procedures in at-risk family members [12, 45, 47]. Early case detection reduces cardiovascular morbidity and mortality due to chronic untreated CMN secretion [12, 43, 44, 47].
PGLs distributed along the parasympathetic chains of the head and neck (glossopharyngeal and vagal nerves) tend to be silent or pseudo-silent tumors (up to 15% of PPGLs). Usually, they do not produce CMNs, have a low metastatic potential, present as a head and neck mass with symptoms related to tumor bulk and local compression, or are incidentally discovered on imaging studies done for other purposes [12, 45, 47].
Recently, PPGLs have been classified based on translational clinical-biochemical-gene mutation cluster features into five groups: silent, biochemically pseudo-silent, noradrenergic, adrenergic and dopaminergic phenotypes [37, 48].
PPGLs associated with hereditary predisposition syndromes are more likely to be recurrent, multifocal and bilateral; occur at a younger age [12, 45, 47]; have a higher malignancy potential; and are associated with genetically driven comorbidities. Table 4 summarizes the main screening indications for PPGLs.
Clinical severity and prognosis are marked by the malignancy potential of PPGLs and the risk of aggressive metastatic evolution. Malignancy, defined as the presence of metastasis in non-chromaffin tissues [49], has a prevalence of 10% in PCCs and reaches 35–40% in PGLs [12, 45, 47], with symptoms and disease burden depending on the affected tissue. Translational risk scores are currently focusing on the identification of tumors with an aggressive outcome.
Hereditary predisposition syndromes and genetic counseling and testing
A hereditary form of PPGL should always be ruled out following the diagnoses of PPGL. Syndromic PPGL is strongly suspected in an individual with multiple, multifocal, recurrent, early onset of the disease and/or a family history of PPGL or related tumors (see predisposition syndromes associated with germinal mutations in Table 3). There are currently over 22 susceptibility genes identified. Among them, genes related to syndromic presentation drive nearly half of the cases. PPGLs present the highest rate of germline susceptibility in cancer genetics, at almost 40% [50,51,52]. These include genes encoding neurofibromin 1 (NF1), RET, VHL, menin (MEN1), SDH complex (SDHx: SDHA, SDHB, SDHC, SDHD), SDH complex assembly factor 2 (SDHAF2), TMEM127, MAX, FH, hypoxia-inducible factor 2A (EPAS1/HIF2A), EGLN1/PHD2, SLC25A11 and DLST. These genes are inherited in an autosomal-dominant manner. However, pathogenic variants in SDHD cause disease only when the pathogenic variant is inherited from the father. Similarly, SDHAF2 and possibly MAX follow an autosomal dominant inheritance, modified by maternal imprinting. Taking into account this genetic heterogeneity, next-generation sequencing (NGS) has been currently established as the new standard screening tool for genetic testing in patients with PPGL. Susceptibility genes and familial PPGL syndromes have been comprehensively reviewed recently [37], and their main features are summarized in Table 3.
The genotype–phenotype correlation is a useful tool to assess the clinical outcome of patients. These include tumor location and CMN secretion profile, as well as the presence of metachronous tumors, aggressive behavior and overall prognosis. Regarding the biochemical profile, genes involved in the pseudohypoxia pathway (Cluster 1) typically are associated with NE and its main metabolite, normetanephrine (NMN) [53, 54] and tumors are commonly located outside the adrenal glands [53, 54]. When located in the head and neck region (HNPGLs), PPGLs have been traditionally classified as biochemically “silent” tumors [55, 56]. However, HNPGLs can also produce high levels of DA and its main metabolite, 3-MT, with normal or near-normal levels of NE/NMN [57, 58]. Elevated levels of DA/3MT/NE have been reported in approximately 65% of patients with SDHxMut tumors [58, 59]. Hereditary forms related to Cluster 1 genes often present with variable expressivity and incomplete penetrance. For instance, patients with a driver mutation in SDHA/C usually lack a positive family history and rarely present with more than one tumor. Finally, regarding recurrence or aggressive disease, almost 90% of patients with metastatic PGLs present a SDHA/BMut or FHMut tumor [33, 60,61,62]. On the other hand, genes involved in the kinase signaling group pathway (Cluster 2) commonly present with adrenal tumors that produce either purely elevated E or its main metabolite, metanephrine (MN) [54], or both E/MN and NE/NMN. This hereditary form often presents with higher expressivity, and complete penetrance occurs more frequently.
If the pathogenic variant has been identified in an affected family member, prenatal testing for a pregnancy at increased risk and preimplantation genetic diagnosis could be considered in certain syndromes, such as VHL disease. Furthermore, genetic identification provides valuable information for the establishment of a treatment plan and for appropriate guidance for follow-up surveillance.
Diagnostic procedures
The diagnosis of PPGLs includes clinical suspicion, biochemical hormonal detection of excess CMN secretion, imaging studies for tumor localization and staging, genetic screening and, if a genetic germline mutation is confirmed, additional diagnostic procedures for genetic syndromic features as appropriate. PPGLs diagnosed as incidental masses in imaging studies require the same approach. A proposed diagnostic algorithm is provided in Fig. 1.

Diagnostic algorithm for PPGLs. CT computed tomography, FDG fluorodeoxyglucose, HNPGL head and neck paraganglioma, MN metanephrine, Mtx metastasis, NMN normetanephrine, MIBG metaiodobenzylguanidine, 3-MT 3-methoxytyramine, MRI magnetic resonance imaging, PCC pheochromocytomas, PGL paragangliomas, PPGL pheochromocytomas and paragangliomas, SSTRI somatostatin receptor imaging, VHL von Hippel-Lindau. a Plasma 3-MT: only in high clinical suspicion of dopamine-secreting tumors/hereditary syndromes associated with HNPGL. b Chromogranin A: nonspecific neuroendocrine tumor marker that may be considered if high clinical suspicion of silent PPGLs. c Recommended at diagnosis only in cases of high suspicion of metastasis, particular if there is family history or silent tumor. d 123-I-MIBG versus 111In/99mTc/68 Ga SSTRI, is recommended before MIBG versus radionuclide-SSTR analogs treatment
Biochemical testing
The biochemical diagnosis and follow-up of PPGLs rely on the quantification of CMN metabolites, such as plasma-free NMN, MN, and 3-MT or urinary fractionated MNs (e.g., MN, NMN). CMN measurement is less informative. Reference intervals (supine position; children) and standard preanalytical conditions (supine position for plasma determination, diet and medication interferences for urine testing) have to be carefully followed. Biochemical testing for PPGLs should be performed before imaging studies. Prior to the 24-h urine and plasma CMN metabolite determination, a 3-day diet without caffeine, black tea, nicotine, alcohol, bananas, cheese, almonds, nuts, chocolate, eggs, or vanilla is recommended. Some drugs, such as MAO inhibitors, ephedrine, cocaine, tricyclic antidepressants, serotonin reuptake inhibitors, morphine, amoxicillin, levodopa, sulfasalazine, acetaminophen, methyldopa and buspirone, can also cause false-positive results and should be avoided if possible [12, 63,64,65].
Guidelines accept both plasma-free and urinary MNs for the screening of PCCs and PGLs, with both determinations being considered to have similar sensitivities (97%) and specificities (91%) [12]. Some reports showed a better sensitivity (99% versus 80%) and a lower specificity (85% versus 98%) for plasma versus urinary MNs. Urinary creatinine is required to normalize urinary metanephrine excretion to renal function (metanephrine-to-creatinine ratio). Recent results, conversely, reported that plasma-free MNs in the supine position have higher specificity than 24-h urinary fractionated MNs (95% versus 90%), with the highest accuracy (95%) for liquid chromatography–mass spectrometry (LC–MS) methods compared with immunoassays [65,66,67]. LC–MS or electrochemical detection [liquid chromatography electron capture dissociation (LC-ECD)] is considered the gold standard, avoiding drug interference [12, 63,64,65,66].
Plasma levels of NMNs, MNs or 3-MT more than twice the upper cutoff value of the reference interval indicate a high probability of PPGL, and further imaging studies are recommended. Combined increases in two or more metabolites also suggest a high probability of a PPGL.
In the case of borderline elevated values, false-positive results due to an inappropriate preanalytical preparation, testing method, CMN-metabolism-interfering medication, intense physical stress, severe illness or laboratory error must be considered, and the test shall be repeated upon condition optimization [64].
In patients with PPGLs, MN levels are generally greater than CMNs (A/NA/D) due to continuous production and release of MNs by tumor cells. Patients with false-positive results usually have larger increments in CMNs than in plasma-free MNs because of sympathoadrenal activation [67,68,69].
The clonidine suppression test is indicated in inconclusive situations with borderline elevated NMN levels. A persistently increased level and a lack of a decrease in plasma-free NMN (< 40%) 3 h after the administration of clonidine support the diagnosis of PPGL (sensitivity of 100% and specificity of 96%, respectively) [12, 67]. Provocative testing (e.g., glucagon) can be dangerous, adds no value to other current testing methods and is not recommended [12, 70].
In case the probability of a tumor diagnosis is low and there are borderline elevated values, intrapatient longitudinal serial assessments can be useful (e.g., retesting patients 6 months later or more), as the disease growth rate is slow in most cases and involves a doubling time of over 2 years [12, 71]. The measurement of urinary fractionated MNs or concomitant measurement of plasma-free MNs and urinary MN, and CgA should be considered as follow-up tests [12, 72]. In the setting of prospective screening in hereditary forms of the disease, even low increased values have to be considered positive.
False-negative results are less frequent but can be observed in microscopic asymptomatic tumors, DA-producing tumors and tumors with CMN synthesis and/or metabolism defects (e.g., in a silent SDHBMut subtype, the enzyme that catalyzes the initial and rate-limiting step in CMN biosynthesis is missing). Additional measurements including CgA (after stopping proton pump inhibitors for at least 10 days) and nonspecific neuroendocrine secretory proteins, as well as imaging studies, are strongly indicated in cases of nonfunctional PPGLs, particularly in SDHBMut carriers [69, 73].
The measurement of 3-MT, the main metabolite of DA, should be considered in patients in whom extra-adrenal HNPGLs are strongly suspected despite normal plasma and urinary MN levels or when metastatic disease is suspected. High-elevated levels of plasma 3-MT indicate the need for preoperative staging, if possible, by radionuclide imaging [12, 59, 68, 73].
Recently, the CMN secretion profile has also been related to the recent molecular cluster classification (see Sect. 3, risk classifications): Cluster 1: pseudohypoxic Krebs cycle-related (10–15%): NE/NMN and DA/3-MT secretion; VHL/EPAS1-related (15–20%): NE/NMN; Cluster 2: kinase signaling-related (50–60%): E/MN (especially RET) or both E/MN and NE/NMN; and Cluster 3 (5–10%): Wnt signaling-related: E/MN and NE/NMN and chromogranin A [37, 48].
Imaging studies
The diagnosis of PPGL relies on the imaging identification of an appropriately located mass with consistent clinical and biochemical features. Once the diagnosis is clinically and biochemically confirmed, imaging studies should be performed to localize and stage the tumor [74]. However, PCGLs, particularly PCCs, are sometimes encountered incidentally on imaging procedures performed for other causes [75].
CT is the most common imaging method used because it is widely available, less expensive and offers better spatial resolution than MRI. PPGLs are usually solid and hypervascular, well-circumscribed masses, ranging from 1 to 15 cm (Fig. 2). Smaller tumors are usually homogeneous, and larger tumors tend to have central necrosis. Some PPGLs can have macroscopic fat simulating adenomas or may have very high attenuation due to hemorrhage or calcifications. There is also a pure cystic form [76, 77].

Typical morphological and functional imaging of PPGLs. a, b Axial contrast-enhanced CT portal (a) and delayed phase (b) of the upper abdomen showing the pheochromocytoma in the right adrenal gland (yellow arrowhead). Intravenous contrast administration typically enhances avidly due to the capillary-rich framework of the tumor. c, d Coronal T2-weighted MRI images revealed a homogeneous pheochromocytoma (c) in the right adrenal gland (yellow arrowhead), and other pheochromocytomas in the left adrenal gland (yellow arrowhead) with central necrosis are characteristically “light-bulb” bright lesions on T2-weighted imaging (d). Pheochromocytomas are potentially malignant (10%), and the only reliable criterion for the diagnosis of malignancy is metastatic spread. e A 61-year-old woman with metastatic cervical paraganglioma. 68 Ga-DOTATOC PET/CT study showing bilateral laterocervical lymph nodes, mediastinal involvement and multiple bone metastases. f A 56-year-old man was diagnosed with a 44 × 39-mm right adrenal incidentaloma. After right adrenalectomy, a histological study showed pheochromocytoma without evidence of malignancy. Negative genetic study. During follow-up, he presented with recurrence. Body scan with 123-I-MIBG shows lesions in the right renal cell and multiple peritoneal implants, some in contact with the liver surface without being able to rule out secondary infiltration. The patient has received treatment with 131I-MIBG with stabilization of the disease
MRI is not a first-choice imaging tool, but it has the advantage of being free of ionizing radiation and is suitable for children, pregnant women and patients with adverse reactions to iodinated contrast medium. Cystic PPGLs with central necrosis are characteristically “light-bulb” bright lesions on T2-weighted imaging, with low signal intensity at T1. The signal intensity of hemorrhage is predominantly high in T1. If PPGLs contain macroscopic fat, they may also be dark on T2 MR images [78] (Fig. 2).
PPGL cells express different transporters on their surface that allow images to be obtained by different radiotracers depending on the capture mechanisms (NE transporters [123I/131I- Metaiodobenzylguanidine (MIBG)], type of transporters (glucose transporters (GLUT (18F-FDG)), amino acid transporters (18F-DOPA) or membrane surface receptors [somatostatin (SST) receptors (111In/99mTc/68 Ga SST analogs)], thus yielding different functional information [79]. The most sensitive functional image for each tumor will depend on the clinical and biochemical profiles and the location of the primary tumor, which are also predictors of the underlying genotype [80].
Positron emission tomography (PET)/CT technology has been shown to be superior to scintigraphy with single photon emission computed tomography (SPECT/CT), with higher spatial resolution, greater sensitivity and fewer indeterminate or equivocal studies [37, 79].
I123−MIBG-sensitivity and specificity reach 83–100% and 95–100%, respectively, for the diagnosis of sporadic PCCs. The sensitivity of I123−MIBG decreases to 52–75% for the diagnosis of PGLs and to 18–50% for HNPGLs.
68Galium (68Ga)-DOTA peptide PET showed an overall detection rate of 98.6% in patients with metastatic SDHBMut PPGLs. In HNPGLs, this is considered the functional image of choice. In polycythemia-related PPGLs and in FHMut or MAXMut PPGLs, 18F-DOPA PET is the functional imaging of choice, and if not available, I123−MIBG-SPECT/CT is recommended [12, 79, 81,82,83].
Generally, when facing metastatic disease, better results are reported with the use of 18F-FDG PET/CT [12, 79, 81, 83]. In these cases, I123−MIBG and studies with SST analogs would be reserved for patients with metastatic disease for whom radiometabolic treatment with 131I-MIBG and/or 90Ytrium (90Y)/177Lutethium (177Lu)-DOTA peptides —peptide receptor radionuclide therapy (PRRT)—is being considered. Recommended PPGL functional imaging studies according to genotype and anatomic location are summarized in Fig. 1.
Treatment
Therapeutic strategy
The therapeutic strategy for PPGLs should be discussed by an expert multidisciplinary team based on patient characteristics (e.g., age, performance status, comorbidities) and tumor features (i.e., primary tumor site, local and distant spread, hormone secretion profile, tumor growth rate, functional imaging and genetic profile). Surgical resection is the mainstay of therapy for the majority of localized PPGLs, with adequate perioperative CMN blockade and cardiovascular monitoring in PCCs and functional PGLs. Adequate timing for surgery and optimal surgical approach are still a matter of debate. Advanced, unresectable disease is not curable, and treatment goals are to slow tumor progression and maintain quality of life. Medical treatment of secretory PPGLs is mandatory to prevent life-threatening events (Fig. 3). Treatment options include watch and wait strategies for indolent tumors, radiometabolic therapy, radiotherapy, chemotherapy, targeted therapy (i.e., antiangiogenic tyrosine kinase inhibitors) and PRRT. Indications and contraindications of these therapeutic options are discussed below, and a therapeutic algorithm is proposed in Fig. 3.

Therapeutic algorithm of metastatic paragangliomas. CVD, Cyclophosphamide, Vincristine, Dacarbazine; MIBG, 123-I-Metaiodobenzylguanidine; MGMT, O-methylguanine-DNA methyltransferase; RF, radiofrequency; RT, radiotherapy; SDH, Succinate dehydrogenase; SSTRI, somatostatin receptor imaging; TKI, Tyrosine kinase inhibitors; TMZ, Temozolomide
Catecholamine blockade
Anesthesia, tumor manipulation during surgery, tumor biopsy, adrenal venography and arteriography with ionic contrast can induce excess CMN secretion, hyperadrenergic symptoms and hypertensive crises in patients with PCCs and functional PGLs (FPGLs) [84]. To prevent this potentially fatal phenomenon, the European Endocrine Society Guidelines recommend that patients with PCCs and FPGLs should undergo a 7- to 14-day preoperative preparation with adrenergic receptor blockers as the first choice. Some authors have questioned the universal indication of preoperative α-adrenoceptor blockade given the potential side effects of this therapy, but prospective studies are needed to identify in which patients’ preoperative therapy may be safely avoided. α1 selective blockers (e.g., doxazosin: initiate with 4 mg/day and then titrate up to 16–32 mg/day) are associated with fewer adverse effects, such as reactive tachycardia and sustained postoperative hypotension, compared to nonselective α-blockers (e.g., phenoxybenzamine: initiate with 10 mg/day and then titrate up to 1 mg/kg/day). Calcium channel blockers are the most often used add-on drug class to further improve blood pressure (BP) control. Only after administration of α-adrenergic receptor blockers may a β-adrenergic receptor antagonist (e.g., propranolol or atenolol) be added 2 or 3 days before surgery if the heart rate (HR) exceeds 80/minute (bpm) [85]. Phone or e-mail-based preoperative monitoring of BP and HR, orthostatic hypotension, and pharmacological side effects may be implemented in the clinic to avoid multiple visits [86]. Presurgical antihypertensive treatment has been also advised for patients with PPGL and a normal blood pressure[87].
A high-sodium diet and fluid intake are also recommended to reverse CMN-induced blood volume contraction preoperatively and thereby reduce orthostatic hypotension and minimize the risk of severe hypotension after surgical tumor removal [12]. Saline infusion (1–2 L) the evening before surgery is also helpful for this purpose. Esmolol β-adrenergic blockade can be employed during surgery. After surgery, BP and HR monitoring is needed to detect postoperative hypotension requiring vasopressor support. Alpha blockade is not specifically required before iv administration of nonionic contrast media in patients with suspected or known PPGL or related tumors [84].
Surgery
Surgical resection is the cornerstone of therapy for most localized PPGLs, as it is the only potentially curative therapeutic modality. Careful preoperative planning is required to select the most appropriate surgical technique. This includes precise anatomical characterization of the primary tumor (or tumors if multifocal) location and extension to adjacent structures and/or distant organs and adequate perioperative medical management. The goal of surgery is to achieve complete tumor resection without rupture, including en bloc resection of adjacent infiltrated organs if needed.
The complete resection of HNPGLs, indicated in young patients, usually requires previous embolization and may be performed in one or more steps depending on the extent of intradural space (IDS) and/or internal carotid artery (ICA) involvement [88]. Elderly or frail patients and those with bilateral multicentric lesions or residual disease may be considered for watch and wait strategies or alternative nonsurgical treatment options [89].
For cervical and mediastinal PGLs, a transcervical approach is generally used, while rarely being associated with a transmandibular, transmastoid or infratemporal approach. Carotid body PGLs with ICA involvement have a higher incidence of complications [90]. For vagal PGLs, a cervical or posterior tear hole approach is recommended. A 2-stage surgery may be required if there is significant intradural extension. The resection of jugulotympanic PGLs [91] has different degrees of complexity. Tympanic PGLs are resected through a low-morbidity transcanal, microscopic/endoscopic approach. Tympanomastoid PGLs implicate transmastoid, transcanal, and infralabyrinthic techniques, occasionally with middle ear removal, that have low morbidity over facial and lower cranial nerves. Jugular PGLs can compromise the ICA and the lower cranial nerves and extend to the IDS. The infratemporal approach employed in these cases entails important technical challenges (protection of the ICA with a stent or its occlusion may be required for complete resection) and functional morbidity, particularly if the IDS is involved or the low facial and hypoglossal nerves are affected (> 30%). There is no consensus on the systematic rerouting of the facial nerve. Partial resection may be a valid option in tumors that reach the external auditory canal (EAC) and generate recurrent hemorrhages in elderly patients.
For abdominal localized PPGLs, a complete surgical resection (PGL) or adrenalectomy (PCC) is indicated. Bilateral adrenal involvement requires bilateral adrenalectomy. Subtotal adrenalectomy with cortical preservation prevents adrenal insufficiency and the need for hormonal supplementation in up to 90% of patients. This procedure is recommended only for cases with a low risk of malignancy, such as MEN2 or VHL syndrome, and not for other genetic syndromes with a greater risk of distant spread or local relapse due to remnant microscopic disease (e.g., SDHxMut or MAXMut) [92,93,94]. In the presence of metastatic disease, total or partial palliative resection may be considered to reduce the disease burden and improve hormonal syndrome control [95].
PCC resection may be open or laparoscopic. Laparoscopic adrenalectomy (LA) is recommended for most PCCs because it is associated with lower morbidity and a shorter postoperative stay than open adrenalectomy (OA). Recurrence rates do not differ between the 2 surgical approaches, with a rate of conversion to OA between 5 and 12% [96]. Minimal tumor manipulation is recommended to avoid excessive CMN release, and anesthetists must also be aware that adrenal vein ligation may induce sudden hypotension. Potential hemodynamic instability is not a contraindication for LA, and the time when the adrenal vein is ligated does not seem to be relevant [92, 96, 97]. LA can be performed transabdominally or retroperitoneally. Both LA approaches achieve adequate resection and have minimal morbidity, with no clear hemodynamic benefits of one over the other. The retroperitoneal approach is especially favorable for simultaneous bilateral adrenalectomy [98, 99]. OA is indicated in bulky (> 6–8 cm) PCCs or in PCCs with a high suspicion of malignancy and/or involvement of neighboring organs or complex locations [92].
PGL resection may be more challenging because such tumors are usually located in complex sites (e.g., retroperitoneum, paravertebral, para-aortic in the Zuckerkandl organ and along the inferior hypogastric plexuses adjacent to the urogenital organs) and have a higher risk of malignancy and recurrence. Therefore, an open approach is generally recommended. Non-infiltrative PGLs in favorable locations can be resected by endoscopic surgery [98].
Acute postoperative complications, such as hemodynamic and metabolic instability with hyper- or hypotension and hypoglycemia, can be avoided with appropriate CMN blockade and fluid replacement.
Acute and chronic adrenal insufficiency should be assessed, and hormonal replacement therapy should be appropriately administered in bilateral total and cortical sparing adrenalectomy or unilateral cortical sparing adrenalectomy of a sole remaining adrenal gland.
Radiotherapy and radiometabolic therapy
Radiotherapy
The greatest experience of PGL radiotherapy (RT) comes from the treatment of glomus jugular tumors, as RT constitutes a noninvasive therapeutic option that is appropriate in locations with high surgical risk or when patients are not candidates for surgery (patients with carotid or intracranial involvement) [100].
Conventional RT achieved modest responses (20–30%) that were surpassed by radiosurgical techniques administered in single doses (12–15 Gy) and, thereafter, by stereotaxic ablative radiotherapy (SABR), with doses of 20–25 Gy in 3–5 fractions, leading to tumor control rates of 90–100% and symptomatic improvement in 80% of patients [101].
Radiometabolic therapy
Systemic radiometabolic treatment is an option for disease control in patients with inoperable locally advanced or metastatic disease and documented tumor uptake of the corresponding radioisotope. It is generally considered for symptomatic patients with slow-growing tumors and significant tumor volume or disease progression [102, 103].
Functional imaging studies should be carried out with 123I-MIBG and/or radiolabeled SST analogs to assess the affinity of the tumor for the radiotracer and to choose the most appropriate radiopharmaceutical for each case before peptide receptor radionuclide therapy (PRRT) [104].
The largest accumulated experience with 123I-MIBG shows that over 50% of patients with mPPGL are candidates for radiometabolic treatment. There is no consensus regarding the preferred treatment protocol, optimal dose and time interval between doses, or response criteria. With the administration of a medium–high activity (200–275 mCi) of 123I-MIBG, repeated every 3 months depending on the achieved response, objective responses have been documented in 30–60% of cases, a hormonal response in 10–71% of patients and a symptomatic response in 23–90% of patients [105, 106]. The main side effects are related to cumulative dose-dependent bone marrow toxicity and renal toxicity. Thyroid radiotracer uptake should be blocked prior to therapy to prevent hypothyroidism, and blood counts and renal function should be monitored.
More recently, a novel 123I-MIBG derivative has been developed, Iobenguane 131I or high-specific-activity (HSA)- 123I-MIBG, produced from a solid-phase ultratrace precursor that eliminates the presence of cold MIBG and is able to deliver a high radioactivity level per dose (~ 2500 mCi/mg; 92.5 MBq/μg). With conventional 123I-MIBG cold MIBG competes with radiolabeled MIBG for the NE transporter, reducing labeled MIBG uptake by the tumor cell, thus limiting efficacy and increasing the levels of circulating NE that can lead to life-threatening acute hypertensive crisis during or shortly after drug administration. A phase 2 trial showed that 17 of 68 PPGL patients (25%) treated with HSA-123I-MIBG had a durable reduction in baseline antihypertensive medication use, and 92% achieved a partial response or stable disease as the best objective response within 12 months. The median OS was 36.7 months, and no patients had drug-related acute hypertensive events. Based on these data, HAS-123I-MIBG received FDA breakthrough therapy designation and was approved in July 2018 for the treatment of patients with iobenguane scan-positive, advanced or mPPGLs who require systemic anticancer therapy [107].
Experience in the use of PRRT for the treatment of PPGLs is limited, although early results look promising. Radiological control has been described in 80% of patients with metastatic SDHBMut PPGL treated with 177Lu-DOTATATE [108, 109]. However, prospective studies are necessary to determine the role of PRRT in the control of patients with inoperable advanced or mPPGLs.
Systemic therapy
Metastatic disease, unless amenable to complete surgical resection, is incurable. Systemic treatment options are limited but can offer symptom palliation and disease control. However, due to the relatively indolent nature of PPGLs, these therapies are generally reserved for patients with clear disease progression or severe symptoms caused by hormone secretion or mass effects. Evidence to support treatment decisions is poor, although increasing data suggest that different molecular subtypes driven by distinctive oncogenic pathways may have unique sensitivity profiles to specific drugs [49, 110]. International collaborative efforts are key to make adequately sized prospective trials feasible.
Chemotherapy is considered the treatment of choice for patients with advanced PPGLs who have progressed to or are not suitable candidates for MIBG or PRRT. Cyclophosphamide, vincristine and dacarbazine (CVD) chemotherapy is the most widely used regimen and is considered the standard of care despite the lack of prospective trials [49, 111, 112]. A systematic review of four retrospective series that included 50 patients reported an objective tumor response rate of 41% (4% complete and 37% partial responses) and a biochemical response rate of 54% (14% complete) [112]. Two of these studies reported median durations of response of 20 and 40 months, respectively. In the largest single-institution experience with chemotherapy (54 patients), 33% of patients achieved a response, defined as improved BP control and/or reduced tumor size. OS was 6.4 years for responders vs 3.7 years for non-responders, a difference that was statistically significant in multivariate analysis [111]. The most common toxicities include myelosuppression, peripheral neuropathy and gastrointestinal toxicity, which may occasionally be severe but are generally transient and manageable. A retrospective study of 15 patients treated with temozolomide (150–200 mg/m2/day d1–5 q28 days), 8 of whom had received prior chemotherapy, documented 5 partial responses (33%) that occurred only in patients with SDHB mutations [113]. The median progression-free survival (PFS) was 13.3 months (19.7 vs 2.9 months in SDHBMut vs noncarriers). SDHB-germline mutations were associated with O-methylguanine-DNA methyltransferase (MGMT) promoter hypermethylation and low MGMT protein expression in a cohort of 190 samples of the French national PPGL network [113]. These findings suggest that MGMT epigenetic silencing in SDHBMut carriers may render them particularly sensitive to this alkylating agent. Successful outcomes have also been reported in two patients with SDHBMut metastic PGLs treated with temozolomide metronomic schedules (75 mg/m2/day × 21/28 days) following progression to prior CVD therapy [114]. More recently, an increased activity in the Poly(ADP-ribose)polymerase (PARP) DNA repair system has been described in SDHBMut PPGLs, associated with chemo-resistance[115]. The PARP-inhibitor olaparib was shown to markedly potentiate the therapeutic effect of TMZ with prolonged overall survival of mice with SDHB knockdown PPGL allograft [115]. Based on these findings, a trial investigating the synergistic effect of the addition Olaparib to TMZ is currently undergoing (NCT04394858).
A number of tyrosine kinase inhibitors (TKIs) are being explored due to the key role that angiogenesis regulation plays in PPGLs, particularly in Cluster 1 (SDH- and VHL-driven PPGLs) and some Cluster 2 tumors (i.e., RET). The phase II SNIPP trial evaluated sunitinib in 25 patients with progressive PPGLs [116]. The overall response rate was low (13%) in the overall unselected population, although all three partial responses occurred in patients with germline mutations in SDHA, SDHB and RET (with this last patient remaining on treatment 7 years later). The disease control rate (DCR) was 83%, meeting the study primary endpoint, and the median PFS was 13.4 months. The most common severe side effects were fatigue and thrombocytopenia (16% each), and three patients discontinued treatment due to cardiovascular adverse events. Sunitinib is currently being assessed in the first randomized, placebo-controlled trial ever conducted in PPGLs, the FIRSTMAPPP trial. A phase II trial with pazopanib was terminated early due to poor patient accrual. One of the six evaluable patients achieved a partial response (17%), and the median PFS and OS periods were 6.5 and 14.8 months, respectively [117]. Similarly, preliminary data of a phase II trial with axitinib reported an objective response in three of nine treated patients (33%) and some degree of tumor shrinkage that did not qualify for partial response in five additional patients, which was associated with biochemical response [118]. Other TKIs (cabozantinib, lenvatinib, etc.) are currently being evaluated in clinical trials (https://clinicaltrials.gov/).
Finally, some other drugs active in the treatment of NETs, such as ‘cold’ SST analogs and interferon, have been poorly addressed in this setting although they are also used for the treatment of PPGLs [114, 119]. Currently, a phase II prospective trial is assessing the role of the SST lanreotide in patients with advanced disease (NCT03946527).
Local ablative therapy and supportive care
In patients with progressive advanced or mPPGLs, the treatment goals are to manage hormone-related symptoms, control tumor growth and prolong OS. The use of local ablative therapies in this setting can improve local control and palliate symptoms [120]. The indication must be individualized and discussed within the multidisciplinary team and carefully balanced versus other treatment options for patients with mPPGLs.
There is no prospective study to assess differences in outcome for patients receiving different ablative treatments for advanced disease. However, two recent retrospective studies published by the Mayo Clinic have analyzed the outcome of patients with mPPGLs receiving local therapies. The first study showed median OS and PFS rates of 24.6 and 33.7 years, respectively, at a median follow-up of 8.2 years (range, 0.01 to 54.1 years). Among the 272 patients analyzed, 97% underwent additional surgical resection (for primary tumors or metastases). In addition, palliative RT, radiofrequency ablation, embolization procedures, stereotactic radiotherapy, cryoablation and percutaneous ethanol injection were performed in 47%, 9%, 8.8%, 5.8%, 4.7% and 2% of the patients, respectively. Almost half of the patients (45%) survived > 10 years [121]. The second study reported the efficacy and safety of radiofrequency ablation, cryoablation and percutaneous ethanol injection in these patients. Radiographic local control was achieved in 69/80 (86%) lesions. Improvement in metastasis-related pain or symptoms of CMN excess was achieved in 12/13 (92%) patients. Thirty-three (67%) procedures had no reported complications [120].
PPGLs are among the solid tumors that most frequently spread to the skeleton and cause skeletal-related events (SRE) (i.e. pain, bone fracture, and spinal cord compression are commonly the first manifestation of metastatic disease (31%). SREs should, therefore, be properly addressed, as they compromise survival and can seriously impair patients’ quality of life. A multidisciplinary approach with specialists in endocrinology, oncology, palliative care, radiotherapy, orthopedic surgery and neurosurgery is of utmost importance.
Follow-up recommendations
Short-term postoperative follow-up should include clinical and biochemical evaluation (MNs, 3MT/CgA if other markers are negative) 2–6 weeks after recovery. Imaging is recommended 3 months after recovery in patients with persistent postoperatively altered biochemical markers, silent PPGLs and absence of preoperative biochemical evaluation [122].
Long-term follow-up is mandatory in all patients, as they are all considered at risk of tumor recurrence, and the clinical behavior of PPGLs is remarkably variable, especially in PPGLs associated with hereditary syndromes. Ten-year follow-up is recommended for all patients with resected PPGLs and lifelong personalized follow-up for patients with hereditary forms of the disease; such follow-up should be performed by a multidisciplinary team at a tertiary center whenever possible. Whereas in some patients with mPPGLs, the discovery of metastases may precede the discovery of the primary tumor, others may develop metastases many years after the initial diagnosis [121, 123].
Candidates for intensified surveillance have to be identified. Male sex, older age at primary tumor diagnosis (≥ 76 years), larger tumor size (> 4.5–5 cm), failure to undergo complete surgical resection of the primary tumor, DA hypersecretion and synchronous metastases are associated with shorter survival 15–21. Tumor size, extra-adrenal location and germline SDHB mutations are independent risk factors for mPPGLs [13, 60, 61, 124,125,126].
Currently, no specific follow-up protocols are established. The frequency of surveillance should be based on a number of factors, such as the affected gene, genotype–phenotype correlation, symptomatic or silent pattern of the disease, potential severity of the disease, penetrance and family history. Overall, it is recommended to carry out annual clinical anamnesis, physical exam (including blood pressure control) and biochemical monitoring (MNs, ± 3-MT and optional CgA in MNs/3-MT negative PPGLs). Imaging studies are recommended yearly in suspected cases (based on clinical or biochemical evaluation) or every 2–3 years in silent PPGLs. To avoid cumulative irradiation, body or head/neck MRI should be considered the imaging procedure of choice for surveillance, especially in children and during pregnancy, reserving CT and functional NM imaging to characterize pathological findings in cases of relapse. Specific monitoring of the other diseases associated with each syndrome should also be performed [12, 122].
Regarding the role of functional imaging during follow-up, experts [12] recommend the use of 123I-MIBG scintigraphy when the risk of metastasis or disease recurrence is high, and 18F-FDG PET/CT is only indicated in established metastatic disease. The use of more than one functional imaging modality may be considered in selected cases, such as the use of both 68 Ga-DOTATATE and 18F-FDG PET/CT in patients with small lesions when there is a high likelihood of metastatic disease and in SDHxMut patients [37, 127].
Follow-up of asymptomatic carriers There is no sufficient clinical epidemiological evidence from clinical data to perform general recommendations for surveillance. The main aim of surveillance programs in healthy mutation carriers, especially in SDHX-mutation carriers, is to identify disease at an early stage in order to allow a successful intervention at the appropriate time, improve cure rates and limit the chance of malignant transformation and metastasis. Modality and frequency of screening that individual centers adopt will be dependent on local expertise, availability and costs. The appropriate age to start screening will vary according to the specific hereditary syndrome including the malignant and metastatic potential associated with the identified genetic mutation. In children it is generally recommended between 5 and 10 years of age or 5 years before the youngest clinical manifestation in the family. Biochemical and clinical monitoring follows diagnosis recommendations mentioned above. Debate is ongoing regarding the frequency and type of the functional image probe to be done. Most tumors are diagnosed in the first screening image performed. The Endocrine Society Guidelines [12] emphasized that consideration for any imaging modality requires prior positive clinical or biochemical evidence of disease, except in case of a personal or family history of HNPGL related or not to a hereditary form. More recent studies and meta-analysis report the need of periodical image evaluation, the recommended frequency varying generally between 2 and 3 years [47, 128,129,130,131,132]. Currently, translational research stratification scores have been developed to estimate the risk of new PPGL events and the frequency of metastatic disease [133]; however, evidence from longitudinal studies is still needed, and guidelines for follow-up continue to evolve. National and international registries are fundamental to collect information necessary to deliver updates that permit the elaboration of clinical guidelines.
References
Leung AA, Pasieka JL, Hyrcza MD, Pacaud D, Dong Y, Boyd JM, et al. Epidemiology of pheochromocytoma and paraganglioma: population-based cohort study. Eur J Endocrinol. 2021;184:19–28.
Rodriguez-Cuevas H, Lau I, Rodriguez HP. High-altitude paragangliomas diagnostic and therapeutic considerations. Cancer. 1986;57:672–6.
Berends AMA, Buitenwerf E, de Krijger RR, Veeger N, van der Horst-Schrivers ANA, Links TP, et al. Incidence of pheochromocytoma and sympathetic paraganglioma in the Netherlands: a nationwide study and systematic review. Eur J Intern Med. 2018;51:68–73.
Lack EE, Cubilla AL, Woodruff JM, Farr HW. Paragangliomas of the head and neck region: a clinical study of 69 patients. Cancer. 1977;39:397–409.
Lloyd RV, Osamura YR, Kloppel G, Rosai J. WHO classification of tumours of endocrine organs. Geneva: WHO Press; 2017.
Waguespack SG, Rich T, Grubbs E, Ying AK, Perrier ND, Ayala-Ramirez M, et al. A current review of the etiology, diagnosis, and treatment of pediatric pheochromocytoma and paraganglioma. J Clin Endocrinol Metab. 2010;95:2023–37.
Pamporaki C, Hamplova B, Peitzsch M, Prejbisz A, Beuschlein F, Timmers H, et al. Characteristics of pediatric vs adult pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2017;102:1122–32.
Curras-Freixes M, Pineiro-Yanez E, Montero-Conde C, Apellaniz-Ruiz M, Calsina B, Mancikova V, et al. PheoSeq: a targeted next-generation sequencing assay for pheochromocytoma and paraganglioma diagnostics. J Mol Diagn. 2017;19:575–88.
Turchini J, Cheung VKY, Tischler AS, De Krijger RR, Gill AJ. Pathology and genetics of phaeochromocytoma and paraganglioma. Histopathology. 2018;72:97–105.
Chen H, Sippel RS, O’Dorisio MS, Vinik AI, Lloyd RV, Pacak K, et al. The North American neuroendocrine tumor society consensus guideline for the diagnosis and management of neuroendocrine tumors: pheochromocytoma, paraganglioma, and medullary thyroid cancer. Pancreas. 2010;39:775–83.
Jochmanova I, Abcede AMT, Guerrero RJS, Malong CLP, Wesley R, Huynh T, et al. Clinical characteristics and outcomes of SDHB-related pheochromocytoma and paraganglioma in children and adolescents. J Cancer Res Clin Oncol. 2020;146:1051–63.
Lenders JW, Duh QY, Eisenhofer G, Gimenez-Roqueplo AP, Grebe SK, Murad MH, et al. Pheochromocytoma and paraganglioma: an endocrine society clinical practice guideline. J Clin Endocrinol Metab. 2014;99:1915–42.
Amar L, Baudin E, Burnichon N, Peyrard S, Silvera S, Bertherat J, et al. Succinate dehydrogenase B gene mutations predict survival in patients with malignant pheochromocytomas or paragangliomas. J Clin Endocrinol Metab. 2007;92:3822–8.
Hescot S, Curras-Freixes M, Deutschbein T, van Berkel A, Vezzosi D, Amar L, et al. Prognosis of malignant pheochromocytoma and paraganglioma (MAPP-Prono Study): a European network for the study of adrenal tumors retrospective study. J Clin Endocrinol Metab. 2019;104:2367–74.
Cascón A, Remacha L, Calsina B, Robledo M. Pheochromocytomas and paragangliomas: bypassing cellular respiration. Cancers. 2019;11:683.
Goffredo P, Sosa JA, Roman SA. Malignant pheochromocytoma and paraganglioma: a population level analysis of long-term survival over two decades. J Surg Oncol. 2013;107:659–64.
Oudijk L, de Krijger RR, Pacak K, Tischler AS. Adrenal medulla and extra-adrenal paraganglia. In: Mete O, Asa SL, editors. Endocrine pathology. Cambridge: Cambridge University Press; 2016. p. 628–76.
Erickson D, Kudva YC, Ebersold MJ, Thompson GB, Grant CS, van Heerden JA, et al. Benign paragangliomas: clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86:5210–6.
Thompson LD. Pheochromocytoma of the adrenal gland scaled score (PASS) to separate benign from malignant neoplasms: a clinicopathologic and immunophenotypic study of 100 cases. Am J Surg Pathol. 2002;26:551–66.
Kimura N, Watanabe T, Noshiro T, Shizawa S, Miura Y. Histological grading of adrenal and extra-adrenal pheochromocytomas and relationship to prognosis: a clinicopathological analysis of 116 adrenal pheochromocytomas and 30 extra-adrenal sympathetic paragangliomas including 38 malignant tumors. Endocr Pathol. 2005;16:23–32.
Curras-Freixes M, Inglada-Perez L, Mancikova V, Montero-Conde C, Leton R, Comino-Mendez I, et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet. 2015;52:647–56.
Papathomas TG, Oudijk L, Persu A, Gill AJ, van Nederveen F, Tischler AS, et al. SDHB/SDHA immunohistochemistry in pheochromocytomas and paragangliomas: a multicenter interobserver variation analysis using virtual microscopy: a multinational study of the European network for the study of adrenal tumors (ENS@T). Mod Pathol. 2015;28:807–21.
Pierre C, Agopiantz M, Brunaud L, Battaglia-Hsu SF, Max A, Pouget C, et al. COPPS, a composite score integrating pathological features, PS100 and SDHB losses, predicts the risk of metastasis and progression-free survival in pheochromocytomas/paragangliomas. Virchows Arch. 2019;474:721–34.
Jimenez C, Libutti SK, Landry CS, Lloyd RV, McKay RR, Rohren E, et al. Adrenal-neuroendocrine tumors. In: Mete O, Asa S, editors., et al., AJCC cancer staging manual. New York: Springer; 2017. p. 919–27.
Langerman A, Athavale SM, Rangarajan SV, Sinard RJ, Netterville JL. Natural history of cervical paragangliomas: outcomes of observation of 43 patients. Arch Otolaryngol Head Neck Surg. 2012;138:341–5.
Amato B, Bianco T, Compagna R, Siano M, Esposito G, Buffone G, et al. Surgical resection of carotid body paragangliomas: 10 years of experience. Am J Surg. 2014;207:293–8.
Lima J, Feijao T, da Silva AF, Pereira-Castro I, Fernandez-Ballester G, Maximo V, et al. High frequency of germline succinate dehydrogenase mutations in sporadic cervical paragangliomas in northern Spain: mitochondrial succinate dehydrogenase structure-function relationships and clinical-pathological correlations. J Clin Endocrinol Metab. 2007;92:4853–64.
Burnichon N, Abermil N, Buffet A, Favier J, Gimenez-Roqueplo AP. The genetics of paragangliomas. Eur Ann Otorhinolaryngol Head Neck Dis. 2012;129:315–8.
Gimenez-Roqueplo AP, Dahia PL, Robledo M. An update on the genetics of paraganglioma, pheochromocytoma, and associated hereditary syndromes. Horm Metab Res. 2012;44:328–33.
Kantorovich V, Pacak K. New insights on the pathogenesis of paraganglioma and pheochromocytoma. F1000Res. 2018;7:F1000 Faculty Rev–500.
Qin Y, Yao L, King EE, Buddavarapu K, Lenci RE, Chocron ES, et al. Germline mutations in TMEM127 confer susceptibility to pheochromocytoma. Nat Genet. 2010;42:229–33.
Burnichon N, Cascon A, Schiavi F, Morales NP, Comino-Mendez I, Abermil N, et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clin Cancer Res. 2012;18:2828–37.
Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, et al. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum Mol Genet. 2014;23:2440–6.
Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31:181–93.
Remacha L, Pirman D, Mahoney CE, Coloma J, Calsina B, Currás-Freixes M, et al. Recurrent germline DLST mutations in individuals with multiple pheochromocytomas and paragangliomas. Am J Hum Genet. 2019;104:1008–10.
Buffet A, Morin A, Castro-Vega LJ, Habarou F, Lussey-Lepoutre C, Letouzé E, et al. Germline mutations in the mitochondrial 2-oxoglutarate/malate carrier SLC25A11 gene confer a predisposition to metastatic paragangliomas. Cancer Res. 2018;78:1914–22.
Alrezk R, Suarez A, Tena I, Pacak K. Update of pheochromocytoma syndromes: genetics, biochemical evaluation, and imaging. Front Endocrinol (Lausanne). 2018;9:515.
Bernardo-Castiñeira C, Valdés N, Celada L, Martinez ASJ, Sáenz-de-Santa-María I, Bayón GF, et al. Epigenetic deregulation of protocadherin PCDHGC3 in pheochromocytomas/paragangliomas associated with SDHB mutations. J Clin Endocrinol Metab. 2019;104:5673–92.
Lopez-Jimenez E, Gomez-Lopez G, Leandro-Garcia LJ, Munoz I, Schiavi F, Montero-Conde C, et al. Research resource: transcriptional profiling reveals different pseudohypoxic signatures in SDHB and VHL-related pheochromocytomas. Mol Endocrinol. 2010;24:2382–91.
Kimura N, Miura Y, Nagatsu I, Nagura H. Catecholamine synthesizing enzymes in 70 cases of functioning and non-functioning phaeochromocytoma and extra-adrenal paraganglioma. Virchows Arch A Pathol Anat Histopathol. 1992;421:25–32.
Whalen RK, Althausen AF, Daniels GH. Extra-adrenal pheochromocytoma. J Urol. 1992;147:1–10.
Lam AK. Update on adrenal tumours in 2017 world health organization (WHO) of endocrine tumours. Endocr Pathol. 2017;28:213–27.
Parikh PP, Rubio GA, Farra JC, Lew JI. Nationwide review of hormonally active adrenal tumors highlights high morbidity in pheochromocytoma. J Surg Res. 2017;215:204–10.
Zhang R, Gupta D, Albert SG. Pheochromocytoma as a reversible cause of cardiomyopathy: analysis and review of the literature. Int J Cardiol. 2017;249:319–23.
Else T, Greenberg S, Fishbein L. Hereditary paraganglioma-pheochromocytoma syndromes. In: Adam M, Ardinger H, Pagon R, editors. GeneReviews. Seattle: University of Washington; 2018. p. 1993–2020.
Geroula A, Deutschbein T, Langton K, Masjkur J, Pamporaki C, Peitzsch M, et al. Pheochromocytoma and paraganglioma: clinical feature-based disease probability in relation to catecholamine biochemistry and reason for disease suspicion. Eur J Endocrinol. 2019;181:409–20.
Neumann HPH, Young WF, Eng C. Pheochromocytoma and paraganglioma. N Engl J Med. 2019;381:552–65.
Nolting S, Ullrich M, Pietzsch J, Ziegler CG, Eisenhofer G, Grossman A, et al. Current management of pheochromocytoma/paraganglioma: a guide for the practicing clinician in the era of precision medicine. Cancers (Basel). 2019;11:1505.
Crona J, Taieb D, Pacak K. New perspectives on pheochromocytoma and paraganglioma: toward a molecular classification. Endocr Rev. 2017;38:489–515.
Castro-Vega LJ, Letouze E, Burnichon N, Buffet A, Disderot PH, Khalifa E, et al. Multi-omics analysis defines core genomic alterations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6044.
Jochmanova I, Pacak K. Genomic landscape of pheochromocytoma and paraganglioma. Trends Cancer. 2018;4:6–9.
Valdés N, Navarro E, Mesa J, Casterás A, Alcázar V, Lamas C, et al. RET Cys634Arg mutation confers a more aggressive multiple endocrine neoplasia type 2A phenotype than Cys634Tyr mutation. Eur J Endocrinol. 2015;172:301–7.
van der Harst E, de Herder WW, de Krijger RR, Bruining HA, Bonjer HJ, Lamberts SW, et al. The value of plasma markers for the clinical behaviour of phaeochromocytomas. Eur J Endocrinol. 2002;147:85–94.
Eisenhofer G, Lenders JW, Goldstein DS, Mannelli M, Csako G, Walther MM, et al. Pheochromocytoma catecholamine phenotypes and prediction of tumor size and location by use of plasma free metanephrines. Clin Chem. 2005;51:735–44.
Righini C, Pecher M, Halimi S, Magne JL, Reyt E. Malignant carotid paraganglioma. A case report. Ann Otolaryngol Chir Cervicofac. 2003;120:103–8.
van Duinen N, Corssmit EP, de Jong WH, Brookman D, Kema IP, Romijn JA. Plasma levels of free metanephrines and 3-methoxytyramine indicate a higher number of biochemically active HNPGL than 24-h urinary excretion rates of catecholamines and metabolites. Eur J Endocrinol. 2013;169:377–82.
Pacak K, Eisenhofer G, Ilias I. Diagnosis of pheochromocytoma with special emphasis on MEN2 syndrome. Hormones (Athens). 2009;8:111–6.
Van der Horst-Schrivers AN, Osinga TE, Kema IP, Van der Laan BF, Dullaart RP. Dopamine excess in patients with head and neck paragangliomas. Anticancer Res. 2010;30:5153–8.
Eisenhofer G, Lenders JW, Timmers H, Mannelli M, Grebe SK, Hofbauer LC, et al. Measurements of plasma methoxytyramine, normetanephrine, and metanephrine as discriminators of different hereditary forms of pheochromocytoma. Clin Chem. 2011;57:411–20.
Neumann HP, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA. 2004;292:943–51.
Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, et al. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8.
Jha A, de Luna K, Balili CA, Millo C, Paraiso CA, Ling A, et al. Clinical, diagnostic, and treatment characteristics of SDHA-related metastatic pheochromocytoma and paraganglioma. Front Oncol. 2019;9:53.
Neary NM, King KS, Pacak K. Drugs and pheochromocytoma–don’t be fooled by every elevated metanephrine. N Engl J Med. 2011;364:2268–70.
Weise M, Merke DP, Pacak K, Walther MM, Eisenhofer G. Utility of plasma free metanephrines for detecting childhood pheochromocytoma. J Clin Endocrinol Metab. 2002;87:1955–60.
Casey R, Griffin TP, Wall D, Dennedy MC, Bell M, O’Shea PM. Screening for phaeochromocytoma and paraganglioma: impact of using supine reference intervals for plasma metanephrines with samples collected from fasted/seated patients. Ann Clin Biochem. 2017;54:170–3.
Darr R, Kuhn M, Bode C, Bornstein SR, Pacak K, Lenders JWM, et al. Accuracy of recommended sampling and assay methods for the determination of plasma-free and urinary fractionated metanephrines in the diagnosis of pheochromocytoma and paraganglioma: a systematic review. Endocrine. 2017;56:495–503.
Eisenhofer G, Goldstein DS, Walther MM, Friberg P, Lenders JW, Keiser HR, et al. Biochemical diagnosis of pheochromocytoma: how to distinguish true- from false-positive test results. J Clin Endocrinol Metab. 2003;88:2656–66.
van Berkel A, Lenders JW, Timmers HJ. Diagnosis of endocrine disease: biochemical diagnosis of phaeochromocytoma and paraganglioma. Eur J Endocrinol. 2014;170:R109–19.
van Berkel A, Rao JU, Kusters B, Demir T, Visser E, Mensenkamp AR, et al. Correlation between in vivo 18F-FDG PET and immunohistochemical markers of glucose uptake and metabolism in pheochromocytoma and paraganglioma. J Nucl Med. 2014;55:1253–9.
Lenders JW, Pacak K, Huynh TT, Sharabi Y, Mannelli M, Bratslavsky G, et al. Low sensitivity of glucagon provocative testing for diagnosis of pheochromocytoma. J Clin Endocrinol Metab. 2010;95:238–45.
Olson SW, Yoon S, Baker T, Prince LK, Oliver D, Abbott KC. Longitudinal plasma metanephrines preceding pheochromocytoma diagnosis: a retrospective case-control serum repository study. Eur J Endocrinol. 2016;174:289–95.
Algeciras-Schimnich A, Preissner CM, Young WF, Singh RJ, Grebe SK. Plasma chromogranin A or urine fractionated metanephrines follow-up testing improves the diagnostic accuracy of plasma fractionated metanephrines for pheochromocytoma. J Clin Endocrinol Metab. 2008;93:91–5.
Rao D, Peitzsch M, Prejbisz A, Hanus K, Fassnacht M, Beuschlein F, et al. Plasma methoxytyramine: clinical utility with metanephrines for diagnosis of pheochromocytoma and paraganglioma. Eur J Endocrinol. 2017;177:103–13.
Ctvrtlik F, Koranda P, Schovanek J, Skarda J, Hartmann I, Tudos Z. Current diagnostic imaging of pheochromocytomas and implications for therapeutic strategy. Exp Ther Med. 2018;15:3151–60.
Rogowski-Lehmann N, Geroula A, Prejbisz A, Timmers H, Megerle F, Robledo M, et al. Missed clinical clues in patients with pheochromocytoma/paraganglioma discovered by imaging. Endocr Connect. 2018;7:1168–77.
Havekes B, King K, Lai EW, Romijn JA, Corssmit EP, Pacak K. New imaging approaches to phaeochromocytomas and paragangliomas. Clin Endocrinol (Oxf). 2010;72:137–45.
Leung K, Stamm M, Raja A, Low G. Pheochromocytoma: the range of appearances on ultrasound, CT, MRI, and functional imaging. AJR Am J Roentgenol. 2013;200:370–8.
Sargar KM, Khanna G, Hulett BR. Imaging of nonmalignant adrenal lesions in children. Radiographics. 2017;37:1648–64.
Taieb D, Hicks RJ, Hindie E, Guillet BA, Avram A, Ghedini P, et al. European association of nuclear medicine practice guideline/society of nuclear medicine and molecular imaging procedure standard 2019 for radionuclide imaging of phaeochromocytoma and paraganglioma. Eur J Nucl Med Mol Imaging. 2019;46:2112–37.
Mercado-Asis LB, Wolf KI, Jochmanova I, Taieb D. Pheochromocytoma: a genetic and diagnostic update. Endocr Pract. 2018;24:78–90.
Pappachan JM, Raskauskiene D, Sriraman R, Edavalath M, Hanna FW. Diagnosis and management of pheochromocytoma: a practical guide to clinicians. Curr Hypertens Rep. 2014;16:442.
Carty SE, Young WF. Paragangliomas: epidemiology, clinical presentation, diagnosis and histology. UptoDate. 2018
Kan Y, Zhang S, Wang W, Liu J, Yang J, Wang Z. (68)Ga-somatostatin receptor analogs and (18)F-FDG PET/CT in the localization of metastatic pheochromocytomas and paragangliomas with germline mutations: a meta-analysis. Acta Radiol. 2018;59:1466–74.
Bessell-Browne R, O’Malley ME. CT of pheochromocytoma and paraganglioma: risk of adverse events with i.v. administration of nonionic contrast material. AJR Am J Roentgenol. 2007;188:970–4.
Weingarten TN, Welch TL, Moore TL, Walters GF, Whipple JL, Cavalcante A, et al. Preoperative levels of catecholamines and metanephrines and intraoperative hemodynamics of patients undergoing pheochromocytoma and paraganglioma resection. Urology. 2017;100:131–8.
Heslin MJ, Liles JS, Moctezuma-Velazquez P. The use of telemedicine in the preoperative management of pheochromocytoma saves resources. Mhealth. 2019;5:27.
Berends AMA, Kerstens MN, Lenders JWM, Timmers H. Approach to the patient: perioperative management of the patient with pheochromocytoma or sympathetic paraganglioma. J Clin Endocrinol Metab. 2020;105:3088–102.
Álvarez-Morujo RJG-O, Ruiz MÁA, Belisario JDC, Guirado TM, Yurrita BS. Head and neck paragangliomas: experience in 126 patients with 162 tumours. Acta Otorrinolaringol. 2015;66:332–41.
Alvarez-Morujo RJ, Ruiz MA, Serafini DP, Delgado IL, Friedlander E, Yurrita BS. Management of multicentric paragangliomas: review of 24 patients with 60 tumors. Head Neck. 2016;38:267–76.
Shamblin WR, Reine WH, Sheps SG, Harrison EG. Carotid body tumor (chemodectoma). Clinicopathologic analysis of ninety cases. Am J Surg. 1971;122:732–9.
Prasad SC, Mimoune HA, Khardaly M, Piazza P, Russo A, Sanna M. Strategies and long-term outcomes in the surgical management of tympanojugular paragangliomas. Head Neck. 2016;38:871–85.
Shen WT, Grogan R, Vriens M, Clark OH, Duh QY. One hundred two patients with pheochromocytoma treated at a single institution since the introduction of laparoscopic adrenalectomy. Arch Surg. 2010;145:893–7.
Rossitti HM, Soderkvist P, Gimm O. Extent of surgery for phaeochromocytomas in the genomic era. Br J Surg. 2018;105:e84-98.
Vidal O, Delgado-Oliver E, Diaz Del Gobbo R, Hanzu F, Squarcia M, Martinez D, et al. Functional adrenal cortex preservation: a good reason for posterior retroperitoneal endoscopic approach. Cir Esp. 2018;96:488–93.
Zarnegar R, Kebebew E, Duh QY, Clark OH. Malignant pheochromocytoma. Surg Oncol Clin N Am. 2006;15:555–71.
Heger P, Probst P, Huttner FJ, Goossen K, Proctor T, Muller-Stich BP, et al. Evaluation of open and minimally invasive adrenalectomy: a systematic review and network meta-analysis. World J Surg. 2017;41:2746–57.
Del Moral JMV, Gonzalez JMR, Llorente PM, Martinez JMM, Barrasate ADLQ, Rodriguez AE, et al. Adrenal surgery in Spain: final results of a national survey. Cir Esp. 2011;89:663–9.
Walz MK, Alesina PF, Wenger FA, Koch JA, Neumann HP, Petersenn S, et al. Laparoscopic and retroperitoneoscopic treatment of pheochromocytomas and retroperitoneal paragangliomas: results of 161 tumors in 126 patients. World J Surg. 2006;30:899–908.
Conzo G, Tartaglia E, Gambardella C, Esposito D, Sciascia V, Mauriello C, et al. Minimally invasive approach for adrenal lesions: systematic review of laparoscopic versus retroperitoneoscopic adrenalectomy and assessment of risk factors for complications. Int J Surg. 2016;28(Suppl 1):S118–23.
Jansen TTG, Timmers H, Marres HAM, Kaanders J, Kunst HPM. Results of a systematic literature review of treatment modalities for jugulotympanic paraganglioma, stratified per Fisch class. Clin Otolaryngol. 2018;43:652–61.
Gigliotti MJ, Hasan S, Liang Y, Chen D, Fuhrer R, Wegner RE. A 10-year experience of linear accelerator-based stereotactic radiosurgery/radiotherapy (SRS/SRT) for paraganglioma: a single institution experience and review of the literature. J Radiosurg SBRT. 2018;5:183–90.
Giammarile F, Chiti A, Lassmann M, Brans B, Flux G. EANM EANM procedure guidelines for 131I-meta-iodobenzylguanidine 131I-mIBG) therapy. Eur J Nucl Med Mol Imasging. 2008;35:1039–47.
Kayano D, Kinuya S. Current consensus on I-131 MIBG therapy. Nucl Med Mol Imaging. 2018;52:254–65.
Baudin E, Habra MA, Deschamps F, Cote G, Dumont F, Cabanillas M, et al. Therapy of endocrine disease: treatment of malignant pheochromocytoma and paraganglioma. Eur J Endocrinol. 2014;171:R111–22.
van Hulsteijn LT, Niemeijer ND, Dekkers OM, Corssmit EP. (131)I-MIBG therapy for malignant paraganglioma and phaeochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf). 2014;80:487–501.
Carrasquillo JA, Pandit-Taskar N, Chen CC. I-131 metaiodobenzylguanidine therapy of pheochromocytoma and paraganglioma. Semin Nucl Med. 2016;46:203–14.
Pryma DA, Chin BB, Noto RB, Dillon JS, Perkins S, Solnes L, et al. Efficacy and safety of high-specific-activity (131)I-MIBG therapy in patients with advanced pheochromocytoma or paraganglioma. J Nucl Med. 2019;60:623–30.
Kong G, Grozinsky-Glasberg S, Hofman MS, Callahan J, Meirovitz A, Maimon O, et al. Efficacy of peptide receptor radionuclide therapy for functional metastatic paraganglioma and pheochromocytoma. J Clin Endocrinol Metab. 2017;102:3278–87.
Kolasinska-Cwikla A, Peczkowska M, Cwikla JB, Michalowska I, Palucki JM, Bodei L, et al. A clinical efficacy of PRRT in patients with advanced, nonresectable, paraganglioma-pheochromocytoma, related to SDHx gene mutation. J Clin Med. 2019;8:952.
Pang Y, Liu Y, Pacak K, Yang C. Pheochromocytomas and paragangliomas: from genetic diversity to targeted therapies. Cancers (Basel). 2019;11:436.
Ayala-Ramirez M, Feng L, Habra MA, Rich T, Dickson PV, Perrier N, et al. Clinical benefits of systemic chemotherapy for patients with metastatic pheochromocytomas or sympathetic extra-adrenal paragangliomas: insights from the largest single-institutional experience. Cancer. 2012;118:2804–12.
Niemeijer ND, Alblas G, van Hulsteijn LT, Dekkers OM, Corssmit EP. Chemotherapy with cyclophosphamide, vincristine and dacarbazine for malignant paraganglioma and pheochromocytoma: systematic review and meta-analysis. Clin Endocrinol (Oxf). 2014;81:642–51.
Hadoux J, Favier J, Scoazec JY, Leboulleux S, Al Ghuzlan A, Caramella C, et al. SDHB mutations are associated with response to temozolomide in patients with metastatic pheochromocytoma or paraganglioma. Int J Cancer. 2014;135:2711–20.
Tena I, Gupta G, Tajahuerce M, Benavent M, Cifrian M, Falcon A, et al. Successful second-line metronomic temozolomide in metastatic paraganglioma: case reports and review of the literature. Clin Med Insights Oncol. 2018;12:1179554918763367.
Pang Y, Lu Y, Caisova V, Liu Y, Bullova P, Huynh TT, et al. Targeting NAD(+)/PARP DNA repair pathway as a novel therapeutic approach to SDHB-mutated cluster I pheochromocytoma and paraganglioma. Clin Cancer Res. 2018;24:3423–32.
O’Kane GM, Ezzat S, Joshua AM, Bourdeau I, Leibowitz-Amit R, Olney HJ, et al. A phase 2 trial of sunitinib in patients with progressive paraganglioma or pheochromocytoma: the SNIPP trial. Br J Cancer. 2019;120:1113–9.
Jasim S, Suman VJ, Jimenez C, Harris P, Sideras K, Burton JK, et al. Phase II trial of pazopanib in advanced/progressive malignant pheochromocytoma and paraganglioma. Endocrine. 2017;57:220–5.
Pichun MEB, Edgerly M, Velarde M, Bates SE, Daerr R, Adams K, et al. Phase II clinical trial of axitinib in metastatic pheochromocytomas and paraganlgiomas (P/PG): preliminary results. J Clin Oncol. 2015;33:457.
Hadoux J, Terroir M, Leboulleux S, Deschamps F, Al Ghuzlan A, Hescot S, et al. Interferon-alpha treatment for disease control in metastatic pheochromocytoma/paraganglioma patients. Horm Cancer. 2017;8:330–7.
Kohlenberg J, Welch B, Hamidi O, Callstrom M, Morris J, Sprung J, et al. Efficacy and safety of ablative therapy in the treatment of patients with metastatic pheochromocytoma and paraganglioma. Cancers (Basel). 2019;11:195.
Kittah NE, Iñiguez-Ariza NM, Hamidi O, Tamhane S, Young WF Jr, Bancos I, et al. Malignant pheochromocytoma and paraganglioma: 272 patients over 55 years. J Clin Endocrinol Metab. 2017;102:3296–305.
Plouin PF, Amar L, Dekkers OM, Fassnacht M, Gimenez-Roqueplo AP, Lenders JW, et al. European society of endocrinology clinical practice guideline for long-term follow-up of patients operated on for a phaeochromocytoma or a paraganglioma. Eur J Endocrinol. 2016;174:G1-10.
van den Broek PJ, de Graeff J. Prolonged survival in a patient with pulmonary metastases of a malignant pheochromocytoma. Neth J Med. 1978;21:245–7.
Ayala-Ramirez M, Feng L, Johnson MM, Ejaz S, Habra MA, Rich T, et al. Clinical risk factors for malignancy and overall survival in patients with pheochromocytomas and sympathetic paragangliomas: primary tumor size and primary tumor location as prognostic indicators. J Clin Endocrinol Metab. 2011;96:717–25.
King KS, Prodanov T, Kantorovich V, Fojo T, Hewitt JK, Zacharin M, et al. Metastatic pheochromocytoma/paraganglioma related to primary tumor development in childhood or adolescence: significant link to SDHB mutations. J Clin Oncol. 2011;29:4137–42.
Schovanek J, Martucci V, Wesley R, Fojo T, Del Rivero J, Huynh T, et al. The size of the primary tumor and age at initial diagnosis are independent predictors of the metastatic behavior and survival of patients with SDHB-related pheochromocytoma and paraganglioma: a retrospective cohort study. BMC Cancer. 2014;14:523.
Janssen I, Blanchet EM, Adams K, Chen CC, Millo CM, Herscovitch P, et al. Superiority of [68Ga]-DOTATATE PET/CT to other functional imaging modalities in the localization of SDHB-associated metastatic pheochromocytoma and paraganglioma. Clin Cancer Res. 2015;21:3888–95.
Muth A, Crona J, Gimm O, Elmgren A, Filipsson K, Stenmark Askmalm M, et al. Genetic testing and surveillance guidelines in hereditary pheochromocytoma and paraganglioma. J Intern Med. 2019;285:187–204.
MacFarlane J, Seong KC, Bisambar C, Madhu B, Allinson K, Marker A, et al. A review of the tumour spectrum of germline succinate dehydrogenase gene mutations: Beyond phaeochromocytoma and paraganglioma. Clin Endocrinol. 2020;93:528–38.
Tufton N, Sahdev A, Drake WM, Akker SA. Can subunit-specific phenotypes guide surveillance imaging decisions in asymptomatic SDH mutation carriers? Clin Endocrinol (Oxf). 2019;90:31–46.
Castinetti F, Waguespack SG, Machens A, Uchino S, Hasse-Lazar K, Sanso G, et al. Natural history, treatment, and long-term follow up of patients with multiple endocrine neoplasia type 2B: an international, multicentre, retrospective study. Lancet Diabetes Endocrinol. 2019;7:213–20.
Casey R, Neumann HPH, Maher ER. Genetic stratification of inherited and sporadic phaeochromocytoma and paraganglioma: implications for precision medicine. Hum Mol Genet. 2020;29:R128–37.
Papathomas TG, Suurd DPD, Pacak K, Tischler AS, Vriens MR, Lam AK, et al. What have we learned from molecular biology of paragangliomas and pheochromocytomas? Endocr Pathol. 2021;32:134–53.
Bryant J, Farmer J, Kessler LJ, Townsend RR, Nathanson KL. Pheochromocytoma: the expanding genetic differential diagnosis. J Natl Cancer Inst. 2003;95:1196–204.
Müller U, Troidl C, Niemann S. SDHC mutations in hereditary paraganglioma/pheochromocytoma. Fam Cancer. 2005;4:9–12.
Bayley JP, Kunst HP, Cascon A, Sampietro ML, Gaal J, Korpershoek E, et al. SDHAF2 mutations in familial and sporadic paraganglioma and phaeochromocytoma. Lancet Oncol. 2010;11:366–72.
Burnichon N, Brière JJ, Libé R, Vescovo L, Rivière J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet. 2010;19:3011–20.
Gaal J, Burnichon N, Korpershoek E, Roncelin I, Bertherat J, Plouin PF, et al. Isocitrate dehydrogenase mutations are rare in pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1274–8.
Yao L, Barontini M, Niederle B, Jech M, Pfragner R, Dahia PLM. Mutations of the metabolic genes IDH1, IDH2, and SDHAF2 are not major determinants of the pseudohypoxic phenotype of sporadic pheochromocytomas and paragangliomas. J Clin Endocrinol Metab. 2010;95:1469–72.
Comino-Méndez I, Gracia-Aznárez FJ, Schiavi F, Landa I, Leandro-García LJ, Letón R, et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet. 2011;43:663–7.
Clark GR, Sciacovelli M, Gaude E, Walsh DM, Kirby G, Simpson MA, et al. Germline FH mutations presenting with pheochromocytoma. J Clin Endocrinol Metab. 2014;99:E2046–50.
Gupta G, Pacak K, Aace Adrenal Scientific Committee. Precision medicine: an update on genotype/biochemical phenotype relationships in pheochromocytoma/paraganglioma patients. Endocr Pract. 2017;23:690–704.
Zhuang Z, Yang C, Lorenzo F, Merino M, Fojo T, Kebebew E, et al. Somatic HIF2A gain-of-function mutations in paraganglioma with polycythemia. N Engl J Med. 2012;367:922–30.
Pacak K, Jochmanova I, Prodanov T, Yang C, Merino MJ, Fojo T, et al. New syndrome of paraganglioma and somatostatinoma associated with polycythemia. J Clin Oncol. 2013;31:1690–8.
Yang C, Sun MG, Matro J, Huynh TT, Rahimpour S, Prchal JT, et al. Novel HIF2A mutations disrupt oxygen sensing, leading to polycythemia, paragangliomas, and somatostatinomas. Blood. 2013;121:2563–6.
Adari H, Lowy DR, Willumsen BM, Der CJ, McCormick F. Guanosine triphosphatase activating protein (GAP) interacts with the p21 ras effector binding domain. Science. 1988;240:518–21.
Crona J, Verdugo AD, Maharjan R, Stalberg P, Granberg D, Hellman P, et al. Somatic mutations in H-RAS in sporadic pheochromocytoma and paraganglioma identified by exome sequencing. J Clin Endocrinol Metab. 2013;98:E1266–71.
Toledo RA, Qin Y, Cheng ZM, Gao Q, Iwata S, Silva GM, et al. Recurrent mutations of chromatin-remodeling genes and kinase receptors in pheochromocytomas and paragangliomas. Clin Cancer Res. 2016;22:2301–10.
Yang C, Zhuang Z, Fliedner SM, Shankavaram U, Sun MG, Bullova P, et al. Germ-line PHD1 and PHD2 mutations detected in patients with pheochromocytoma/paraganglioma-polycythemia. J Mol Med (Berl). 2015;93:93–104.
Cascon A, Comino-Mendez I, Curras-Freixes M, de Cubas AA, Contreras L, Richter S, et al. Whole-exome sequencing identifies MDH2 as a new familial paraganglioma gene. J Natl Cancer Inst. 2015;107:53.
Fishbein L, Khare S, Wubbenhorst B, DeSloover D, D’Andrea K, Merrill S, et al. Whole-exome sequencing identifies somatic ATRX mutations in pheochromocytomas and paragangliomas. Nat Commun. 2015;6:6140.
Pang Y, Gupta G, Yang C, Wang H, Huynh TT, Abdullaev Z, et al. A novel splicing site IRP1 somatic mutation in a patient with pheochromocytoma and JAK2(V617F) positive polycythemia vera: a case report. BMC Cancer. 2018;18:286.
Acknowledgements
The authors would like to thank Fernando Rico-Villademoros (COCIENTE SL, Madrid, Spain) for editorial assistance in the preparation of this manuscript. His participation has been funded by the participating scientific societies.
Funding
The Spanish Societies of Endocrinology and Nutrition (SEEN), Medical Oncology (SEOM), Medical Radiology (SERAM), Nuclear Medicine and Molecular Imaging (SEMNIM), Otorhinolaryngology (SEORL), Pathology (SEAP), Radiation Oncology (SEOR), and Surgery (AEC) equally funded this project.
Author information
Authors and Affiliations
Contributions
Conception and design: RGC, FH. Manuscript writing: All authors. Final approval of manuscript: All authors. Accountable for all aspects of the work: All authors.
Corresponding author
Ethics declarations
Conflict of interest
RGC has provided scientific advice and/or received honoraria or funding for continuous medical education from AAA, Advanz Pharma, Amgen, Bayer, BMS, HMP, Ipsen, Merck, Midatech Pharma, MSD, Novartis, PharmaMar, Pfizer, Pierre Fabre, Roche, Servier and Sanofi, and has received research support from Pfizer, MSD and BMS. EM-C has received honoraria for educational event from Medtronic Iberica S.A. CA-E has received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing or educational events and support for attending meetings and/or travel from AAA (Advanced Accelerator Aplications, a Novartis Company), Novartis, Ipsen and Recordati. She has participated Participation on a Data Safety Monitoring Board or Advisory Board of Recordati. She has received medical writing services from Ipsen. FAH has provided scientific advice and/or received honoraria or funding for continuous medical education from Ipsen, Novartis and Pfizer. FMT, MM-C, MR, IT, MA, MRB-C and CF-A have no conflicts of interest to declare.
Informed consent
Informed consent is not required for this type of study.
Ethical approval
Ethical approval is not required for this type of study.
Research involving human participants and/or animals
The manuscript does not contain any original study with human participants or animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Garcia-Carbonero, R., Matute Teresa, F., Mercader-Cidoncha, E. et al. Multidisciplinary practice guidelines for the diagnosis, genetic counseling and treatment of pheochromocytomas and paragangliomas. Clin Transl Oncol 23, 1995–2019 (2021). https://doi.org/10.1007/s12094-021-02622-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12094-021-02622-9