
Abstract
Aims/hypothesis
We quantified the glomerular expression of thrombospondin-1 (THBS1, also known as TSP-1), transforming growth factor beta 1 (TGFB1, also known as TGF-β1) and connective tissue growth factor (CTGF) at each stage of diabetic nephropathy. We also examined the roles of THBS1 and CTGF in mediating high-glucose- and glycated-albumin-induced synthesis of the matrix protein, fibronectin, by mesangial cells.
Methods
THBS1, latent and active TGFB1, and CTGF, were detected by immunohistochemistry and in situ hybridisation in biopsies from 19 insulin-dependent diabetic patients with incipient, manifest and advanced diabetic nephropathy, and in 11 control kidneys. Findings were quantified by image analysis. Human mesangial cells were cultured with normal or high glucose, albumin or glycated albumin (Amadori product), +/−THBS1 or CTGF antisense oligonucleotides, or with peptide W, an inhibitor of TGFB1 bioactivation by THBS1. Proteins were measured by western blot analysis or ELISA.
Results
In glomeruli of normal kidneys, mRNA and protein levels for THBS1, latent-TGFB1 and CTGF were low. They were increased in the incipient stage of diabetic nephropathy, predominantly in mesangial areas, with further increases at later stages of the disease. Little or no active TGFB1 immunostaining was detected prior to manifest diabetic nephropathy. In contrast to high-glucose conditions, increases in fibronectin synthesis that were stimulated by glycated albumin were not dependent on THBS1 activation of latent TGFB1. However, increased fibronectin synthesis in both conditions required CTGF.
Conclusions/interpretation
Increased glomerular expression of all three factors occurs from the earliest stage of diabetic nephropathy. In contrast to THBS1, CTGF is required for mesangial synthesis of fibronectin stimulated by high glucose or glycated albumin, and is thus a potential therapeutic target.
Similar content being viewed by others

Introduction
Diabetic nephropathy is the leading cause of end-stage renal failure in the Western world [1–3]. Morgensen et al. have defined incipient, manifest and advanced stages of the disease, which usually develops over a period of years [4]. The later stages of diabetic nephropathy are accompanied by a pronounced increase in glomerular extracellular matrix, giving rise to diffuse or nodular glomerular sclerosis.
Prolonged hyperglycaemia stimulates the development of diabetic nephropathy in man and in animals [2, 3, 5] via (1) the effects of elevated glucose and its metabolites on cells, (2) hyperosmolarity, (3) oxidative stress, and (4) by the Amadori products and advanced glycated end-products, formed by non-enzymatic glycosylation of proteins [6]. Transforming growth factor beta 1 (TGFB1, also known as TGF-β) and connective tissue growth factor (CTGF) appear to be key players in the pathogenesis of diabetic nephropathy [7–9]. Both are upregulated by high glucose and by glycated proteins, and both promote mesangial cell hypertrophy and extracellular matrix accumulation [5, 10–13]. CTGF is rapidly induced by TGFB1 and appears to mediate some of the fibrogenic effects of this growth factor [9, 13–15], but no systematic study of its expression at the protein level, or the relationship of this to TGFB1 expression at various stages of diabetic nephropathy, has been performed.
TGFB1, synthesised as a latent precursor, must be activated to exert its effects [16]. Mesangial thrombospondin-1 (THBS1, also known as TSP-1) expression is increased in high-glucose conditions and is a key activator of latent TGFB1 (L-TGFB1) in vitro [17], and in mesangial proliferative glomerulonephritis in live rats [18]. Moreover, THBS1 null mice have a similar phenotype to TGFB1 null mice [19] suggesting that the THBS1 activation mechanism generally plays an important role in vivo. To achieve a better understanding of the relationship between THBS1, TGFB1 and CTGF in diabetic nephropathy, we investigated their expression levels in the glomerulus in the incipient, manifest and advanced stages of disease, and whether, in mesangial cells exposed to glycated proteins, THBS1-dependent activation of TGFB1 is required for CTGF induction and subsequent fibronectin expression, as occurs in high-glucose conditions.
Subjects, materials and methods
Materials
Human serum albumin (HSA) (99%, essentially fatty-acid- and globulin-free, cat. no. A3782) and glycated albumin (cat. no. A8301) were from Sigma (Poole, Dorset, UK). The glycated albumin was generated by incubating HSA with glucose for 6 days (information supplied by Sigma). The enzyme immunoassay kit for fibronectin was from Takara Biomedicals (Shiga, Japan). Assays were carried out in triplicate. Primary normal adult human mesangial cells (CC-2259, lot F1507) were purchased from Biowhittaker (Wokingham, Berkshire, UK), cultured [12], and used for experiments between passage numbers 9 and 10.
Tissues for histological investigations
Human kidney tissues were obtained from patients after they had given their written, informed consent and in compliance with permissions obtained from the local ethical committee. The study was approved by the Ethical Committee, Medical Faculty, University of Munster (registration number 178/98). For controls, tumour-free kidney tissues were obtained from 12 patients (five females and seven males) undergoing nephrectomy for renal carcinoma. Diabetic changes were examined in tissues from kidney biopsies of eight diabetic patients with no evidence of kidney cancer, and in tumour-free tissue of renal carcinoma kidneys from 14 patients with insulin-dependent diabetes mellitus. This provided kidney tissue from patients with incipient (n=3), manifest (n=13) and advanced diabetic nephropathy (n=6) (details, see Table 1), as described by Mogensen [4, 20]. From these tissues, RNase-free sections were available for ten control, two incipient, eight manifest, and four advanced diabetic nephropathy kidneys, for in situ hybridisation (ISH). All 22 diabetic patients were treated with an ACE inhibitor, Ramipril 5 mg per 24 h. If blood pressure control was not satisfactory, the loop diuretic torasemide, the β-receptor antagonist metoprolol and the dihydropyridin calcium channel blocker amlodipine were added one at a time, while keeping the dose of Ramipril constant. Although ACE inhibitors can be effective in reducing disease progression in diabetic nephropathy, the criterion for entry to the study was that the patient had a well-defined histological diagnosis of one of the stages of diabetic nephropathy (Fig. 1a–d).

Glomerular morphology of diabetic patients and non-diabetic control subjects. a Glomerulus from a non-diabetic control subject with normal cellularity and normal mesangium. b Glomerulus from a patient with incipient disease showing early, diffuse expansion of mesangial matrix, normal cellularity and vasculature. c Glomerulus with pronounced nodular and diffuse intercapillary sclerosis and somewhat reduced cellularity (from a patient with manifest nephropathy). d Glomerulus from a patient with end-stage disease showing advanced, diffuse and nodular sclerosis, reduced cellularity, and encroachment of the mesangium on capillary lumina. Magnification ×400
Tissues were fixed immediately with 4% formaldehyde in 18 mmol/l sodium phosphate, pH 7.4, containing 0.15 mol/l NaCl (PBS), dehydrated and embedded in paraffin wax under RNase-free conditions. Serial sections (2–6 μm) were stained with periodic acid–Schiff reaction to ascertain the absence of tumour infiltration and processed for immunohistochemistry or ISH.
Glomerular immunostaining and ISH were quantified by a computer-aided image analysis system (Soft Imaging System, Münster, Germany) where all glomeruli per section were analysed. The lowest number of glomeruli per section was four (in the case of biopsy material), the highest was 32 (in sections derived from tumour nephrectomies). Results were expressed as proportional area (percentage) of positive staining per glomerulus. Bowman’s capsule was not counted as a part of the glomerulus. All morphometric evaluations were performed by a blinded observer.
Western blotting
Cells were lysed in reducing SDS-PAGE loading buffer, scraped off the plate, sonicated for 10 s to shear DNA, boiled for 5 min, and resolved on 4–12% gradient gels by SDS-PAGE followed by western blotting [9].The primary antibodies were anti-human CTGF (1/2000; a kind gift from FibroGen, South San Francisco, CA, USA) and anti-human fibronectin (1/2000, F3648; Sigma) with a secondary horseradish peroxidase-conjugated antibody. Bound antibodies were visualised using Luminol (Autogen Bioclear, Calne, Wiltshire, UK).
In situ hybridisation
In situ hybridisation was performed as described previously [21]. The sense and antisense riboprobes were transcribed from linearised plasmids containing human thrombospondin-1 (662–1155 bp), TGFB1 (601–1582 bp) and CTGF (116–492 bp) cDNA, respectively, using digoxigenin-labelled UTP and an SP6/T7-RNA polymerase kit (Roche Diagnostics, Mannheim, Germany). Prehybridisation (2 h) was carried out in a humid chamber at 54°C (thrombospondin-1 and CTGF) or at 51°C (TGFB1). Hybridisation with the appropriate riboprobe at a final concentration of 7.5 ng/ml was carried out for 16 h at the same temperatures, respectively. All sections were hybridised with the sense and anti-sense riboprobes in parallel and under the same conditions.
Immunohistochemistry
The alkaline phosphatase anti-alkaline phosphatase technique was used as described previously [22]. Various procedures for demasking tissue antigens (enzymatically and by heating) were used prior to incubation with the first antibodies. The primary antibodies used were: mouse anti-human thrombospondin-1 (1:50; Calbiochem-Novabiochem, Bad Soden, Germany), rabbit anti-human CTGF [9] (1:1500; FibroGen), and an affinity-purified rabbit antibody to human L-TGFB1 (1:50; Santa Cruz, Heidelberg, Germany) raised against a peptide mapping to the carboxy terminus of the precursor form of human TGFB1. The active form of TGFB1 was detected with affinity-purified chicken anti-human TGFB1 (1:50, AF-101-NA; R&D Systems, Wiesbaden, Germany) raised against purified active rhTGFB1. This antibody neutralises TGFB1 inhibition of mouse IL-4-dependent [3H]-thymidine incorporation by HT-2 cells in a dose-dependent manner [23] and specifically recognises active TGFB1 in tissue sections without cross-reactivity to L-TGFB1 [24]. Secondary antibodies have been described previously [21]. The specificity of immunostaining was tested by omitting the primary antibody, by using non-immune serum-‘unspecific’ IgG, and by pre-absorption of appropriate antisera with rCTGF or with a synthetic TGFB1 peptide (Santa Cruz).
Confocal microscopy
Paraffin sections were incubated with mouse anti-human THBS1 antibody (1:50; Calbiochem-Novabiochem) and visualised by a Texas Red-conjugated goat anti-mouse IgG (1:500; Dianova, Hamburg, Germany). Sections were washed extensively with PBS, blocked with PBS/1% (w/v) BSA/20% (v/v) goat serum, and stained for α-smooth muscle actin (α-SMA; mesangial cell marker) with fluorescein-conjugated mouse anti-α-SMA antiserum (1:200, clone 1A4; Sigma). Colocalisation of THBS1 with Wilms’ tumour protein-1 (WT-1), a marker recognising a podocyte-specific nuclear epitope, was determined in frozen sections by application of the mouse anti-human thrombospondin-1 antibody (1:20) together with rabbit anti-human WT-1 (1:100; Santa Cruz). Unspecific staining was blocked with PBS/1% (w/v) BSA/20% (v/v) goat serum. For visualisation, Texas Red-conjugated goat anti-mouse IgG and fluorescein-conjugated goat anti-rabbit IgG were used (both 1:500; Dianova). Laser scan microscopy was carried out with a confocal microscope (PCM 2000; Nikon, Duesseldorf, Germany). Non-specific staining was determined by the use of secondary antibodies alone.
Other procedures
Serum creatinine levels were measured with an autoanalyser (Hitachi 747; Roche Diagnostics), serum HbA1c with a BN2 autoanalyser (Dade-Behring, Schwalbach, Germany) and albuminuria with the Merck-Hitachi L-9100 autoanalyser (Merck, Darmstadt, Germany).
Statistical analysis
Data are given as means±SEM and analysed by ANOVA, with Dunnett’s significance correction test (SPSS software), or by an unpaired Student’s t-test, as appropriate. Differences were considered significant at a pvalue of<0.05.
Results
Clinical characteristics and glomerular morphology of diabetic patients and non-diabetic control subjects
Patients were classified as controls or as having incipient, manifest or advanced diabetic nephropathy according to Mogensen [4, 20]. Classification was based on the duration of diabetic nephropathy, clinical signs and symptoms (Table 1) and renal morphology (Fig. 1a–d). The glomerular morphology shown in Fig. 1a–d was representative of the glomeruli depicted in Figs 2, 3, 4.

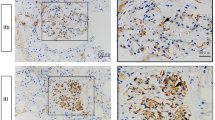
Glomerular thrombospondin-1 in the normal kidney and in kidneys from patients with incipient, manifest or advanced diabetic nephropathy. Immunohistochemistry (IHC) (a–e) and mRNA by in situ hybridisation (ISH) (f–j) are shown. Arrows point to THBS1 expression in the mesangium. Arrowheads point to THBS1 expression in podocytes. The negative controls for IHC (e) and ISH (j), shown in tissue sections from a patient with manifest diabetic nephropathy, were performed by omitting the primary antiserum (e) or using a digoxigenin-labelled sense riboprobe for THBS1 (j). Original magnification ×400

Precursor and active forms of TGFB1 in glomeruli of normal kidneys and in kidneys from patients at the disease stages indicated. Immunohistochemistry (IHC) for latent TGFB1 (a–e) and active forms of TGFB1 protein (f–j). Nodular changes (c, h) (manifest disease) are visible, as is diffuse glomerulosclerosis (d, i) (advanced disease). k–o In situ hybridisation (ISH) was used to detect TGFB1 mRNA in the same tissue samples. Arrows indicate mesangial cells; arrowheads indicate podocytes. The inset (g) shows strong staining for active TGFB1 in tubular epithelial cells as a positive control. The negative controls for IHC (e, j) and ISH (o), shown in tissue sections from a patient with manifest diabetic nephropathy, were performed by pre-adsorbing anti-precursor TGFB1 antiserum with a synthetic TGFB1 peptide (e), by omitting the primary antibodies (j), or by using a digoxygenin-labelled sense riboprobe for TGFB1 (o). Original magnification ×400

Glomerular CTGF in normal kidney and in renal biopsies from patients with incipient, manifest or advanced diabetic nephropathy. (a–e) Immunohistochemistry (IHC) for CTGF protein. (f–j) mRNA detected by in situ hybridisation (ISH). Nodular changes (c, h) are visible in manifest disease, as is diffuse glomerulosclerosis (d, i) in advanced disease. Arrows indicate mesangial cells; arrowheads indicate podocytes. Negative controls for IHC (e) and ISH (j), shown in tissue sections from a patient with manifest diabetic nephropathy, were performed by preabsorption of the anti-CTGF antiserum with rCTGF (e) or by using a digoxigenin-labelled sense riboprobe for CTGF (j). Original magnification ×400
THBS1, TGFB1 and CTGF in glomeruli in normal kidney and in diabetic nephropathy
THBS1 protein and mRNA are almost undetectable in the tuft of normal glomeruli, but are expressed in Bowman’s capsule (Fig. 2). Double-staining for THBS1 and α-SMA (mesangial marker), or WT-1 (podocyte marker), confirmed that only a few mesangial cells and podocytes within normal glomeruli show weak expression of THBS1 (not shown). In incipient diabetic nephropathy, intraglomerular cells show an increased in situ signal for THBS1 and immunostaining for the protein, mainly in mesangial areas, but also in a few podocytes. Bowman’s capsule continues to stain strongly (Fig. 2b,g). A similar pattern of THBS1 expression is evident in manifest diabetic nephropathy and in the advanced stages of the disease (Fig. 2c,d,h,i). Overall the results indicate that glomerular THBS1 expression increases with progression of diabetic nephropathy. Colocalisation of THBS1 with α-SMA provided further evidence of increased mesangial deposition of THBS1 protein in diabetic nephropathy (not shown). Only trace amounts of THBS1 were associated with podocytes. Semiquantification of glomerular immunostaining and ISH clearly indicate a progressive increase of THBS1 mRNA and the corresponding protein in the glomerular tuft with the advancement of diabetic nephropathy (Table 2).
No TGFB1 mRNA was detectable within the glomerulus in normal kidneys, but a weak signal associated with Bowman’s capsule was observed (Fig. 3k) as well as a stronger signal in certain tubular epithelial cells (not shown). The tubular epithelium also showed weak immunostaining for L-TGFB1, while glomerular structures were generally devoid of stain (Fig. 3a). Likewise there was no staining for active TGFB1 in the normal glomerulus and weak staining was only occasionally detected in tubular epithelial cells (Fig. 3f).
With the development of incipient diabetic nephropathy, the ISH signal for TGFB1 mRNA increased and clear immunostaining was seen for L-TGFB1 in mesangial cells and podocytes, as well as in tubular epithelial cells (Fig. 3b,l). However, there was very little immunostaining of active TGFB1 in the glomerulus at this stage (Fig. 3g). This contrasts with strong staining for the active factor in tubular epithelial cells in the same section (Fig. 3g, inset). With progression to manifest diabetic nephropathy, glomerular staining for TGFB1 mRNA and for L-TGFB1 was increased and was mainly associated with the mesangium and some podocytes. More active TGFB1 was also seen in the glomerulus in manifest diabetic nephropathy, especially in mesangial cells, but the intensity of staining was low when compared with that in tubular epithelial cells (Fig. 3c,h,m). With progression to advanced diabetic nephropathy, immunostaining for active TGFB1 became more prominent in the glomerulus. The staining had a diffuse appearance, suggesting that active TGFB1 is present in mesangial as well as epithelial cells (Fig. 3i). A very similar expression pattern for active TGFB1 was obtained using the anti-LC (1-30) antibody, which also recognises intracellular active TGFB1 [25]. Due to the limited availability of this antibody, only a few sections were examined (data not shown). Semiquantification of the immunostainings and ISH is given in Tables 2 and 3.
Only low levels of CTGF were found in the glomeruli of normal kidneys (Fig. 4a,f). However, even though the ISH signal for CTGF was low, there was some immunostaining of the protein in podocytes. Both mRNA and protein CTGF were also detected in distal tubules and collecting ducts (not shown). In contrast, proximal tubules showed weak protein staining but no detectable mRNA.
Glomeruli of patients with incipient diabetic nephropathy showed definite positive signals for both CTGF mRNA and protein (Fig. 4b,g). There was some CTGF immunostaining in mesangial areas and in podocytes at this early stage of disease. The pattern of CTGF expression in manifest and advanced diabetic nephropathy was similar to that in incipient disease, but both the mRNA and protein staining were more intense. In addition to staining of podocytes, there was strong immunostaining in the mesangium and, in some glomeruli, in juxtaglomerular areas (not shown). Prominent CTGF staining was also detected in Bowman’s capsule. Semiquantification of the immunostaining and ISH for CTGF is given in Tables 2 and 3.
Interdependency between THBS1, TGFB1 and CTGF in mediating effects of diabetic stimuli on mesangial cells
Many cellular responses to hyperglycaemia are TGFB1-dependent [5, 10]. THBS1 is a key activator of L-TGFB1 expressed in human mesangial cells (HMC) exposed to high glucose [17] and TGFB1-dependent effects in this environment were suppressed either by THBS1 antisense oligonucleotides, or by a peptide (GGWSHW, peptide W) blocking the THBS1 activation mechanism [26]. To test whether THBS1 activation of L-TGFB1 is required for induction of CTGF by high glucose or glycated albumin, we cultured HMC for 7 days in media containing either 4 mmol/l d-glucose and 30 mmol/l d-glucose, or 4 mmol/l d-glucose and 30 mmol/l d-glucose with either 0.6 mg/ml HSA or 0.6 mg/ml glycated albumin, in the presence or absence of THBS1 antisense oligonucleotide or of peptide W. Serum-free conditioned medium was collected for day 7 and analysed for CTGF [9] (Fig. 5). As expected, both high glucose and glycated albumin increased the level of secreted CTGF compared to normal glucose by about 4.6-fold each. No additive increase was observed with high glucose and glycated albumin together. HSA alone stimulated CTGF secretion slightly (Fig. 6, lane 3), but 0.6 mg/ml HSA contained no detectable TGFB1 bioactivity when tested with the mink lung cell assay [17] (result not shown). Interestingly, THBS1 antisense oligonucleotide alone and peptide W alone blocked the increase of CTGF expression induced by high glucose, but not that induced by glycated albumin (Fig. 5). Neither a control oligonucleotide, nor a control peptide had any effect under otherwise identical conditions (data not shown). Thus CTGF expression induced by high glucose is dependent on THBS1 activation of L-TGFB1, while upregulation induced by glycated albumin does not require THBS1.

Effect of high glucose and glycated albumin on synthesis of CTGF by human mesangial cells in vitro, and selective inhibition of the high-glucose effect by THBS1 peptide W, or THBS1 antisense (AS) oligonucleotide. Primary HMC were maintained for 6 days in medium containing 10% fetal calf serum and either 4 mmol/l d-glucose (lane 1), 30 mmol/l d-glucose (lane 2), or 4 mmol/l glucose supplemented with 0.6 mg/ml human serum albumin (HSA) (lane 3) or 0.6 mg/ml glycated albumin (lane 4), or with both 30 mmol/l glucose and glycated albumin together (lane 5). Cultures were maintained for a further 24 h (day 7) in the same media but without fetal calf serum. A further set of cultures was treated as above but with the addition of 1 μmol/l peptide W, or 2 μmol/l THBS1 antisense oligonucleotide, throughout. The media were then analysed for CTGF by SDS-PAGE, Western blotting (a) and scanning densitometry (b–d). Numbers (x-axes) correspond to lane numbers (a). The results (b–d) represent the means±SEM of three independent experiments. Statistical significance was analysed using a two-tail unpaired Student’s t-test. * p<0.0001 (n=6) compared with 4 mmol/l d-glucose; +p<0.001 (n=6) compared with HSA

Effect of high glucose and glycated albumin on synthesis of fibronectin by human mesangial cells in vitro, and selective inhibition of the high-glucose effect by THBS1 peptide W or THBS1 antisense (AS) oligonucleotide. Primary HMC were maintained for 6 days in medium containing 10% fetal calf serum and either 4 mmol/l d-glucose (d-glc), 30 mmol/l d-glc, or 4 mmol/l glucose supplemented with 0.6 mg/ml human serum albumin (HSA) or 0.6 mg/ml glycated albumin (GA), or with 30 mmol/l d-glc and GA together. Cultures were maintained for a further 24 h (day 7) in the same medium but without fetal calf serum. The media were analysed for fibronectin by ELISA (a). A further set of cultures was maintained with either high glucose (b) or glycated albumin (c), but with the addition of 1 μmol/l peptide W or 2 μmol/l THBS1 antisense oligonucleotide throughout. Statistical significance was analysed using a two-tailed unpaired Student’s t-test. *P≤0.05 (n=4). Error bars (b) were too small to show. A second independent experiment gave very similar results
High glucose treatment for 7 days increased fibronectin levels in culture medium by 1.4-fold, while glycated albumin stimulated them by about three-fold (Fig. 6), but there was no additive effect when both were present together. HSA alone stimulated fibronectin levels 1.6-fold more than 4 mmol/l d-glucose. Cell layer fibronectin levels, measured by western blotting, correlated with those in the medium under all studied conditions (data not shown). When high-glucose cultures were treated with THBS1 antisense oligonucleotide or with peptide W, they were reduced by 60 and 52% respectively (Fig. 6b), in accordance with our previous results [17]. In marked contrast, elevated fibronectin levels in cultures maintained with glycated albumin were not significantly affected when treated with THBS1 antisense oligonucleotide or peptide W (Fig. 6c). Thus, as for CTGF, elevated synthesis of fibronectin in a high glucose environment is, to a large extent, dependent on THBS1 activation of L-TGFB1, while elevated synthesis stimulated by glycated albumin is not.
CTGF antisense oligonucleotide abolishes TGFB1-dependent induction of fibronectin in HMC [9]. To compare the dependency of upregulated fibronectin expression on CTGF in response to glycated albumin, we maintained HMC cultures for 7 days with either 30 mmol/l d-glucose, or 0.6 mg/ml glycated albumin, in the presence or absence of 2 μmol/l CTGF antisense oligonucleotide, and measured fibronectin levels in the serum-free medium from the last 24 h and in the cell layer. The CTGF-antisense oligonucleotide reduced the level of secreted CTGF protein to almost zero, both under high-glucose and glycated albumin conditions (Fig. 7a, lanes 5 and 6), and also inhibited the increase in fibronectin levels in the culture medium (Fig. 7b) and cell layer in both conditions (Fig. 7c). Thus fibronectin synthesis induced in mesangial cells by high glucose is dependent on CTGF expression, which depends, in turn, on THBS1 activation of L-TGFB1. In contrast, while the increase in fibronectin synthesis stimulated by glycated albumin is also totally dependent on CTGF, induction of the latter is independent of THBS1 activation of L-TGFB1 in cells exposed to this stimulus.

Effect of a CTGF antisense oligonucleotide on fibronectin levels in human mesangial cell cultures treated with different diabetic conditions. Primary HMC were maintained as describes in Fig. 6. Some cultures were treated throughout with 2.0 μmol/l CTGF antisense oligonucleotide (AS CTGF). The effectiveness of antisense treatment was assessed by analysing CTGF levels in the media from day 7 by SDS-PAGE and western blotting (a). Media from day 7 were also analysed for fibronectin by ELISA (b). Cell layers were analysed for fibronectin by SDS-PAGE and western blot analysis (c). Statistical significance was analysed using a two-tail unpaired Student’s t-test. *P≤0.05 (n=4)
Discussion
It is well established that TGFB1 is a major driver of fibrosis in the glomerulus and elsewhere in the renal cortex in diabetic nephropathy [5, 10, 27, 28] and neutralising antibodies to its isoforms attenuate matrix gene expression in murine models [29, 30]. However, while our study found marked increases in glomerular L-TGFB1 in incipient diabetic nephropathy, only very low levels of active growth factor were detected at this stage. Active TGFB1 is present in the urine of diabetic rats [31] and human patients with diabetic nephropathy excrete larger amounts of TGFB1 in the urine [21], so it is possible that, upon activation, glomerular TGFB1 is rapidly lost by this route. In the more advanced stages of diabetic nephropathy, Type I collagen is present in scarred glomeruli and active TGFB1 could become sequestered in the tissue by formation of a tertiary complex with it and decorin, whose expression also increases with disease progression [21]. Previous in vitro studies have shown that THBS1 expression increased in mesangial cells exposed to high glucose [32] and suggested that it may be a key activator of TGFB1 in diabetic nephropathy [17, 33]. The present study extends these observations by showing a marked increase in glomerular THBS1 expression in incipient diabetic nephropathy, with further increases as the disease progresses. THBS1 is a major activator of TGFB1 in vivo [19] and it is therefore likely to play an important role in the development of glomerulosclerosis in diabetic nephropathy.
Although expression of CTGF mRNA has been reported in a diffuse mesangial proliferative lesion in diabetic nephropathy [8], this is the first semiquantitative study demonstrating its upregulation at both the mRNA and protein levels in the glomerulus in the incipient stage of the disease, and its increasing expression level with progression of the disease. Upregulation of CTGF was also detected in murine glomeruli soon after the onset of hyperglycaemia in the non-obese diabetic mouse [9]. Moreover, it can be induced by stretching mesangial cells in vitro [34], suggesting that increases in its expression early in diabetic nephropathy may be a response to haemodynamic changes which occur at that stage of the disease [35], as well as to TGFB1 [36]. Moreover, once induced, CTGF can upregulate its own expression [9] and is able to initiate a number of changes in mesangial cells that are characteristic of those found in diabetic nephropathy. These include increased expression of fibronectin [9] and its enhanced assembly into an insoluble matrix [37], induction of plasminogen activator inhibitor-1 [9], cell cycle arrest and hypertrophy [13], and rearrangement of the actin cytoskeleton [38]. The effects on matrix protein synthesis and the cell cycle are similar to those induced by TGFB1. Moreover TGFB1-induced effects can be blocked by CTGF antisense oligonucleotides [9, 13, 37], indicating that CTGF is involved in some way in mediating the effects of TGFB1. Recently we reported that CTGF binds and activates a tyrosine kinase receptor, NTRK1 (also known as TRKA), triggering intracellular signalling pathways and inducing the expression of Kruppel-like factor 10 (KLF10, previously known as TGFB inducible early growth response [TIEG]) [39]. KLF10 is a transcriptional repressor for SMAD7 [40], an inhibitor of the SMAD signalling pathway that mediates the effects of TGFB1 [41]. Thus we proposed that CTGF works in concert with TGFB1 to promote the latter’s profibrotic actions, rather than downstream of it [42, 43]. Hence, progressively increased CTGF expression during diabetic nephropathy, as reported above, would probably decrease the inhibitory action of SMAD7, promoting ever increasing TGFB1 signalling in the mesangial cell and glomerulosclerosis.
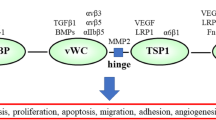
Many different metabolic, haemodynamic, growth and cellular signalling factors are involved in the response to hyperglycaemia [44]. While in vitro the stimulatory effect of high glucose and glycated albumin on fibronectin synthesis by mesangial cells required CTGF, there were two noteworthy differences in the actions of these diabetic stimuli. Firstly, typical diabetic concentrations of glycated albumin (0.6 mg/ml) induced much higher fibronectin expression than high concentrations of glucose (30 mmol/l). Secondly, increased fibronectin synthesis was highly dependent on THBS1 activation of TGFB1 in high glucose, but virtually independent of it with glycated albumin. Both conditions upregulated CTGF to similar levels. The reasons for these differences are not clear although it is known that high glucose and glycated albumin act on mesangial cells by different routes. High glucose leads to increased levels of its intracellular metabolites including hexosamines [45], polyols and diacylglycerol, the latter activating protein kinase C isoforms, which induce TGFB1 expression [46, 47], upregulating CTGF [9]. Glucose–protein adducts form in high-glucose environments (the major circulating factor in diabetes being an albumin-Amadori product like the commercial glycated albumin used in this study). These interact with a specific ligand-receptor system on mesangial cells [48], activating protein kinase C isoforms and leading to increased expression of TGFB1 type II receptor [35, 49]. They also generate reactive oxygen species, which can activate latent TGFB1 and induce CTGF [50], upon which, some actions of TGFB1 appear to be critically dependent. Thus, both diabetic stimuli lead by different pathways to the activation of TGFB1 and CTGF to promote its signalling (Fig. 8).

Proposed roles of THBS1, TGFB1, and CTGF in upregulating fibronectin expression in mesangial cells in response to high glucose or glycated albumin. THBS1 is required for activation of L-TGFB1 in response to high glucose but not in response to glycated albumin, where activation may be by reactive oxygen species (ROS). TGFB1 upregulates CTGF in either condition. CTGF is obligatory for the upregulation of fibronectin expression in response to either high glucose or glycated albumin. The pathways can be blocked by THBS1 antisense oligonucleotide, peptide W, or CTGF antisense oligonucleotide, at the points indicated
This paper provides evidence that glomerular CTGF expression is elevated from the earliest stages of diabetic nephropathy, that it is involved in mediating the adverse effects of hyperglycaemia and glycated albumin on human mesangial cells, and supports the view that CTGF is a potential therapeutic target in diabetic nephropathy [44].
Duality of interest:
The authors declare that they have no conflict of interest.
Abbreviations
- CTGF:
-
connective tissue growth factor
- HMC:
-
human mesangial cells
- HSA:
-
human serum albumin
- ISH:
-
in situ hybridisation
- L-TGFB1:
-
latent TGFB1
- α-SMA:
-
α-smooth muscle actin
- TGFB1:
-
transforming growth factor beta
- THBS1:
-
thrombospondin-1
- WT-1:
-
Wilms’ tumour protein-1
References
Mogensen CE, Christensen CK (1984) Predicting diabetic nephropathy in insulin-dependent patients. N Engl J Med 311:89–93
The Diabetic Control and Complications Trial Research Group (1993) The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 329:977–986
UK Prospective Diabetes Study (UKPDS) Group (1998) Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 352:837–853
Mogensen CE, Christensen CK, Vittinghus E (1983) The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 32(Suppl. 2):64–78
Sharma K, Ziyadeh FN (1995) Hyperglycemia and diabetic kidney disease. Diabetes 44:1139–1146
Brownlee M (2001) Biochemistry and molecular cell biology of diabetic complications. Nature 414:813–820
Border WA, Noble NA (1997) TGF-beta in kidney fibrosis: a target for gene therapy. Kidney Int 51:1388–1396
Ito Y, Aten J, Bende RJ et al (1998) Expression of connective tissue growth factor in human renal fibrosis. Kidney Int 53:853–861
Abdel-Wahab N, Yevdokimova N, Weston BS et al. (2001) Role of connective tissue growth factor in the pathogenesis of diabetic nephropathy. Biochem J 359:77–87
Sharma K, Jin Y, Guo J, Ziyadeh FN (1996) Neutralization of TGFβ by anti-TGFβ antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STR-induced diabetic mice. Diabetes 45:522–530
Twigg SM, Chenn MM, Joly AH et al (2001) Advanced glycosylation end-products upregulate connective tissue growth factor (insulin-like growth factor-binding protein-related protein 2) in human fibroblasts: a potential mechanism for expansion of extracellular matrix in diabetes mellitus. Endocrinology 142:1760–1769
Abdel Wahab N, Harper K, Mason R (1996) Expression of extracellular matrix molecules in human mesangial cells in response to prolonged hyperglycaemia. Biochem J 316:985–992
Abdel-Wahab N, Weston BS, Roberts T, Mason RM (2002) Connective tissue growth factor and regulation of the mesangial cell cycle: role in cellular hypertrophy. J Am Soc Nephrol 13:2437–2445
Grotendorst GR, Okochi H, Hayashi N (1996) A novel transforming growth factor beta response element controls the expression of the connective tissue growth factor gene. Cell Growth Differ 7:469–480
Duncan MR, Frazier KS, Abramson S et al (1999) Connective tissue growth factor mediates transforming growth factor beta-induced collagen synthesis: down-regulation by cAMP. FASEB J 13:1774–1786
Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J (2001) Latency, activation, and binding proteins of TGF-beta. Microsc Res Tech 52:354–362
Yevdokimova N, Abdel-Wahab N, Mason RM (2001) Thrombospondin-1 is the key activator of TGF-β1 in human mesangial cells exposed to high glucose. J Am Soc Nephrol 12:703–712
Daniel C, Wiede J, Krutzsch HC et al (2004) Thrombospondin-1 is a major activator of TGF-beta in fibrotic renal disease in the rat in vivo. Kidney Int 65:459–468
Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM et al (1998) Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell 93:1159–1170
Mogensen CE (1987) Microalbuminuria as a predictor of clinical diabetic nephropathy. Kidney Int 31:673–689
Schaefer L, Raslik I, Gröne HJ et al (2001) Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican and fibromodulin. FASEB J 15:559–561
Schaefer L, Hausser H, Altenburger M et al (1998) Decorin, biglycan and their endocytosis receptor in rat renal cortex. Kidney Int 54:1529–1541
Tsang ML, Zhou L, Zheng BL et al (1995) Characterization of recombinant soluble human transforming growth factor-β receptor type II (rhTGF-β sRII).Cytokine 7:389–397
Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA (1994) Transforming growth factor-β activation in irradiated murine mammary gland. J Clin Invest 93:892–899
Flanders KC, Thompson NL, Cissel DS et al (1989) Transforming growth factor-β1: histochemical localization with antibodies to different epitopes. J Cell Biol 108:653–660
Murphy-Ullrich JE, Poczatek M (2000) Activation of latent TGF-beta by thrombospondin-1: mechanisms and physiology. Cytokine Growth Factor Rev 11:59–69
Border WA, Yamamoto T, Noble NA (1996) Transforming growth factor beta in diabetic nephropathy. Diabetes Metab Rev 12:309–339
Hong SW, Isono M, Chen S et al (2001) Increased glomerular and tubular expression of transforming growth factor-beta1, its type II receptor, and activation of the Smad signaling pathway in the db/db mouse. Am J Pathol 158:1653–1663
Ziyadeh FN, Hoffman BB, Han DC et al (2000) Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proc Natl Acad Sci USA 97:8015–8020
Hill C, Flyvbjerg A, Rasch R, Bak M, Logan A (2001) Transforming growth factor-beta2 antibody attenuates fibrosis in the experimental diabetic rat kidney. J Endocrinol 170:647–651
Wang SN, Hirschberg R (2000) Growth factor ultrafiltration in experimental diabetic nephropathy contributes to interstitial fibrosis. Am J Physiol Renal Physiol 278:F554–F560
Holmes DI, Abdel Wahab N, Mason RM (1997) Identification of glucose-regulated genes in human mesangial cells by mRNA differential display. Biochem Biophys Res Commun 238:179–184
Poczatek MH, Hugo C, Darley-Usmar V, Murphy-Ullrich JE (2000) Glucose stimulation of transforming growth factor-beta bioactivity in mesangial cells is mediated by thrombospondin-1. Am J Pathol 157:1353–1363
Riser BL, Denichilo M, Cortes P et al (2000) Regulation of connective tissue growth factor activity in cultured rat mesangial cells and its expression in experimental diabetic glomerulosclerosis. J Am Soc Nephrol 11:25–38
Raptis AE, Viberti G (2001) Pathogenesis of diabetic nephropathy. Exp Clin Endocrinol Diabetes 109(Suppl 2):S424–S437
Blom IE, Goldschmeding R, Leask A (2002) Gene regulation of connective tissue growth factor: new targets for antifibrotic therapy? Matrix Biol 21:473–482
Weston BS, Abdel Wahab N, Mason RM (2003) CTGF mediates TGF-β induced fibronectin matrix deposition by upregulating active α5β1 integrin in human mesangial cells. J Am Soc Nephrol 14:601–610
Crean JK, Finlay D, Murphy M et al (2002) The role of p42/44 MAPK and protein kinase B in connective tissue growth factor induced extracellular matrix protein production, cell migration, and actin cytoskeletal rearrangement in human mesangial cells. J Biol Chem 277:44187–44194
Wahab NA, Weston BS, Mason RM (2004) Connective tissue growth factor (CTGF, CCN2) interacts with and activates the tyrosine kinase receptor TrkA. J Am Soc Nephrol 16:340–351
Johnsen SA, Subramaniam M, Janknecht R, Spelsberg TC (2002) TGF beta inducible early gene enhances TGFbeta/Smad-dependent transcriptional responses. Oncogene 21:5783–5790
Kavsak P, Rasmussen RK, Causing CG et al (2000) Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell 6:1365–1375
Wahab NA, Mason RM (2004) Connective tissue growth factor and renal diseases: some answers, more questions. Curr Opin Nephrol Hypertens 13:52–58
Wahab NA, Weston BS, Mason, RM (2005) Modulation of TGFβ/Smad signaling pathway by CTGF/CCN2. Exp Cell Res 307:305–314
Schrijvers BF, De Vriese AS, Flyvbjerg A (2004) From hyperglycaemia to diabetic kidney disease: the role of metabolic, haemodynamic, intracellular factors and growth factors/cytokines. Endocr Rev 25:971–1010
Schleicher ED, Weigert C (2000) Role of the hexosamine biosynthetic pathway in diabetic nephropathy. Kidney Int Suppl 77:S13–S18
Kapor-Drezgic J, Zhou X, Babazono T, Dlugosz JA, Hohman T, Whiteside C (1999) Effect of high glucose on mesangial cell protein kinase C-delta and -epsilon is polyol pathway-dependent. J Am Soc Nephrol 10:1193–1203
Whiteside CI, Dlugosz JA (2002) Mesangial cell protein kinase C isozyme activation in the diabetic milieu. Am J Physiol Renal Physiol 282:F975–F980
Wu VY, Cohen MP (1995) Evidence for a ligand receptor system mediating the biologic effect of glycated albumin in glomerular mesangial cells. Biochem Biophys Res Commun 207:521–528
Cohen MP, Ziyadeh FN, Lautenslager GT, Cohen JA, Shearman CW (1999) Glycated albumin stimulation of PKC-β activity is linked to increased collagen IV in mesangial cells. Am J Physiol Renal Physiol 276:F684–F690
Mason RM, Wahab NA (2003) Extracellular matrix metabolism in diabetic nephropathy. J Am Soc Nephrol 14:1358–1373
Acknowledgements
We are grateful to the Medical Research Council UK, the Deutsche Forschungsgemeinschaft (SFB 492, Project B10) and the Interdisciplinary Centre for Clinical Research, University of Münster (Projects D18 and Schae2/004/04) for financial support as well as to Diabetes UK for a postgraduate studentship (to O. Yiannikouris). We thank FibroGen (South San Francisco, CA, USA) for the anti-CTGF antibody and Dr K. C. Flanders (NIH, Bethesda, MD, USA) for the LC (1-30) anti-active TGFB1 antibody.
Author information
Authors and Affiliations
Corresponding author
Additional information
N. A. Wahab and L. Schaefer made an equal contribution to the work reported in this paper
Rights and permissions
About this article
Cite this article
Wahab, N.A., Schaefer, L., Weston, B.S. et al. Glomerular expression of thrombospondin-1, transforming growth factor beta and connective tissue growth factor at different stages of diabetic nephropathy and their interdependent roles in mesangial response to diabetic stimuli. Diabetologia 48, 2650–2660 (2005). https://doi.org/10.1007/s00125-005-0006-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00125-005-0006-5