
Abstract
Connective tissue growth factor (CTGF/CCN2) is a member of the CCN family of matricellular proteins. Its expression is induced by a number of factors including TGF-β. It has been associated with fibrosis in various tissues including the kidney. Diabetic nephropathy (DN) develops in about 30% of patients with diabetes and is characterized by thickening of renal basement membranes, fibrosis in the glomerulus (glomerulosclerosis), tubular atrophy and interstitial fibrosis, all of which compromise kidney function. This review examines changes in CTGF expression in the kidney in DN, the effects they have on glomerular mesangial and podocyte cells and the tubulointerstitium, and how these contribute to driving fibrotic changes in the disease. CTGF can bind to several other growth factors modifying their function. CTGF is also able to interact with receptors on cells, including integrins, tyrosine receptor kinase A (TrkA), low density lipoprotein receptor-related protein (LRP) and heparan sulphate proteoglycans. These interactions, the intracellular signalling pathways they activate, and the cellular responses evoked are reviewed. CTGF also induces the expression of chemokines which themselves have pharmacological actions on cells. CTGF may prompt some responses by acting through several different mechanisms, possibly simultaneously. For example, CTGF is often described as an effector of TGF-β. It can promote TGF-β signalling by binding directly to the growth factor, promoting its interaction with the TGF-β receptor; by triggering intracellular signalling on binding the TrkA receptor, which leads to the transcriptional repression of Smad7, an inhibitor of the TGF-β signalling pathway; and by binding to BMP-7 whose own signalling pathway opposing TGF-β is inhibited, leading to enhanced TGF-β signalling.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Connective Tissue Growth factor (CTGF, CCN2) is a secreted 36-38 kDa protein and a prototypical member of the CCN (Cyr61/CTGF/Nov) family of matricellular proteins. Family members have four domains, each of which can bind several ligands. Amongst these are (a) other growth factors (e.g. TGF-β, BMP-4 and -7, IGF-1, VEGF), whose function is then modified; (b) cell surface proteins (e.g. integrins, LRP-1, heparan sulphate proteogycans, tyrosine receptor kinase A), through which intracellular signalling may be initiated; and (c) extracellular matrix proteins (e.g. fibronectin) which may act as a sink for CCN proteins and modify their turnover. With so many potential interactions it is predictable that CCN proteins will influence many different biological events (Leask and Abraham 2006). CTGF is typical in this respect. Many responses triggered by it, or by fragments derived proteolytically from it, have been described since it was discovered in 1991 (Bradham et al. 1991, de Winter et al. 2008)). However it was recognised early on that CTGF stimulates the production of extracellular proteins by mesenchymal cells and that its expression in such cells is induced by the profibrotic cytokine TGF-β (Frazier et al. 1996), suggesting that it may have an important role in fibrosis.
About 30-40% of diabetics develop diabetic nephropathy (DN). In this life threatening disease, the basement membranes of the glomeruli (filtration units of the kidney) and of the renal tubules draining the urinary filtrate from them become thickened. Later the mesangium, an extracellular matrix which supports the glomerular capillary loops, and the interstitial areas between the tubules become fibrosed (Mason and Wahab 2003). Expanding mesangial fibrosis “crushes” the capillaries compromising filtration, and expanding fibrosis in the interstitium correlates closely with declining renal function (Steffes et al. 1989, Gilbert and Cooper 1999). It is widely accepted that these processes are mediated by TGF-β (Sharma and Ziyadeh 1995, Border et al. 1996) whose expression is elevated in renal tissues in DN (Yamamoto et al. 1996). Treatment of diabetic mice (db/db) with a neutralising antibody to TGF-β1 attenuated glomerular hypertrophy and prevented increased expression of collagen and fibronectin and glomerular mesangial matrix expansion (Ziyadeh et al. 2000). However CTGF has many of the same profibrotic actions as TGF-β and may act as a mediator of TGF-β actions on mesenchymal cells (Grotendorst 1997). Indeed, treatment with a CTGF- antisense oligonucleotide attenuates progression of diabetic nephropathy in diabetic mice (Guha et al. 2007). Moreover high plasma CTGF levels are an independent predictor of end-stage renal disease and mortality in DN in human Type 1 diabetes (Nguyen et al. 2008a). Several publications have proposed that CTGF could be an alternative therapeutic target to TGF-β in DN (e.g. Crean et al. 2001, Wahab et al. 2001, Blom et al. 2002, Twigg and Cooper 2004).
Renal CTGF expression in diabetic nephropathy
Each glomerulus comprises a series of capillary loops, through which blood circulates, and is enclosed by a Bowman’s capsule. Its supporting core structure, the mesangium, is composed of mesangial cells surrounded by their extracellular matrix. The capillary lumens are lined with fenestrated endothelial cells sitting on a basement membrane – the glomerular basement membrane (gbm). Water and small solutes in the blood diffuse across the gbm into the urinary space on the abluminal side, forming the urinary filtrate. This side of the gbm is covered by the foot processes of podocytes, also termed glomerular visceral epithelial cells. The filtrate then passes into the tubular system where molecules are both absorbed from and added to the tubular fluid by the lining epithelial cells, finally forming the urine. The tubules occupy both outer cortical and inner medullary areas of the kidney and are juxtaposed to intertubular blood vessels and a small amount of interstitium which contains a few fibroblast-like cells. This tubulointerstitium accounts for approximately 90% of kidney volume (Border et al. 1996).
Only low levels of glomerular CTGF are found in normal human glomeruli, but both mRNA and protein levels of the factor increase during the early (incipient) stage of DN and continue to do so as the disease progresses. Both glomerular podocytes and mesangial cells express CTGF in DN (Adler et al. 2001, Wahab et al. 2005a). Similarly, glomerular CTGF mRNA and protein levels increase in murine models of DN, initially in podocytes and later in parietal epithelia (lining the Bowman’s capsule) and mesangial cells (Roestenberg at al. 2006). Distal to the glomerulus, CTGF protein levels are increased in proximal tubular epithelial cells (PTECs) in rodent models of DN in both type 1diabetes (STZ-rats after 30 weeks) and type 2 diabetes (db/db mouse) (Wang et al. 2001, Guha et al. 2007). Interestingly, CTGF mRNA expression was induced in mouse PTEC cultures by proximal tubular fluid collected by micropuncture from STZ-diabetic rats (Wang et al. 2001). PTEC’s would be exposed to low molecular weight growth factors in the tubular fluid in vivo, such as TGF-β originating in the glomerular ultrafiltrate. CTGF protein was also detected in distal tubules of diabetic rats but controls also stained positively (Wang et al. 2001) so it is not clear whether its level are increased in the disease state. Although it is well established that declining renal function during progressive human DN correlates with interstitial expansion and fibrosis (reviewed by Gilbert and Cooper 1999), few studies have specifically addressed the expression of CTGF in interstitial cells in DN. One reason for this may be because rodent models of DN show little or no interstitial fibrosis even though glomerular changes are well developed. However STZ-diabetic mice show some increase in CTGF mRNA in tubulointerstitial areas and in vessel walls (Roestenberg et al. 2006) and CTGF mRNA expression is elevated in human renal interstitial fibrosis in a variety of kidney diseases, including DN. The interstitial cells over-expressing CTGF were indentified as myofibroblasts by their co-expression of α-smooth muscle actin (Ito et al. 1998).
Increased CTGF expression in renal cells in DN is likely to be induced by several different factors, including the high glucose environment, mechanical strain, TGF-β, Advanced Glycation End Product (AGE) – proteins and reactive oxygen species (ROS) ((Murphy et al. 1999, Riser et al. 2000, Wahab et al. 2001, Mason and Wahab 2003, Burns et al. 2006). AGE-proteins and ROS form in tissues in high glucose conditions and mesangial cells are subject to mechanical strain due to elevated systemic and intraglomerular pressure in diabetes (Forbes et al. 2007). CTGF is also able to auto-induce further CTGF gene expression in mesangial cells (Riser et al. 2000). It should be noted that one group reported a decrease in glomerular CTGF mRNA in DN in type 2 diabetes. This correlated with a decrease in a podocyte specific marker and mRNAs indicating podocyte loss (Baelde et al. 2007). Thus following increases in expression, overall glomerular levels of CTGF mRNA may decrease with increasing podocyte loss.
CTGF and mesangial cells
CTGF induces many responses in human mesangial cells in vitro which are relevant to the development of glomerulopathy in DN in vivo. It stimulates fibronectin and plasminogen activator inhibitor-1 synthesis, thus contributing to the mesangial extracellular matrix and, potentially, opposing turnover of the matrix since plasminogen activator is an important activator of matrix metalloproteinases (Wahab et al. 2001). CTGF also upregulates expression of α5β1 integrin, a plasma membrane anchor to which fibronectin must attach to permit the formation of an extracellular fibronectin fibrillar network. Increased levels of ligand-occupied β1 integrin appear on the cell surface in cells treated with CTGF for 48 h, promoting both increased cell adhesion and mesangial matrix expansion (Weston et al. 2003). However over short periods (12 h) CTGF induced dissolution of mesangial cell focal adhesions and a transient disassembly of the actin cytoskeleton, which reformed after 24 h. In vitro, these cytoskeletal changes may facilitate increased mesangial cell migration which was maximal after 4 h of CTGF treatment (Crean et al. 2002). Interestingly mesangial cell migration in response to CTGF was abrogated in a high glucose (30 mM) environment. Migration requires activation of PKCζ and CTGF was unable to stimulate further increases in phosphorylation of PKCζ which was already highly phosphorylated after exposure to high glucose. Other CTGF responses remained intact in high glucose (Furlong et al. 2007). These results illustrate that some CTGF-induced cell responses are likely to be time and context dependent.
CTGF induces mesangial cell cycle arrest in late G1 phase. This occurs by induction of Cyclin D1, which initiates the G1 phase, and by stimulating the expression of the cyclin-dependent kinase inhibitors p15 INK4, p21Cip1 and p27Kip1 which bring about arrest in late G1. This arrest is accompanied by mesangial cell hypertrophy as the cells continue to synthesize proteins at a high level (Wahab et al. 2002). Furthermore, CTGF blocks mesangial cell death by inducing MAP kinase phosphatase-1(MKP-1), which prevents MAPK p38 phosphorylation and apoptosis (Wahab et al. 2007). Persistence of myofibroblast-like cells producing high levels of extracellular matrix proteins is one of the hallmarks of fibrosis, in contrast to apotosis of such cells in damaged tissue undergoing normal wound healing and repair. TGF-β also induces these changes in mesangial cells. However such TGF-β-induced effects are blocked by treating the cells with a CTGF-antisense oligonucleotide, or CTGF-siRNA. Since mesangial matrix expansion, mesangial cell hypertrophy, and prolonged cell survival, all occur in the mesangium in DN, it is likely that TGF-β plays a key role in orchestrating these events in the disease, but that CTGF facilitates its actions (Wahab et al. 2007), as discussed below.
CTGF and podocytes
The earliest sign of onset of DN is microalbuminuria. Glomerular permeability to plasma proteins increases progressively as the disease advances and persistent proteinuria may contribute to tubular damage further along the nephron (Strutz 2008). The change in permeability is due predominantly to podocyte injury and loss, and to changes in the slit diaphragms, regions of special protein composition between adjacent gbm attachment sites of podocyte foot processes (Wolf et al. 2005, Li et al. 2007). Recent studies of STZ-induced diabetes in mice which are transgenic for specific expression of CTGF in podocytes point to an important role for CTGF in microalbuminuria. After 12 weeks of diabetes CTGF protein levels were higher in podocytes of transgenic mice than in non-transgenic mice and urinary albumin excretion was enhanced significantly in the former compared to the latter. The increased albuminuria may relate to podocyte injury in the diabetic transgenics since their podocytes were vacuolated, reduced in number, and levels of podocin, a protein known to be critical for slit diaphragm function, were partly reduced (Yokoi et al. 2008). The biochemical mechanism underlying the link between podocyte CTGF expression and microalbuminuria is currently unknown. However it is noteworthy that whilst treatment of diabetic mice (db/db) with an anti-TGF-β antibody prevented increased kidney matrix protein expression and mesangial expansion, it did not reduce albuminuria (Ziyadeh et al. 2000), raising the possibility that CTGF may have a TGF-β-independent role in the latter. In agreement with this notion, CTGF antisense oligonucleotide treatment of db/db mice or STZ-induced diabetic mice did attenuate albuminuria (Guha et al. 2007).
Podocyte-specific CTGF-transgenic mice have two other interesting characteristics. First, endogenous glomerular CTGF mRNA levels are also enhanced in diabetic transgenic mice, but mainly in the mesangial area, suggesting induction of mesangial cell CTGF expression by transgenic–CTGF from podocytes. Thus CTGF expressed by one cell type may have paracrine effects on CTGF expression in another cell type. It was proposed that the enhanced mesangial expansion seen in these diabetic transgenics may be due to decreased glomerular MMP2 expression/activity and therefore decreased matrix degradation, rather than to enhanced matrix synthesis (Yokoi et al. 2008). Second, non-diabetic transgenic mice had normal renal histology even though their glomerular CTGF protein levels were five times higher than in non-transgenic mice. Thus either CTGF levels can vary widely without affecting tissues or, more likely, CTGF acts on some cells in concert with one or more other factors, for example TGF-β, and has no effect if such factors are absent.
CTGF and the tubulointerstitium
Tubular basement membrane thickening, like gbm thickening (Osterby 1972), occurs early in type1 diabetes. At a later stage tubular atrophy and loss may occur and the interstitium becomes fibrotic and greatly expanded (Fioretto and Maur 2007). This is due to excessive deposition of extracellular matrix by myofibroblastic type cells, probably accompanied by decreased matrix degradation. The underlying mechanisms of renal interstitial fibrosis have similarities in the different kidney diseases in which it develops (Nangaku 2004).
Tubular epithelial cells synthesize the matrix proteins of the tubular basement membrane. Proximal tubular epithelial cells not only express CTGF in DN but are also likely to be exposed to CTGF from the glomerulus. Small proteins like CTGF may filter across the gbm and be present in the tubular fluid in pharmacologically active concentrations (see Wang et al. 2001 for example). Mouse PTEC cultures treated with rCTGF increased expression of fibronectin mRNA, but not that of Collagen IIIα1 or collagen1α2 chains (Wang et al. 2001). This may be relevant to the mechanism of basement membrane thickening since fibronectin is a tbm component (Brees et al. 1995) whilst the collagens are components of the interstitial matrix. CTGF may also play a role in early gbm thickening in DN (Thompson et al. 2007).
Potentially, CTGF secreted by PTECs has important paracrine actions on renal interstitial fibroblasts in DN. rCTGF synergised with IGF-1 action on normal renal fibroblasts (NRK-49F cells), stimulating upregulation of collagen IIIα1 mRNA. IGF-1 alone had only a modest stimulatory action whilst CTGF alone had no effect (Wang et al. 2001). CTGF module 1 has a binding site for IGF-1 which is likely to be important in this synergistic action. Moreover Lam et al. (2003) showed that human renal interstitial fibroblasts synthesize both IGF-1 and CTGF and that their expression increased in hyperglycaemic conditions. This study also recorded that CTGF alone had no effect on collagen types I and III secretion by these cells but it enhanced that induced by IGF-1. It was also reported that an anti-CTGF neutralizing antibody reduced high glucose-induced secretion of types I and III collagen by the fibroblasts, indicating the importance of CTGF in interstitial matrix production by these cells in diabetic conditions. It seems probable that CTGF secreted into the interstitium by PTECs would contribute to this effect on fibroblasts.
Co-culture experiments of murine interstitial fibroblast monolayers with murine PTECs grown on cell culture inserts were used to directly test the paracrine effect of the latter. The inserts allow diffusion of PTEC-secreted products into the fibroblast culture. When fibroblasts alone were stimulated with low doses of TGF-β there was a modest increase in mRNA levels of proteins involved in fibrosis, namely collagen I α1, fibronectin (EIIIA form), PAI-1 and TIMP-1. However in the presence of both TGF-β and PTECs these fibroblast mRNA levels were greatly enhanced. The enhancement was blocked in the presence of a neutralizing anti-CTGF antibody (Okada et al. 2005). Since rCTGF stimulated increases in the same mRNA’s in the fibroblast cultures, the enhanced mRNA levels in the co-culture experiments could be due to either an additive effect from the PTEC CTGF, or to this CTGF promoting the effects of TGF-β (see below), or to both actions. Irrespective of this, the results demonstrate a paracrine action of epithelial cell CTGF on interstitial fibroblasts.
The origin of the myofibroblasts which lay down excessive extracellular matrix in interstitial fibrosis in DN and other renal diseases is not well understood. Some may derive from the small number of endogenous interstitial fibroblasts in normal kidneys which become activated by exogenous factors such as TGF-β, CTGF, and FGF-2, secreted by infiltrating inflammatory cells in disease (Strutz 2008). Normal rat kidney fibroblasts (NRK clone 49F) proliferate and differentiate to myofibroblasts (α-SMA positive and incorporate 3H-proline into collagen) in response to CTGF. The proliferative activity resides in the C-terminal domain of CTGF whilst the myofibroblast differentiation activity is in the N-terminal domain (Grotendorst and Duncan 2005). Interestingly another group reported that TGF-β induced proliferation and differentiation of NRK-49F cells to myofibroblasts and that this effect was greatly enhanced if CTGF was present. In these experiments CTGF alone had no effect (Yang et al. 2004). However Gao et al. (2008) reported that primary cultures of rat renal cortical fibroblasts also proliferate and up-regulate their collagen and fibronectin expression in response to CTGF alone.
Renal interstitial myofibroblasts may also arise from other cell types such as tubular epithelial cells, bone marrow stem cells, vascular pericytes and endothelial cells. In one study unilateral ureteric obstruction (UUO) was used to induce rapid renal interstitial fibrosis in transgenic reporter mice and bone marrow chimeras. 10 days after UUO 36% of interstitial fibroblasts (identified by FSP1 expression) were estimated to arise from tubular epithelial cells (tagged with lacZ expression) and15% from bone marrow cells (tagged with GFP expression), the latter being only 3% more than proportionately found in the contralateral unobstructed kidney (Iwano et al. 2002). Both AGE-albumin and TGF-β induced CTGF expression and EMT in a renal epithelial cell line (NRK-52E) in vitro. Partial knock down of CTGF with siRNA attenuated the induction of EMT by AGE or TGF-β in these cells (Burns et al. 2006). In contrast to EMT, a recent report focuses attention on vascular pericytes as a major source of myofibroblasts in the UUO model. Myofibroblasts were indentified by coll1a1-GFP expression (Lin et al. 2008). Interestingly AGE-protein induces CTGF (CCN2) and Cyr 61 (CCN1) expression in retinal pericytes and adenoviral mediated over-expression of both proteins in these cells induced de-adhesion, an event that was followed by apoptosis (Liu et al. 2008).The effect of CTGF on interstitial blood vessel pericytes is not known currently. However the larger interstitial blood vessels remain in human DN and could provide a source of pericytes, even though the number of small peritubular vessels is reduced (Lindenmeyer et al. 2007). A further recent study indicates that endothelial to mesenchymal transition (EndMT) may also contribute interstitial myofibroblasts in interstitial fibrosis. UUO was performed in Tie2-Cre;R26R-stop-EYFP mice in which cells of endothelial lineage are labelled with YFP. Substantial numbers of interstitial cells which were FSP1+ or α-SMA+ also co-expressed YFP(Zeisberg et al. 2008). Similarly 30-50% of cells which stained positive for these mesenchymal cell markers in either murine DN (STZ- induced in CD1 mice), or in Col4a3 knockout mice which develop interstitial fibrosis, also co-expressed the endothelial marker CD31 (Zeisberg et al. 2008). Whilst concerns remain that rodent models of interstitial fibrosis may not precisely mirror that in humans, overall these experiments point to remarkable plasticity of several cell types in the tubulointerstitium in renal disease. The exact role of CTGF in the transition of these various cell types to myofibroblasts remains poorly defined. Nevertheless it is noteworthy that CTGF-antisense oligonucleotide treatment dramatically attenuated the development of myofibroblasts and renal interstitial fibrosis in both the rat UUO model (Yokoi et al 2004).and in the remnant kidney following 5/6 nephrectomy in TGF-β transgenic mice (Okada et al 2005), indicating a key function for the factor in the pathogenic mechanism.
How does CTGF work?
It seems likely that CTGF exerts its effects on cells in several ways including (i) interaction via one of its domains with other bioactive proteins, modifying the latter’s action, (ii) interaction with one or more cell surface tyrosine kinase receptors, triggering intracellular signalling and downstream actions, (iii) interaction with cell surface integrins triggering intracellular signalling and downstream actions, (iv) inducing expression of other secreted molecules with biological actions such as chemokines. There is currently little information about whether one, or another, of these actions is relatively more important than others in DN, or in other disease or biological processes and more than one route of action may be operative in any particular response to CTGF. For example, CTGF promotes many profibrotic effects of TGF-β. It seems to do this by enhancing TGF-β-Smad2/3 signalling by several mechanisms. One mechanism may involve CTGF binding directly to TGF-β, facilitating the latter’s interaction with its receptors (Abreu et al 2002). Another mechanism, operative in mesangial cells, is by CTGF binding to the tyrosine receptor kinase complex TrkA/p75 NTR. This triggers intracellular signalling which results in the expression of TIEG-1(TGF-β immediate early gene-1), a known repressor of Smad7 transcription. The latter is a negative regulator of TGF-β signalling (Wahab et al 2005b, c). Thus this results in enhanced transcription of TGF-β responsive genes including those encoding extracellular matrix proteins, which could be important in mesangial expansion in DN. A third potential mechanism involves BMP-7, a factor which counteracts TGF-β – mediated fibrosis (Patel and Dressler 2005). Nguyen et al (2008b) showed that CTGF binds to BMP-7 with high affinity. Moreover renal cortical levels of CTGF were much higher in STZ-induced DN in CTGF +/+ mice than in CTGF+/- mice and DN changes were attenuated in the latter (Nguyen et al 2008b). Higher levels of CTGF were shown to inhibit BMP-7 signalling activity through the Smad1/5 pathway. Normally Smad5 signalling up-regulates Smad6, which in turn blocks phospho-Smad2/3 translocation to the nucleus, reducing TGF-β signalling (Patel and Dressler 2005). Thus with higher CTGF levels this would not occur, promoting TGF-β –phospho-smad2/3 signalling.
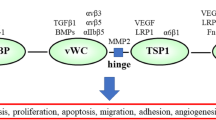
CTGF binds other growth factors including BMP-4, whose actions it antagonises by preventing BMP-4 - receptor interaction (Abreu et al 2002). Effects of another interaction, that between CTGF and IGF-1, were discussed above. CTGF also binds VEGF165, one of the commonly expressed splice variants of VEGF-A, interrupting its interaction with its receptor VEGFR2 and inhibiting angiogenesis (Inoki et al 2002). VEGF-A is expressed in podocytes and has autocrine actions on them. Its expression is increased in both STZ-diabetic rats and db/db mice and this promotes increased glomerular permeability since proteinuria was reduced by 50% or more by treatment with neutralizing anti-VEGF antibodies (reviewed by Wolf et al 2005). Interactions might conceivably occur between podocyte-derived CTGF and VEGF in early DN, perhaps opposing VEGF-driven increases in glomerular protein permeability. The autocrine VEGF-A - VEGFR2 loop also protects podocytes from apoptosis (Guan at al 2006) so the balance between podocyte derived VEGF-A and CTGF levels and the degree of interaction between the two factors is, potentially, also important in determining podocyte survival in DN. In contrast to rodent studies, glomerular levels of both VEGF-A and CTGF decreased in human DN which was attributed to podocyte loss (Baelde et al 2008). These results may reflect the much later time in the human disease when measurements were made.
CTGF and other members of the CCN family bind to a variety of integrins expressed by different cells, for example integrin αvβ3 on endothelial cells (Babic et al 1999), αIIbβ3 on activated platelets (Jedsadayanmata et al 1999), α6β1 on skin fibroblasts (Chen et al 2001), αvβ3 on hepatic stellate cells (Gao and Brigstock 2004), α5β1 on pancreatic stellate cells (Gao and Brigstock 2006) and α6β1 on gingival fibroblasts (Heng et al 2006). In doing so they activate focal adhesion kinase (FAK) and other intracellular signalling molecules (Chen et al 2001) and initiate a variety of biological responses, including cell adhesion, migration, changes in filopodia and lamellipodia, and extracellular matrix protein deposition. Frequently such responses depend on simultaneous binding of a heparan suphate proteoglycan co-receptor. CTGF signalling via integrin receptors clearly has a role in mesangial cells which may be relevant to the changes in DN. Crean et al (2002) reported that CTGF increased fibronectin expression in human mesangial cells in a β3 integrin-dependent manner which, via Src activation, stimulates p42/p44MAPK and PI3K activation. Neutralizing antibody to β3-integrin, or specific inhibition of MAPK, or of PI3K, all reduced the CTGF-induced fibronectin synthesis. Thus both the MAPK and the PI3K/PKB signalling pathways are required to mediate this effect of CTGF.
CTGF also induced a loss of focal adhesions in mesangial cells, but independently of β3 integrin -MAPK/PI3K signalling (Crean et al 2002). Dissolution of focal adhesions is a prerequisite of actin cytoskeletal reorganisation for cell migration. CTGF induces cell migration in vitro by dephosphorylation of focal adhesion kinase and paxillin, loss of Rho A activity, activation of Cdc 42 and phosphorylation of PKCζ and GSK-3β (Crean et al 2004). Mechanisms of mesangial cytoskeletal reorganisation are pertinent to understanding the pathobiology of DN since F-actin reorganisation occurs in these cells in a high glucose environment, mediated by PKCζ, with loss of their contractile response to vasoconstrictors (Dlugosz et al 2002). Moreover mesangial F-actin fibres undergo rearrangement in STZ-diabetic rats (Cortes et al 2000). It was proposed that during dissolution of the focal adhesion complex, FAK and paxillin dephosphorylation may depend on the protein tyrosine phosphatase SHP-2 whose activity was elevated in the CTGF–treated mesangial cells (Crean et al 2004). The TrkA/p75 neurotrophin receptor is expressed in mesangial cells and is activated by CTGF (Wahab et al 2005b). Interestingly, when TrkA was activated in PC12 pheochromocytoma cells by its established ligand, NGF, SHP-2 rapidly and transiently associated with it, was itself activated by phosphorylation, and was then found associated with multiple phospho-proteins (Goldsmith and Koizumi 1997). This raises the speculative question as to whether a TrkA –SHP-2 pathway could also be involved in mediating CTGF- driven dissolution of focal adhesions in mesangial cells. Crean et al (2006) went on to show how CTGF promotes the depolymerization of F-actin in mesangial cells and proposed that following CTGF interaction with unspecified cell surface receptors, the PI3K-PKB pathway is activated. Again, CTGF-TrkA interaction could be involved since it also activates the PI3K-PKB pathway (Wahab et al 2005b). PKB then phosphorylates p27Kip1 at ser10 and thr157. As a result p27Kip1 translocates from the nucleus to the cytoplasm where it binds to and inactivates RhoA, uncoupling it from the LIM kinase/cofilin system which regulates actin polymerization/depolymerization. This role of p27Kip1 is quite separate from its role as a cyclin-dependant kinase inhibitor referred to above in the discussion of CTGF-induced mesangial cell hypertrophy.
Tyrosine receptor kinase A (TrkA) and p75NTR form a dual receptor which transduces nerve growth factor - initiated signals in both neuronal and non- neuronal cells. This receptor is expressed on mesangial cells and, in response to CTGF, TrkA rapidly autophosphorylates several tyrosine residues (490,674/675), triggering association with several effectors and activating several signalling pathways. TrkA/ p75NTR interaction with CTGF was confirmed by cross-linking experiments. Moreover, K252a, a selective inhibitor of TrkA kinase activity (Tapley et al 1992), blocked the rapid phosphorylation of p42/ p44 MAPK (ERK1, 2), JNK, MAPK7 (ERK5) and PKB (Akt) in mesangial cells treated with CTGF, inferring that these signalling kinases and their associated pathways are activated through TrkA (Wahab et al 2005b). CTGF exerts its effects through TrkA-initiated signalling pathways in several ways, each of which is blocked by K252a. First, responses such as upregulated expression of the transcription factor TIEG-1 are a direct result of this signalling (Wahab et al 2005b, 2007). Second, CTGF-enhanced expression of TGF-β responsive genes such as Col III and PAI1 results from a stimulatory effect on the TGFb-Smad signalling pathway. Ligation of TrkA by CTGF initiates MAPK signalling, TIEG-1 expression and transcriptional repression of Smad 7, as discussed above. This increases the levels of phospho-smad2/3 and their nuclear translocation, stimulating transcription of genes having smad binding elements (SBE) in their promoters (Wahab et al 2005c). Third, CTGF induces expression of pharmacologically active proteins in mesangial cells such as MCP-1, RANTES and fracktalkine, these chemokines having important roles in leukocyte chemotaxis and trafficking in inflammation (Wu et al 2008). There are emerging views that inflammation and chemokines play important roles in the pathobiology of DN (Mora and Navarro 2006). Thus, overall, ligation of TrkA by CTGF is likely to be of key importance in the development of the disease.
The low density lipoprotein receptor-related protein (LRP)/α2 –macroglobulin receptor (α2MR) was identified as a CTGF receptor in bone marrow stromal cells (BMS2) following chromatography of solubilized cell membranes on a CTGF-Sepharose affinity column (Segarini et al 2001). The authors confirmed binding of CTGF to this ~620 kDa member of the LDL receptor family by competition studies with known ligands, immunoprecipitation of CTGF-LRP complex with LRP antibodies, and by showing that CTGF could bind LRP+/+ and LRP+/- mouse embryonic fibroblast cells, but not LRP-/- cells. However they were unable to detect phosphorylation of LRP in cells treated with CTGF. LRP was also confirmed as a receptor for adhesion of activated hepatic stellate cells to CTGF (Gao and Brigstock 2003). Subsequently Yang et al (2004) reported that CTGF induced phosphorylation of the cytoplasmic domain of LRP in a renal fibroblast cell line (NRK-49F). Unlike TGF-β, CTGF was unable to stimulate differentiation of these cells to myofibroblasts. However it increased TGF-β-stimulated differentiation, including the level of α-SMA expression (see above). Receptor associated protein (RAP), a known inhibitor of LRP, blocked CTGF-induced phosphorylation of LRP and reduced the increased α-SMA expression seen with co-stimulation with TGF-β and CTGF. These experiments suggest a possible role for CTGF binding to the LRP receptor in interstitial fibrosis in DN. CTGF also binds the Wnt co-receptor LRP-6 in Xenopus embryos and inhibits Wnt signalling (Mercurio et al 2004). The significance of this interaction has not been explored in mammalian renal cells but it is known that high glucose downregulates Wnt signalling in mesangial cells and that this is proapoptotic (Lin et al 2006).
The C-terminal domain (domain 4), and possibly other domains, of CTGF contain a heparin/heparan sulphate binding site which enables it to ligate cell surface heparan sulphate proteoglycans (HSPGs) (Gao and Brigstock 2004). HSPGs act as co-receptors when CTGF binds to receptors such as the αVβ3 integrin, expressed by activated hepatic stellate cells (Gao and Brigstock 2004). This was shown by prior treatment of cells with heparitinase1, an enzyme which specifically degrades the heparan sulphate side chains of HSPG, or by first culturing cells with sodium chlorate, a treatment which interferes with the sulphation of the heparan sulphate chains. In either case the cells were then unable to adhere to CTGF. Binding of CTGF to the LRP receptor is also heparin-dependent (Gao and Brigstock 2003). Current data indicate no structural changes in glomerular cell -associated heparan sulphate in DN (Van den Born et al 2006, Lauer et al 2007) so it seems likely that CTGF should be able to interact normally with HSPG co-receptors in DN.
As discussed above CTGF interacts with several different classes of receptor including integrins, TrkA, LRP and HSPGs. One intriguing speculation is that CTGF-TrkA/p75NTR signalling may require ligation of other cell surface proteins following clustering in the plasma membrane. In an analogous situation, oligodendrocye α6β1integrins cluster in lipid rafts containing PDGFα -receptor on binding their ligand, laminin-2. This enables PDGF to generate a signal within the raft, leading to PI3K/Akt activitation and cell survival (Baron et al 2003). Moreover, TrkA translocates to lipid rafts in PC12 cells on binding NGF, forming receptor-associated signalling complexes which result in ERK1/2 activation (Limpert et al 2007).Could it be that CTGF binds to integrins, HSPGs and TrkA/p75NTR receptors in mesangial cell membranes, promoting clustering into lipid rafts to generate intracellular signaling? Blocking the integrin or HSPG interaction may then prevent CTGF signalling via the neurotrophin receptor. Clearly, much further research is needed before we gain a fuller understanding of how CTGF achieves its effects and how we might modulate them therapeutically in disease states such as DN.
References
Abreu JG, Ketpura NI, Reversade B, De Robertis EM (2002) Connective–tissue growth factor(CTGF) modulates cell signalling by BMP and TGF-β. Nat Cell Biol 4:599–604
Adler SG, Shin-Wook K, Feld S, Cha DR, Barba L, Striker L, Striker G, Riser BL, LaPage J, Nast C (2001) Glomerular mRNAs in human type 1diabetes: Biochemical evidence for microalbuminuria as a manifestation of diabetic nephropathy. Kidney Int 60:2330–2336. doi:10.1046/j.1523-1755.2001.00073.x
Babic AM, Chen CC, Lau LF (1999) Fisp12/mouse connective tissue growth factor mediates endothelial cell adhesion and migration through integrin alphavbeta3, promotes endothelial cell survival, and induces angiogenesis in vivo. Mol Cell Biol 19:2958–2966
Baelde HJ, Eikmans M, Lappin DWP, Doran PP, Hohenadel D, Brinkkoetter P-T, van der Woude FJ, Waldeherr R, Rabelink TJ, de Heer E, Bruijin JA (2007) Reduction of VEGF-A and CTGF expression in diabetic nephropathy is associated with podocyte loss. Kidney Int 71:637–645. doi:10.1038/sj.ki.5002101
Baron W, Decker L, Colognato H, ffrench-Constant C (2003) Regulation of integrin growth factor interactions in oligodendrocytes by lipid raft microdomains. Curr Biol 13:151–155. doi:10.1016/S0960-9822(02) 01437-9
Blom IE, Goldschmeding R, Leask A (2002) Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy? Matrix Biol 21:473–482. doi:10.1016/S0945-053X(02) 00055-0
Border WA, Yahamoto T, Noble NA (1996) Transforming growth factor–β in diabetic nephropathy. Diabetes Metab Rev 12:309–339. doi:10.1002/(SICI) 1099-0895(199612) 12:4<309::AID-DMR171>3.0.CO;2-A
Bradham DM, Igarashi A, Potter RL, Grotendorst GR (1991) Connective tissue growth factor: a cysteine-rich mitogen secreted by human vascular endothelial cells is related to the SRC-induced immediate early gene product CEF-10. J Cell Biol 114:1285–1294. doi:10.1083/jcb.114.6.1285
Brees DK, Ogle RC, Williams JC (1995) Laminin and fibronectin content of mouse glomerular and tubular basement membrane. Ren Physiol Biochem 18:1–11. doi:10.1159/000173893
Burns WC, Twigg SM, Forbes JM, Pete J, Tikellis C, Thallas-Bonke V, Thomas MC, Cooper ME, Kantharidis P (2006) Connective tissue growth factor plays an important role in advanced glycation end product-induced tubular epithelial-to-mesenchymal transition: implications for diabetic renal disease. J Am Soc Nephrol 17:2484–2494. doi:10.1681/ASN.2006050525
Chen CC, Chen N, Lau LF (2001) The angiogenic factors Cyr61 and connective tissue growth factor induce adhesive signalling in primary human skin fibroblasts. J Biol Chem 276:10443–10452. doi:10.1074/jbc.M008087200
Cortes P, Mendez M, Riser BL, Guerin CJ, Rodriguez-Barbero A, Hassett C, Yee J (2000) F-actin fibre distribution in glomerular cells: structural and functional implications. Kidney Int 58:2452–2461. doi:10.1046/j.1523-1755.2000.00428.x
Crean JK, Lappin D, Godson C, Brady HR (2001) Connective tissue growth factor: an attractive therapeutic target in fibrotic renal disease. Expert Opin Ther Targets 5:519–530. doi:10.1517/14728222.5.4.519
Crean JKG, Finlay D, Murphy M, Moss C, Godson C, Martin F, Brady HR (2002) The role of p42/44 MAPK and protein kinase B in connective tissue growth factor induced extracellular matrix protein production, cell migration and actin cytoskeletal rearrangement in human mesangial cells. J Biol Chem 46:44187–44194. doi:10.1074/jbc.M203715200
Crean JK, Furlong F, Finlay D, Mitchell D, Conway B, Brady HR, Godson C, Martin F (2004) Connective tissue growth factor [CTGF]/CCN2 stimulates mesangial cell migration through integrated dissolution of focal adhesion complexes and activation of cell polarization. FASEB J 18:1541–1543
Crean JK, Furlong F, Mitchell D, McArdle E, Godson C, Martin F (2006) Connective tissue growth factor/CCN2 stimulates actin disassembly through Akt/protein kinase B-mediated phosphorylation and cytoplasmic translocation of p27Kip-1. FASEB J 20:E1037–E1048. doi:10.1096/fj.05-5010fje
De Winter P, Leoni P, Abraham D (2008) Connective tissue growth factor: Structure–function relationships of a mosaic, multifunctional protein. Growth Factors 26:80–91. doi:10.1080/08977190802025602
Dlugosz JA, Munk S, Ispanovic E, Goldberg HJ, Whiteside CI (2002) Mesangial cell filamentous actin disassembly and hypocontractility in high glucose are mediated by PKC-zeta. Am J Physiol Renal Physiol 282:F151–F163
Fioretto P, Mauer M (2007) Histopathology of diabetic nephropathy. Semin Nephrol 27:195–207. doi:10.1016/j.semnephrol.2007.01.012
Forbes JM, Fukami K, Cooper ME (2007) Diabetic nephropathy: where haemodynamics meets metabolism. Exp Clin Endocrinol Diabetes 115:69–84. doi:10.1055/s-2007-949721
Frazier K, Williams S, Kothapalli D, Klapper H, Grotendorst GR (1996) Stimulation of fibroblast cell growth, matrix production and granulation tissue by connective tissue growth factor. J Invest Dermatol 107:404–411. doi:10.1111/1523-1747.ep12363389
Furlong F, Crean J, Thornton L, O’Leary R, Murphy M, Martin F (2007) Dysregulated intracellular signalling impairs CTGF-stimulated responses in human mesangial cells exposed to high extracellular glucose. Am J Physiol Renal Physiol 292:F1691–F1700. doi:10.1152/ajprenal.00342.2006
Gao R, Brigstock DR (2003) Low density lipoprotein receptor-related protein (LRP) is a heparin-dependant adhesion receptor for connective tissue growth factor (CTGF) in rat activated hepatic stellate cells. Hepatol Res 27:214–220. doi:10.1016/S1386-6346(03) 00241-9
Gao R, Brigstock DR (2004) Connective tissue growth factor (CCN2) induces adhesion of rat activated hepatic stellate cells by binding of its C-terminal domain to integrin alpha(v) beta(3) and heparan sulphate proteoglycan. J Biol Chem 279:8848–8855. doi:10.1074/jbc.M313204200
Gao R, Brigstock DR (2006) A novel integrin alpha5beta1 binding domain in module 4 of connective tissue growth factor (CCN2/CTGF) promotes adhesion and migration of activated pancreatic stellate cells. Gut 55:856–862. doi:10.1136/gut.2005.079178
Gao X, Jinqzi L, Huang H, Li X (2008) Connective tissue growth factor stimulates renal cortical myfibroblast-like cell proliferation and matrix production. Wound Repair Regen 16:408–415. doi:10.1111/j.1524-475X.2008.00380.x
Gilbert RE, Cooper ME (1999) The tubulointerstitium in progressive diabetic kidney disease: More than an aftermath of glomerular injury. Kidney Int 56:1627–1637. doi:10.1046/j.1523-1755.1999.00721.x
Goldsmith B, Koizumi S (1997) Transient association of the phosphotyrosine phosphatase SHP-2 with TrkA is induced by nerve growth factor. J Neurochem 69:1014–1019
Grotendorst GR (1997) Connective tissue growth factor: a mediator of TGF-beta action on fibroblasts. Cytokine Growth Factor Rev 8:171–179. doi:10.1016/S1359-6101(97) 00010-5
Grotendorst GR, Duncan MR (2005) Individual domains of connective tissue growth factor regulate fibroblast proliferation and myofibroblast differentiation. FASEB J 19:729–738. doi:10.1096/fj.04-3217com
Guan F, Villegas G, Teichman J, Mundel P, Tufro A (2006) Autocrine VEGF-A system in podocytes regulates podocin and its interaction with CD2AP. Am J Physiol Renal Physiol 291:F422–F428. doi:10.1152/ajprenal.00448.2005
Guha M, Xu Z-G, Tung D, Lanting L, Natatajam R (2007) Specific down-regulation of connective tissue growth factor attenuates progression of nephropathy in mouse models of type 1 and type 2 diabetes. FASEB J 21:3355–3368. doi:10.1096/fj.06-6713com
Heng EC, Huang Y, Jn Black SA, Trackman PC (2006) CCN2, connective tissue growth factor, stimulates collagen deposition by gingival fibroblasts via module3 and alpha6- and beta1 integrins. J Cell Biochem 98:409–420. doi:10.1002/jcb.20810
Inoki I, Shiomi T, Hashimoto G, Enomoto H, Nakamura H, Makino K-I, Ikeda E, Takata S, Kobayashi K-I, Okada Y (2002) Connective tissue growth factor binds vascular endothelial growth factor (VEGF) and inhibits VEGF-induced angiogenesis. FASEB J 16:219–221
Ito Y, Aten J, Bende RJ, Oemar BS, Rabelink TJ, Weening JJ, Goldschmeding R (1998) Expression of Connective tissue growth factor in human renal fibrosis. Kidney Int 53:853–861. doi:10.1111/j.1523-1755.1998.00820.x
Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG (2002) Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest 110:341–350
Jedsadayanmata A, Chen CC, Kireeva ML, Lau LF, Lam SC (1999) Activation-dependent adhesion of human platelets to Cyr61 and Fisp12/mouse connective tissue growth factor is mediated through integrin alpha(IIb) beta(3). J Biol Chem 274:24321–24327. doi:10.1074/jbc.274.34.24321
Lam S, van der Geest RN, Verhagen AM, van Nieuwenhoven FA, Blom I, Aten J, Goldschmeding R, Daha MR, van Kootem C (2003) Connective tissue growth factor and IGF-1 are produced by human renal fibroblasts and cooperate in the induction of collagen production by high glucose. Diabetes 52:2975–2983. doi:10.2337/diabetes.52.12.2975
Lauer ME, Hascall VC, Wang A (2007) Heparan sulfate analysis from diabetic rat glomeruli. J Biol Chem 282:843–852. doi:10.1074/jbc.M608823200
Leask A, Abraham DJ (2006) All in the CCN family: essential matricellular signalling modulators emerge from the bunker. J Cell Sci 119:4803–4810. doi:10.1242/jcs.03270
Li JJ, Kwak SJ, Jung DS, Kim J-J, Yoo T-H, Ryu D-R, Han SH, Choi HY, Lee JE, Moon SJ, Kim DK, Han DS, Kang S-W (2007) Podocyte biology in diabetic nephropathy. Kidney Int 72:s536–s542. doi:10.1038/sj.ki.5002010
Limpert AS, Karlo JC, Landreth GE (2007) Nerve growth factor stimulates the concentration of TrkA within lipid rafts and extracellular signal-regulated kinase activation through c-Cbl-associated protein. Mol Cell Biol 27:5686–5698. doi:10.1128/MCB.01109-06
Lin CL, Wang JY, Huang YT, Kuo YH, Surendran K, Wang FS (2006) Wnt/beta-catenin signalling modulates survival of high glucose-stressed mesangial cells. J Am Soc Nephrol 17:2812–2820. doi:10.1681/ASN.2005121355
Lin S-L, Kisseleva T, Brenner DA, Duffield JS (2008) Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173:1617–1627. doi:10.2353/ajpath.2008.080433
Lindenmeyer MT, Kretzler M, Boucherot A, Berra S, Yasuda Y, Henger A, Eichinger F, Gaiser S, Schmid H, Rastaldi MP, Schrier RW, Schlödorff D, Cohen CD (2007) Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol 18:1765–1776. doi:10.1681/ASN.2006121304
Liu H, Yang R, Tinner B, Choudhry A, Schutze N, Chaqour B (2008) Cysteine-rich protein61 and connective tissue growth factor induce deadhesion and anoikis of retinal pericytes. Endocrinology 149:1666–1677. doi:10.1210/en.2007-1415
Mason RM, Wahab NA (2003) Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol 14:1358–1373. doi:10.1097/01.ASN.0000065640.77499.D7
Mercurio S, Latinkic B, Itasaki N, Krumlauf R, Smith JC (2004) Connective-tissue growth factor modulates WNT signalling and interacts with the WNT receptor complex. Development 131:2137–2147. doi:10.1242/dev.01045
Mora C, Navarro JF (2006) Inflammation and diabetic nephropathy. Curr Diab Rep 6:463–468. doi:10.1007/s11892-006-0080-1
Murphy M, Godson C, Cannon S, Cato S, Mackenzie HS, martin F, Brady HR (1999) Suppression subtractive hybridisation identifies high glucose levels as a stimulus for expression of connective tissue growth factor and other genes in human mesangial cells. J Biol Chem 274:5830–5834. doi:10.1074/jbc.274.9.5830
Nangaku M (2004) Mechanisms of tubulointerstitial injury in kidney: Final common pathways to end-stage renal failure. Intern Med 43:9–17. doi:10.2169/internalmedicine.43.9
Nguyen TQ, Tranow L, Jorsal A, Oliver N, Roestenberg P, Ito Y, Parving H-H, Rossing P, van Nieuwenhoven FA, Goldschmeding R (2008a) Plasma connective tissue growth factor is an independent predictor of end-stage renal disease and mortality in type 1 diabetic nephropathy. Diabetes Care 31:1177–1182. doi:10.2337/dc07-2469
Nguyen TQ, Roestenberg P, van Nieuwenhoven FA, Bovenschen N, Li Z, Xu L, Oliver N, Aten J, Joles JA, Vial C, Brandan E, Lyons KM, Goldschmeding R (2008b) CTGF inhibits BMP-7 signalling in diabetic nephropathy. J Am Soc Nephrol 19:2098–2107. doi:10.1681/ASN.2007111261
Østerby R (1972) Morphometric studies of the peripheral glomerular basement membrane in early juvenile diabetes. 1.Development of initial basement membrane thickening. Diabetologia 8:84–92. doi:10.1007/BF01235631
Okada H, Kikuta T, Kobayashi T, Inoue T, Kanno Y, Takigawa M, Sugaya T, Kopp JB, Suzuki H (2005) Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrogenesis. J Am Soc Nephrol 16:133–143. doi:10.1681/ASN.2004040339
Patel SR, Dressler GR (2005) BMP-7 signaling in renal development and disease. Trends Mol Med 11:512–518. doi:10.1016/j.molmed.2005.09.007
Riser BL, Denichlo M, Cortes P, Baker C, Grondin JM, Yee J, Narins RG (2000) Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol 11:25–38. doi:10.1159/000017209
Roestenberg P, van Nieuwenhoven FA, Joles JA, Trischberger C, Martens PP, Oliver N, Aten J, Hoppener JE, Goldschmeding R (2006) Temporal expression profile and distribution pattern indicate a role of connective tissue growth factor in diabetic nephropathy in mice. Am J Physiol Renal Physiol 209:F1344–F1354. doi:10.1152/ajprenal.00174.2005
Segarini PR, Nesbitt JE, Li D, Hays LG, Yates JRIII, Carmichael DF (2001) The low density lipoprotein receptor-related protein/ α2–macroglobulin receptor is a receptor for connective tissue growth factor. J Biol Chem 44:40659–40667. doi:10.1074/jbc.M105180200
Sharma K, Ziyadeh FN (1995) Hyperglycemia and diabetic kidney disease. The case for transforming growth factor-β as a key mediator. Diabetes 44:439–446. doi:10.2337/diabetes.44.10.1139
Steffes MW, Osterby R, Chavers B, Mauer SM (1989) Mesangial expansion as a central mechanism for loss of kidney function in diabetic patients. Diabetes 38:1077–1081. doi:10.2337/diabetes.38.9.1077
Strutz FM (2008) EMT and proteinuria as progression factors. Kidney International epub Aug 20th
Tapley P, Lamballe F, Barbacid M (1992) K252a is a highly selective inhibitor of the tyrosine kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene 7:371–381
Thompson SE, Mclannan SV, Kirwan PD, Heffernan SJ, Hennessy A, Yue DK, Twigg SM (2007) Renal connective tissue growth factor correlates with glomerular basement membrane thickness and prospective albuminuria in a non-human primate model of diabetes: possible predictive marker for incipient diabetic nephropathy. J Diabetes Complications 22:284–294. doi:10.1016/j.jdiacomp. 2007.07.001
Twigg SM, Cooper ME (2004) The time has come to target connective tissue growth factor in diabetic complications. Diabetologia 47:965–968. doi:10.1007/s00125-004-1423-6
Van den Born J, Pisa B, Bakker MAH, Celie JWAM, Thomas S, Viberti GC, Kjellen L, Berden JHM (2006) No change in glomerular heparan sulphate structure in early human and experimental diabetic nephropathy. J Biol Chem 281:29606–29613. doi:10.1074/jbc.M601552200
Wahab NA, Yevdokimova N, Weston BS, Roberts T, Li XJ, Brinkman H, Mason RM (2001) Role of connective tissue growth factor in the pathogenesis of diabetic nephropathy. Biochem J 359:77–87. doi:10.1042/0264-6021:3590077
Wahab NA, Weston BS, Roberts T, Mason RM (2002) Connective tissue growth factor and regulation of the mesangial cell cycle: role in cellular hypertrophy. J Am Soc Nephrol 13:2437–2445. doi:10.1097/01.ASN.0000031828.58276.02
Wahab NA, Schaefer L, Weston BS, Yiannikouris O, Wright A, Babelova A, Schaefer R, Mason RM (2005a) Glomerular expression of thrombospondin -1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial responses to diabetic stimuli. Diabetologia 48:2650–2660. doi:10.1007/s00125-005-0006-5
Wahab NA, Weston BS, Mason RM (2005b) Connective tissue growth factor CCN2 interacts with and activates the tyrosine kinase receptor TrkA. J. Am. Soc. Nephrol.16: 340-351. See also correction J Am Soc 17:314
Wahab NA, Weston BS, Mason RM (2005b) Modulation of the TGFβ/Smad signalling pathway in mesangial cells by CTGF/CCN2. Exp Cell Res 307:305–314. doi:10.1016/j.yexcr.2005.03.022
Wahab NA, Cox D, Witherden A, Mason RM (2007) Connective tissue growth factor (CTGF) promotes activated mesangial cell survival via up-regulation of mitogen-activated protein kinase phosphatase-1 (MKP-1). Biochem J 406:131–138. doi:10.1042/BJ20061817
Wang S, DeNichilo M, Brubaker C, Hirschberg R (2001) Connective tissue growth factor in interstitial injury of diabetic nephropathy. Kidney Int 60:96–105. doi:10.1046/j.1523-1755.2001.00776.x
Weston BS, Wahab NA, Mason RM (2003) CTGF mediates TGF-beta-induced fibronectin matrix deposition by upregulating active alpha5beta1 integrin in human mesangial cells. J Am Soc Nephrol 14:601–610. doi:10.1097/01.ASN.0000051600.53134.B9
Wolf G, Chen S, Ziyadeh FN (2005) From the periphery of the glomerular capillary wall toward the centre of disease. Podocyte injury comes of age in diabetic nephropathy. Diabetes 54:1623–1634. doi:10.2337/diabetes.54.6.1626
Wu S-H, Lu C, Dong L, Chen Z-Q (2008) Signal transduction involved in CTGF-induced production of chemokines in mesangial cells. Growth Factors 26:192–200. doi:10.1080/08977190802227828
Yamamoto T, Noble NA, Cohen AH, Nast CC, Hishida A, Gold LI, Border WA (1996) Expression of transforming growth factor-β isoforms in human glomerular diseases. Kidney Int 49:461–469. doi:10.1038/ki.1996.65
Yang M, Huang H, Li J, Li D, Wang H (2004) Tyrosine phosphorylation of the LDL-receptor related protein and activation of the ERK pathway are required for connective tissue growth factor to potentiate myofibroblast differentiation. FASEB J 18:1920–1921
Yokoi H, Mukoyama M, Nagae T, Mori K, Suganami T, Sawai K, Yoshioka T, Koshikawa M, Nishida T, Sugawara A, Nakao K (2004) Reduction in connective tissue growth factor by antisense treatment ameliorates renal tubulointerstitial fibrosis. J Am Soc Nephrol 15:1430–1440. doi:10.1097/01.ASN.0000130565.69170.85
Yoki H, Mukoyama M, Mori K, Kasahara M, Suganami T, Sawai K, Yosioka T, Saito Y, Ogawa Y, Kuwabara T, Sugawara A, Nakao K (2008) Overexpression of connective tissue growth factor in podocytes worsens diabetic nephropathy in mice. Kidney Int 73:446–455. doi:10.1038/sj.ki.5002722
Zeisberg EM, Potenta SE, Zeisberg M, Kalluri R (2008) Fibroblasts in kidney fibrosis emerge via endothelial-to mesenchymal transition. J Am Soc Nephrol 19:2282–2287. doi:10.1681/ASN.2008050513
Ziyadeh FN, Hoffman BB, Han DC, Iglesias-De La Cruz MC, Hong SW, Isono M, Chen S, McGowan TA, Sharma K (2000) Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal anti-transforming growth factor-β antibody in db/db diabetic mice. Proc Natl Acad Sci USA 97:8015–8020. doi:10.1073/pnas.120055097
Acknowledgements
RMM acknowledges financial support from The Medical Research Council UK and Diabetes UK.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This is an open access article distributed under the terms of the Creative Commons Attribution Noncommercial License (https://creativecommons.org/licenses/by-nc/2.0), which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and source are credited.
About this article
Cite this article
Mason, R.M. Connective tissue growth factor(CCN2), a pathogenic factor in diabetic nephropathy. What does it do? How does it do it?. J. Cell Commun. Signal. 3, 95–104 (2009). https://doi.org/10.1007/s12079-009-0038-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12079-009-0038-6