
Abstract
Synthesis, characterization and theoretical studies of a novel coumarin-triazole-thiophene hybrid 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-6,7-dimethyl-2H-chromen-2-one (1), which was fabricated from 4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol and 4-(chloromethyl)-6,7-dimethyl-2H-chromen-2-one, are reported. The resulting compound was characterized by microanalysis, IR, 1H, and 13C APT NMR spectroscopy. The DFT calculations examined the structure and electronic properties of 1 in gas phase. Its reactivity descriptors and molecular electrostatic potential revealed the reactivity and the reactive centers of 1. ADMET properties of 1 were evaluated using the respective online tools. It was established that 1 exhibit positive gastrointestinal absorption properties and negative human blood-brain barrier penetration. The Toxicity Model Report revealed that 1 belongs to toxicity class 4. Molecular docking was additionally applied to study the interaction of 1 with some SARS-CoV-2 proteins. It was established that the title compound is active against all the applied proteins with the most efficient interaction with Papain-like protease (PLpro). The interaction of 1 with the applied proteins was also studied using molecular dynamics simulations.
Graphical abstract
A novel coumarin-triazole-thiophene hybrid 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-6,7-dimethyl-2H-chromen-2-one (1) is reported. The structure and electronic properties of 1 were examined by the DFT calculations. ADMET properties of 1 were also evaluated. Molecular docking and molecular dynamics simulations were applied to study interactions of 1 with a series of the SARS-CoV-2 proteins.

Similar content being viewed by others

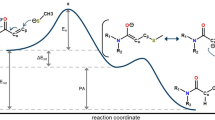

1 Introduction
Throughout its history, humankind has constantly been faced with health problems. All this becomes even more relevant in the emergence of previously unknown diseases that could potentially transform into pandemics. In this regard, designing and producing molecules with potential biological and medicinal properties is likely, one of the most effective ways to counter diseases. Heterocyclic compounds are abundant in nature and are necessary for survival. Suffice it to say that the deoxyribonucleic acid (DNA), which carries genetic instructions for the development, functioning, growth, and reproduction of all known organisms and many viruses, is composed of four nucleobases, viz., cytosine, guanine, adenine, and thymine, which are nitrogen-containing heterocycles. Thus, heterocyclic compounds play a fundamental and pivotal role in nature.
Many pharmacologically active heterocyclic compounds are on the market, many of which are regularly used in clinical practice.1,2,3,4,5,6,7 Of a myriad of heterocyclic compounds, coumarins, containing fused benzene and pyrone fragments, are a large family of particular importance. These compounds are one of the components in many plants. It is not surprising that coumarin was, for the first time, isolated from the tonka bean (Dipteryx odorata Wild) in 1820 by A. Vogel. Since then, coumarins have continuously been the focus of scientists and have extensively been studied for biochemical and pharmaceutical properties.8,9,10 On the other hand, 1,2,4-triazole and thiophene are five-membered nitrogen- and sulfur-containing heterocyclic molecules, respectively. These molecules adopt a planar structure and are aromatic with the corresponding aromaticity index >0.8 11 Both the 1,2,4-triazole and thiophene-based compounds are of great importance for pharmacy and medicine due to their pronounced biological properties.1,2,3,4,5,6,7,12,13,14,15,16 Thus, a combination of the coumarin, 1,2,4-triazole and thiophene functionalities in one molecule is of interest in terms of potential generating of novel biological properties, which, in turn, might be of great value against different diseases and for the design of new therapies, as well as for the design and fabrication of new materials of valuable properties.17
Nowadays, COVID-19 has become one of the world's most crucial problems affecting global economics. Thus, drugs against SARS-CoV-2, a virus that causes COVID-19, are of particular value. It was also reported that the heterocyclic fragments could serve as valuable and important resources for developing coronavirus treatment strategies and therapy.18 Of a variety of heterocycles, coumarin-,19,20,21,22,23,24 1,2,4-triazole-25,26 and thiophene-derived compounds have also been found to be of interest against COVID-19.27,28,29 Thus, a combination of the above-mentioned heterocycles in a single molecule seems to be of value for the fabrication of an active agent used in the COVID-19 therapy.
We have also been continuously interested in the chemistry of heterocycles as well as in computational analyses of compounds with biological activity.30,31,32,33,34,35,36,37,38,39,40,41,42,43 In the present work, we have focused on a novel molecule constructed from the coumarin, 1,2,4-triazole and thiophene frgaments, namely 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-6,7-dimethyl-2H-chromen-2-one (1). Furthermore, to be within the borders of the pink area of the bioavailability radar, which enables a first glance at the drug-likeness of a molecule in the SwissADME online tool,44 ethyl, methyl and ethylene fragments were also incorporated in the structure of 1. We have also performed DFT-based computational studies to probe the most favourable structure of the title compound as well as to examine its electronic properties. Additionally, the interaction of 1 with a series of SARS-CoV-2 proteins was examined using molecular docking and molecular dynamics.
2 Experimental
2.1 Materials
Starting materials 4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol and 4-(chloromethyl)-6,7-dimethyl-2H-chromen-2-one were provided by the Firat University organic laboratory. The other chemicals and solvents were purchased from Merck KGaA and used without further purification.
2.2 Physical measurements
The 1H and 13C APT NMR spectra in DMSO-d6 were recorded with a Bruker AC-400 NMR spectrometer. The IR spectrum was recorded in the KBr pellet with a PerkinElmer Spectrum One FT-IR system. The melting point was determined using the Gallenkamp melting point apparatus. Microanalysis was performed using a LECO 932 CHNS-O elemental analyzer.
2.3 Synthesis of 1
A mixture of 4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol (0.02 mol, 4.226 g), 4-(chloromethyl)-6,7-dimethyl-2H-chromen-2-one (0.02 mol, 4.453 g) and K2CO3 (0.02 mol, 2.764 g) in dry acetone (30 mL) was stirred for 6 h under ambient conditions. Then the resulting white precipitate was filtered off and dried under ambient conditions, followed by recrystallization from EtOH. Yield: 5.963 g (75%). M.p.: 165–167 °C. 1H NMR, δ: 1.18 (t, 3JH,H = 7.2 Hz, CH3, Et), 2.29 (s, CH3–coumarin), 2.33 (s, CH3–coumarin), 4.09 (q, 3JH,H = 7.2 Hz, CH2, Et), 4.61 (s, CH2S), 6.36 (s, H11, coumarin), 7.22 (s, H12, coumarin), 7.63 (s, H13, coumarin), 7.26 (t, 3JH,H = 7.8 Hz, H7, thiophene), 7.55 (t, 3JH,H = 7.8 Hz, H6, thiophene), 7.80 (t, 3JH,H = 7.8 Hz, H8, thiophene), ppm. 13C APT NMR, δ: 15.29 (CH3, Et), 19.32 (CH3–coumarin), 20.07 (CH3–coumarin), 33.85 (CH2, Et), 40.19 (CH2S), 114.52 (C11, coumarin), 115.69 (C18, coumarin), 117.60 (C14, coumarin), 125.58 (C6, thophene), 128.05 (C17, coumarin), 128.09 (C16, coumarin), 128.78 (C7, thophene), 129.61 (C8, thophene), 133.37 (C5, thophene), 142.58 (C15, coumarin), 149.51 (C10, coumarin), 150.32 (C1, triazole), 151.56 (C13, coumarin), 152.23 (C12, coumarin), 160.28 (C2, triazole) ppm. IR, ν: 1722 (C=O), 1622 (C=C), 1559 (C=C), 1418 (C=N), 1130 (C–O), 903 (C–O), 770 (C–S) cm–1. Anal. calc. for C20H19N3O2S2 (397.51) (%): C 60.43, H 4.82 and N 10.57; found: C 60.58, H 4.76 and N 10.62.
2.4 Computational details
The optimized geometry of 1, highest occupied and lowest unoccupied molecular orbital (HOMO and LUMO, respectively) surfaces, and a molecular electrostatic potential (MEP) surface were calculated without symmetry restrictions in gas phase with the GaussView 6.0 molecular visualization program45 and Gaussian 09, Revision D.01 program package46 using the DFT/B3LYP hybrid functional47,48 and cc-pVDZ47,49 basis set.
2.5 Molecular docking
Molecular docking simulations of the optimized structure of 1 with a series of the SARS-CoV-2 proteins were carried out with AutoDock Vina,50,51 using the Lamarckian Genetic Algorithm (LGA) scoring function with a number of GA runs = 100, population size = 500, and a maximum number of evaluations = 25 000 000. The targeted protein structures were subtracted from the RCSB PDB database,52 and were pretreated before the docking, including water removing and inserting hydrogen atoms and missing residues and charges. Gasteiger charges were added to the ligand molecules prior to converting to PDBQT format. AutoDock Tools (v. 1.5.7) was utilized to define the grid box with the dimensions of 30 × 30 × 30 Å with 0.375 Å grid spacing. Semi-flexible docking was performed, keeping the receptor molecule rigid and ligands flexible. During the docking procedure, 200 conformations for each ligand were left flexible, while the protein was held rigid. The lowest binding energy conformers and 2D interactions were filtered from 10 top-ranked poses. Docking results were visualized in BIOVIA Discovery Studio 2020.53
2.6 Molecular dynamics simulation
Molecular dynamics (MD) simulations of complexes of 1 with a series of the SARS-CoV-2 proteins were performed using the WebGRO online service.54 Parameters such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), a radius of gyration (Rg), solvent accessible surface area (SASA) and intermolecular hydrogen bonds were assessed. Complexes were prepared for MD using GROMOS96 43a1 forcefield and were equilibrated using the canonical (NVT) and the isothermal–isobaric (NPT) ensembles. Topology of 1 was generated with the PRODRG tool.55 Simple point charge (SPC) was used as a solvent model (triclinic water box with size 50 × 75 × 70 Å) for complexes.56 These systems were neutralized by adding sodium or chlorine ions based on the total charges. For minimization of the system before MD, the steepest descent algorithm (5000 steps) was applied. The MD simulations were performed in the presence of 0.15 M NaCl using the constant temperature (310 K) and pressure (1.0 bar). The approximate number of frames per simulation was 1000. The simulation time was set to 50 ns.
2.7 In silico drug-likeness analysis
The SwissADME,44 BOILED-Egg57 and ProTox-II58,59 online tools were applied to study ADMET properties of 1.
3 Results and Discussion
Coumarin-triazole-thiophene hybrid 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-6,7-dimethyl-2H-chromen-2-one (1) was obtained with a good yield by reacting 4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol with an equimolar amount of 4-(chloromethyl)-6,7-dimethyl-2H-chromen-2-one in the presence of K2CO3 (Scheme 1).

Synthesis of 1.
The IR spectrum of 1 contains bands typical for characteristic fragments. The bands for the C=O and C=N groups were shown at 1722 and 1418 cm–1, respectively, while the band for the C–O groups were found at 903 and 1130 cm–1. Two bands for the C=C vibrations were revealed at 1559 and 1622 cm–1, and the band for the C–S group was observed at 770 cm–1.
The 1H NMR spectrum of 1 recorded in DMSO-d6 exhibits a single set of peaks (Figure 1). Particularly, the ethyl hydrogen atoms were shown as a triplet at 1.18 ppm and a quartet at 4.09 ppm with the characteristic coupling constant of 3JH,H = 7.2 Hz. The hydrogen atoms of the coumarin-attached methyl groups were observed as two singlets at 2.29 and 2.33 ppm, while the CH2S hydrogen atoms were found as a singlet at 4.61 ppm. The coumarin hydrogen atoms were shown as three singlets at 6.36, 7.22, and 7.63 ppm. Finally, a triplet at 7.26 ppm, and two doublets at 7.55 and 7.80 ppm were attributed to the thiophene hydrogens. The 13C APT NMR spectrum of 1 in the same solvent exclusively exhibits signals typical for the observed carbons (Figure 1). Particularly, the ethyl fragment is shown as two signals at 15.29 and 33.85 ppm for the methyl and methylene carbons, respectively. The coumarin methyl carbon atoms were found in the spectrum as two signals at 19.32 and 20.07 ppm, while the CH2S carbon was observed as a signal at 40.19 ppm. The triazole fragment was shown as two signals at 150.32 and 160.28 ppm, while the thiophen fragment was found as four signals at 125.58, 128.78, 129.61 and 133.37 ppm. Finally, the carbon atoms of the coumarin fragment were observed at 114.52, 115.69, 117.60, 128.05, 128.09, 142.58, 149.51, 151.56 and 152.23 ppm.

The 1H (top) and 13C APT (bottom) NMR spectra of 1 recorded in DMSO-d6 (see Figure 2 for atoms labelling).

Top and side views of the optimized structure of 1, obtained using the DFT/B3LYP/cc-pVDZ method.
The structure of 1 as well as its electronic properties were revealed using the DFT/B3LYP/cc-pVDZ calculations. The structure was first optimized in gas phase at 298.15 K (Figure 2). The calculated energies and thermodynamic parameters of the optimized structure are given in Table S1 (Supplementary Information), while the cartesian coordinates of the atoms are listed in Table S2 (Supplementary Information). The vibration frequencies were also calculated for the optimized structure of 1 in gas phase, and no imaginary frequencies were obtained (Figure S1 and Table S3, Supplementary Information).
The obtained bond lengths and angles in the optimized structure of 1 are typical for certain functionalities. Particularly, the C–C, C–N, C–O and C–S bonds within the thiophene and coumarin fragments are 1.359–1.458, 1.317–1.388, 1.365–1.394 and 1.734–1.755 Å, respectively, indicating their bond order of about one-and-a-half, while remaining C–C, C–N and C–S bonds are 1.454–1.529, 1.464 and 1.768–1.841 Å, respectively, being close to single bonds (Table 1). However, some shortening of the C–C bond between the triazole and thiophene fragments is clearly evidenced due to the conjugation of the π-systems of these rings. The exocyclic carbonyl C–O bond length is 1.207 Å, indicating its double-bond nature. The endocyclic C–N–N and N–C–N bond angles within the triazole ring vary from 107.01° to 111.29°, while the C–N–C bond angle is slightly smaller and of 103.60° (Table 1). The C–C–C and C–C–S bond angles in the thiophene fragments are 110.62–113.20°, while the C–S–C angle is close to the right angle and 91.45° (Table 1). The thioester C–S–C bond angle is slightly larger in comparison to the thiophene analogue and of 96.81°. Finally, all the endocyclic bond angles within the coumarin fragment are similar and vary from 116.23° to 123.18°, clearly indicating their sp2 hybridization (Table 1). The molecule of 1 slightly deviates from planarity as evidenced by the corresponding dihedral angles between the triazole, thiophene and coumarin fragments, while the ethyl fragment is almost orthogonal to the triazole fragment (Figure 2, Table 1).
It was established that the energies of the HOMO and LUMO for 1 are –5.87032 and –1.85582 eV, respectively, with an energy gap of 4.01450 eV (Figure 3). The HOMO is mainly delocalized over the thiophene, triazole and thioester sulfur fragments, while the LUMO is mainly spread over the coumarin fragment (Figure 3).

Energy levels and views on the electronic isosurfaces of the HOMO and LUMO of the ground state of 1, obtained using the DFT/B3LYP/cc-pVDZ method.
According to the ionization potential and the electron affinity value, the optimized structure of 1 exhibits both good electron-donor and electron-acceptor properties (Table 2). Further, as evidenced by the relatively low electronegativity value, 1 is a poor electron attractor, which is additionally supported by the corresponding chemical potential value (Table 2). Values of the chemical hardness and chemical softness indicate that the optimized structure of 1 tends to exchange its electron cloud with the surrounding environment (Table 2). The electrophilicity index value of 1 is about 3.72 eV, which is in the range for strong electrophiles.60 The corresponding ΔNmax value indicates that the reported compound can accept about 1.92 electrons (Table 2).
We have also examined the molecular electrostatic potential (MEP) surface of the optimized structure of 1 to reveal the nucleophilic and electrophilic regions of a molecule. As a result, the carbonyl oxygen atom and the triazole dinitrogen fragment were established to be the most distinguished nucleophilic sites (red colour), while the methyl hydrogen atoms were highlighted as the most pronounced electrophilic sites (blue colour) (Figure 4).

View of the molecular electrostatic potential surface of 1, obtained using the DFT/B3LYP/cc-pVDZ method.
In this work, we have also probed the optimized structure of 1 for its potential ADMET properties. According to ProTox-II,58,59 1 belongs to the fourth class of toxicity with the predicted LD50 of about 1 g/kg (Figure 5). It was also predicted that 1 is an inhibitor of the enzyme, cytochrome P450, nuclear receptor, family A G protein-coupled receptor, phosphatase, hydrolase, oxidoreductase and other membrane protein with the probabilities of 33.3%, 20.0%, 13.3%, 6.7%, 6.7%, 6.7%, 6.7% and 6.7%, respectively (Figure 5). According to the Toxicity Model Report, 1 was revealed to be inactive towards the listed targets (Figure 5).

(top-left and bottom) Toxicity results of 1 calculated by ProTox-II. (top-right) Druggability predictions of 1 calculated by SwissADME.
As evidenced by the SwissADME44 bioavailability radar, the title compound is preferred in all the considered six parameters (Figure 6). Further, one of the efficient approaches to examine molecules for the human blood-brain barrier (BBB) penetration and gastrointestinal absorption (GIA) is the BOILED-Egg method, which is derived from lipohilicity and polarity (Figure 6).57 As such, points in the Egg’s yolk and white correspond to molecules predicted to passively permeate through the BBB and be passively absorbed by the gastrointestinal tract, respectively. Furthermore, molecules found to be effluated (PGP+) and not to be effluated (PGP–) from the central nervous system by the P-glycoprotein are shown as blue and red dots, respectively. Thus, the described compound 1 was predicted to possess negative BBB penetration and positive GIA property with the PGP+ effect (Figure 6).

(left) Bioavailability radar for 1 within the domain borders of ADME properties, calculated by SwissADME. The coloured zone of the radar is the suitable physicochemical space for oral bioavailability. (right) BOILED-Egg model of 1 calculated by SwissADME.
Finally, 1 was examined as a potential inhibitor toward a series of the SARS-CoV-2 proteins (Table 3) using in silico molecular docking. Nowadays, this method is popular for probing the interaction of a biomolecule with a ligand (small molecule) to design and discover new drugs,61,62,63,64 thus being efficient in saving time and money. The target structures of proteins were primarily selected in accordance with the structural features of the virus62,63 as well as based on biological mechanisms and functions that can be utilized to reduce, prevent or treat the virus.64
According to the obtained results, compound 1 efficiently interacts with all the applied proteins and generally shows comparable or slightly higher absolute values of binding scores compared to Remdesivir and almost all redocked initial ligands and even much higher values compared to Favipiravir.34 Compound 1 demonstrates the best binding affinity with Papain-like protease (PLpro) (Table 3), which is defined by two conventional hydrogen bonds with THR75, one π···anion interaction with ASP76, two π···sulfur interactions with TYR154 and HIS175, one π···π stacked interaction with HIS72 and one T-shaped π···π interaction with HIS175 (Figure 7, Table 4). The worst binding affinity of 1 was found for Main protease (Mpro) (Table 3), which is described by three conventional hydrogen bonds with GLN110 and THR111, one π···sigma interaction with ILE106, two π···π stacked interactions with PHE294, three π···alkyl interactions with PHE294 and VAL104 (Figure 7, Table 4). Interestingly, compound 1 binds Nonstructural protein 3 (Nsp3_range 207–379-MES) more efficiently than Nonstructural protein 3 (Nsp3_range 207–379-AMP) (Table 3), which is obviously explained by a greater number of both conventional hydrogen bonds and hydrophobic interactions (Figure 7, Table 4). Notably, it was established that compound 1 interacts with the same efficiency with both Nonstructural protein 16 (Nsp16_GTA site) and Nonstructural protein 16 (Nsp16_SAM site) (Table 3). This is explained by the formation of almost the same intermolecular interactions in the resulting complexes with a marked predominance of hydrogen bonds and electrostatic interactions (Figure 7, Table 4). However, the interaction of compound 1 with Nonstructural protein 16 (Nsp16_MGP site) is less efficient due to a lower number of intermolecular interactions with a predominance of hydrophobic interactions (Figure 7, Table 4).

2D (left) and 3D (right) views on the interaction of 1 with the applied SARS-CoV-2 proteins.
We have also performed molecular dynamics (MD) simulations of 50 ns to evaluate interactions in complexes of 1 with the applied SARS-CoV-2 proteins. Particularly, complexes with Mpro, PLpro, Nsp3_range 207–379-AMP and Nsp3_range 207–379-MES showed an RMSD below 0.5 nm with average values of 0.294, 0.387, 0.386 and 0.370 nm, respectively (Figure 8). However, complexes of 1 with Nsp16_SAM site and Nsp16_MGP site showed a higher RMSD of about 0.5 and 0.6 nm, respectively, with average values of 0.562 and 0.498 nm (Figure S2, Supplementary Information). Furthermore, the complex of 1 with Nsp16_GTA site showed a gradual increase of an RMSD over the whole simulation time, reaching the value of about 0.7 nm with an average value of 0.486 nm (Figure S2, Supplementary Information).

RMSD, RMSF, Rg, SASA and intermolecular hydrogen bonds analysis profiles of complexes of 1 with Main protease (Mpro), Papain-like protease (PLpro), Nonstructural protein 3 (Nsp_range 207–379-AMP) and Nonstructural protein 3 (Nsp_range 207–379-MES).
The RMSF value for complexes of 1 with Mpro, PLpro, Nsp3_range 207–379-AMP and Nsp3_range 207–379-MES was below 0.789, 1.151, 0.365 and 0.431 nm, respectively (Figure 8). The same value for complexes with Nsp16_GTA site, Nsp16_MGP site and Nsp16_SAM site was below 1.841, 1.044, and 0.766 nm, respectively (Figure S2, Supplementary Information). The strongest fluctuations of amino acid residues for each complex are listed in Table S4 (Supplementary Information). Rg values for all the complexes form relatively stable profiles (Figure 8 and Figure S2, Supplementary Information). Particularly, Rg values vary in the ranges 2.186–2.313, 2.631–2.720, 2.310–2.466, 2.319–2.471, 2.258–2.357, 2.224–2.340 and 2.226–2.338 nm for complexes of 1 with Mpro, PLpro, Nsp3_range 207–379-AMP, Nsp3_range 207–379-MES, Nsp16_GTA site, Nsp16_MGP site and Nsp16_SAM site, respectively. The SASA profiles were calculated to predict the interaction between complexes of 1 with the applied proteins and solvents. It was also established that the binding of compound 1 to the applied proteins did not impair the proteins’ interaction with the solvent molecule and the stability of the proteins (Figure 8 and Figure S2, Supplementary Information). During the 50 ns simulation time, the average SASA was calculated as 151.72, 309.74, 164.43, 158.09, 211.46, 203.20 and 207.26 nm2 for complexes of 1 with Mpro, PLpro, Nsp3_range 207–379-AMP, Nsp3_range 207–379-MES, Nsp16_GTA site, Nsp16_MGP site and Nsp16_SAM site, respectively. It was also observed that the complex of compound 1 with Mpro mainly forms 1 intermolecular hydrogen bond during almost the whole simulation time and 2 intermolecular hydrogen bonds at about 12–20 and 24–38 ns (Figure 8). Complex of 1 with PLpro is characterized by forming 1 intermolecular hydrogen bond at about 0–17 and 24–50 ns, and 2 intermolecular hydrogen bonds at about 12–16 ns (Figure 8). Notably, the same complex does not form intermolecular hydrogen bonds at about 17–23 ns (Figure 8). Complexes of 1 with both Nsp3_range 207–379-AMP site and Nsp3_range 207–379-MES site form 1 intermolecular hydrogen bond during the whole simulation time with a much higher frequency in comparison to complex with Mpro (Figure 8). Furthermore, complex of 1 with Nsp3_range 207–379-MES site forms 2 and 3 intermolecular hydrogen bonds during almost the whole simulation time, while complex with Nsp3_range 207–379-AMP site forms 2 and 3 intermolecular hydrogen bonds exclusively at about 26–50 nm (Figure 8). Finally, complexes of compound 1 with Nsp16_GTA site, Nsp16_MGP site and Nsp16_SAM site form 1 intermolecular hydrogen bond at about 12–50, 0–43 and 0–47 ns, respectively, with a relatively rare frequency (Figure S2, Supplementary Information). The formation of 2 intermolecular hydrogen bonds is observed for the same complexes mainly at about 13–22, 1–42 and 0–8 ns, respectively (Figure S2, Supplementary Information).
4 Conclusions
In summary, we report the synthesis, characterization, and theoretical studies of the coumarin-triazole-thiophene hybrid 4-(((4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazol-3-yl)thio)methyl)-6,7-dimethyl-2H-chromen-2-one (1), which was readily obtained with a good yield by reacting 4-ethyl-5-(thiophen-2-yl)-4H-1,2,4-triazole-3-thiol with an equimolar amount of 4-(chloromethyl)-6,7-dimethyl-2H-chromen-2-one in the presence of K2CO3.
The structure of 1 was optimized by the DFT/B3LYP/cc-pVDZ calculations to reveal its geometrical parameters and electronic properties. The obtained bond lengths and angles in the optimized structure of 1 are typical for certain functionalities. The HOMO is mainly delocalized over the thiophene, triazole, and thioester sulfur fragments, while the LUMO is mainly spread over the coumarin fragment of 1. Molecular electrostatic potential surface of the optimized structure of 1 revealed the carbonyl oxygen atom and the triazole dinitrogen fragment as the most distinguished nucleophilic sites, while the methyl hydrogen atoms were found as the most pronounced electrophilic sites. The global chemical reactivity descriptors of the optimized structure of 1 were also calculated, which allowed the revealing its electron-accepting and donating abilities. The optimized structure of 1 exhibits both good electron-donor and electron-acceptor properties and tends to exchange its electron cloud with the surrounding environment. 1 was also found to be a strong electrophile.
Some biological properties of the title compound were evaluated using a set of online tools, which revealed that 1 exhibits negative human blood-brain barrier penetration and positive gastrointestinal absorption property. Furthermore, molecule 1 was predicted to be effluated from the central nervous system by the P-glycoprotein. According to the molecular docking results, compound 1 is active against all the applied SARS-CoV-2 proteins and, in general, shows comparable or slightly higher absolute values of binding scores compared to Remdesivir and almost all the redocked initial ligands and even much higher values compared to Favipiravir. The best binding affinity of 1 was revealed with Papain-like protease (PLpro). According to the molecular dynamics simulation data, compound 1 forms stable complexes with Main protease (Mpro), Papain-like protease (PLpro), Nonstructural protein 3 (Nsp_range 207–379-AMP) and Nonstructural protein 3 (Nsp_range 207–379-MES), while less stable complexes are formed with Nonstructural protein 16 (Nsp16_GTA site), Nonstructural protein 16 (Nsp16_MGP site) and Nonstructural protein 16 (Nsp16_SAM site).
References
Martins P, Jesus J, Santos S, Raposo L R, Roma-Rodrigues C, Baptista P V and Fernandes A R 2015 Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box Molecules 20 16852
Pathania S, Narang R K and Rawal R K 2019 Role of sulphur-heterocycles in medicinal chemistry: An update Eur. J. Med. Chem. 180 486
Jampilek J 2019 Heterocycles in medicinal chemistry Molecules 24 3839
https://www.mdpi.com/journal/molecules/special_issues/Heterocycles_Medicinal_Chemistry
Thematic issue “Heterocyclic Compounds in Medicinal Chemistry” 2020 Chem. Heterocycl. Comp. 56 625
https://link.springer.com/journal/10593/volumes-and-issues/56-6
Heravi M M and Zadsirjan V 2020 Prescribed drugs containing nitrogen heterocycles: an overview RSC Adv. 10 44247
Wu L, Wang X, Xu W, Farzaneh F and Xu R 2009 The structure and pharmacological functions of coumarins and their derivatives Curr. Med. Chem. 16 4236
Medina F G, Marrero J G, Macías-Alonso M, González M C, Córdova-Guerrero I, Teissier Garcíaa A G and Osegueda-Robles S 2015 Coumarin heterocyclic derivatives: chemical synthesis and biological activity Nat. Prod. Rep. 32 1472
Wei H, Ruan J and Zhang X 2016 Coumarin–chalcone hybrids: promising agents with diverse pharmacological properties RSC Adv. 6 10846
Frizzo CP and Martins M A P 2012 Aromaticity in heterocycles: new HOMA index parametrization Struct. Chem. 23 375
Mahajan N S, Manikrao A M, Shinde P N, Jawarkar R D, Khatale P N and Dhawale S C 2012 A Review: Biological Importance of Mercapto Substituted 1,2,4-triazole Derivatives Res. J. Pharm. Tech. 5 863
Banerjee S, Ganguly S and Sen K K 2013 A Review on 1,2,4-Triazoles J. Adv. Pharm. Edu. Res. 3 102
Maddila S, Pagadala R and Jonnalagadda S B 2013 1,2,4-Triazoles: A Review of Synthetic Approaches and the Biological Activity Lett. Org. Chem. 10 693
Kaur P and Chawla A 2017 1,2,4-Triazole: A review of pharmacological activities Int. Res. J. Pharm. 8 10
Sameliuk Y G, Al Zedan F and Kaplaushenko T M 2021 1,2,4-Triazole derivatives in medicine and pharmacy and application prospects J. Fac. Pharm. Ankara 45 598
Pasricha S, Mittal K, Gahlot P, Kaur H, Avasthi N and Shweta 2022 Multicomponent synthetic strategies and perspectives for synthesis of linked or fused coumarin heterocycles J. Iran. Chem. Soc. https://doi.org/10.1007/s13738-022-02603-x
Negi M, Chawla P A, Faruk A and Chawla V 2020 Role of heterocyclic compounds in SARS and SARS CoV-2 pandemic Bioorg. Chem. 104 104315
Özdemir M, Köksoy B, Ceyhan D, Sayın K, Erçağ E, Bulut M and Yalçın B 2020 Design and in silico study of the novel coumarin derivatives against SARS-CoV-2 main enzymes J. Biomol. Struct. Dyn. 27 1
Kumar C S, Ali D, Alarifi S, Radhakrishnan S and Akbar I 2020 In silico molecular docking: Evaluation of coumarin based derivatives against SARS-CoV-2 J. Infect. Public Health 13 1671
Mohamed N M and Eltelbany R F F 2021 Synthetic Coumarin Derivatives as SARS-CoV-2 Major Protease Inhibitors: Design, Synthesis, Bioevaluation and Molecular Docking ChemistrySelect 6 13616
Abdelmohsen UR, Albohy A, Abdulrazik BS, Bayoumi SAL, Malak LG, Khallaf ISA, et al. 2021 Natural coumarins as potential anti-SARS-CoV-2 agents supported by docking analysis RSC Adv. 11 16970
Nejabat M, Ghodsi R and Hadizadeh F 2022 Coumarins and Quinolones as Effective Multiple Targeted Agents Versus Covid-19: An In Silico Study Med. Chem. 18 220
Abdizadeh R, Hadizadeh F and Abdizadeh T 2022 In silico analysis and identifcation of antiviral coumarin derivatives against 3-chymotrypsin-like main protease of the novel coronavirus SARS-CoV-2 Mol. Divers. 26 1053
Seck I and Nguemo F 2021 Triazole, imidazole, and thiazole-based compounds as potential agents against coronavirus Results Chem. 3 100132
Seliem I A, Panda S S, Girgis A S, Moatasim Y, Kandeil A, Mostafa A, et al. 2021 New quinoline-triazole conjugates: Synthesis, and antiviral properties against SARS-CoV-2 Bioorg. Chem. 114 105117
Arikrishnan J, Pazhamalai S, Manikandan H, Sekar S, Kalaivani P and Gopalakrishnan M 2020 Synthesis, Characterization, ADMET Prediction and Molecular Docking Studies Against COVID-19 Proteins of Novel 2-(3-(4-substituted aryl)guanidine-1-yl)-4-phenyl-6-(thiophene-2-yl)pyrimidines Eur. J. Mol. Clin. Med. 7 3930
Abdel-Latif E, Khatab T K, Fekri A and Khalifa M E 2021 Synthesis of New Binary Thiazole-Based Heterocycles and Their Molecular Docking Study as COVID-19 Main Protease (Mpro) Inhibitors Russ. J. Gen. Chem. 91 1767
Tlidjane H, Chafai N, Chafaa S, Bensouici C and Benbouguerra K 2022 New thiophene-derived α-aminophosphonic acids: Synthesis under microwave irradiations, antioxidant and antifungal activities, DFT investigations and SARS-CoV-2 main protease inhibition J. Mol. Struct. 1250 131853
Ay B, Şahin O, Demir B S, Saygideger Y, López-de-Luzuriaga J M, Mahmoudi G and Safin D A 2020 Antitumor effects of novel nickel–hydrazine complexes in lung cancer cells New J. Chem. 44 9064
Alkhimova LE, Babashkina M G and Safin D A 2021 α-Aminophosphonates 4-XC6H4–NH–CH(4-BrC6H4)–P(O)(OiPr)2 (X = H, Br, MeO): Crystal structures, Hirshfeld surface analysis, computational studies and in silico molecular docking with the SARS-CoV-2 proteins Tetrahedron 97 132376
Shiryaev A A, Goncharenko A N, Burkhanova T M, Alkhimova L E, Babashkina M G, Chandrasekaran R and Safin D A 2021 A chiral (1R,2R)-N, N′-bis-(salicylidene)-1,2-diphenyl-1,2-ethanediamine Schiff base dye: Synthesis, crystal structure, Hirshfeld surface analysis, computational study, photophysical properties and in silico antifungal activity J. Iran. Chem. Soc. 18 2897
Babashkina M G, Frontera A, Kertman A V, Saygideger Y, Murugavel S and Safin D A 2022 Favipiravir: Insight into the crystal structure, Hirshfeld surface analysis and computational study J. Iran. Chem. Soc. 19 85
Alkhimova L E, Babashkina M G and Safin D A 2022 Computational analysis of aspirin J. Mol. Struct. 1251 131975
Alkhimova L E, Burkhanova T M, Babashkina M G and Safin D A 2022 A readily available structural analogue of integrastatins A and B: insight into the crystal structure, Hirshfeld surface analysis and computational study Tetrahedron 109 132671
Sharov A V, Burkhanova T M, Taskın Tok T, Babashkina M G and Safin D A 2022 Computational analysis of molnupiravir Int. J. Mol. Sci. 23 1508
Burkhanova T M, Babashkina M G, Taskin-Tok T, Sharov A V and Safin D A 2022 Naphthalene-based bis-N-salicylidene aniline dyes: Crystal structures, Hirshfeld surface analysis, computational study and molecular docking with the SARS-CoV-2 J. Iran. Chem. Soc. 19 1979
Alkhimova LE, Sharov AV, Burkhanova TM, Babashkina MG and Safin DA 2022 Ambroxol: Insight into the Crystal Structure, Hirshfeld Surface Analysis and Computational Study Polycycl. Aromat. Comp. https://doi.org/10.1080/10406638.2022.2049323
Babashkina M G and Safn D A 2022 6-Amino-2-(4-fuorophenyl)-4-(trifuoromethyl)quinoline: insight into the crystal structure, Hirshfeld surface analysis and computational study Polycycl. Aromat. Comp. https://doi.org/10.1080/10406638.2022.2068622
Babashkina M G, Taskin-Tok T, Burkhanova T M and Safin D A 2022 1-hydroxy-6,6-Dimethyl-3-Phenyl-1,6-Dihydropyridine-2,5-Dione as a Promising Inhibitor of the SARS-CoV-2 Proteins: insight into the Crystal Structure, Hirshfeld Surface Analysis and Computational Study Polycycl. Aromat. Comp. https://doi.org/10.1080/10406638.2022.2094420
Babashkina M G, Panova E V, Alkhimova L E and Safn D A 2022 Salen: insight into the crystal structure, Hirshfeld surface analysis, optical properties, DFT, and molecular docking studies Polycycl. Aromat. Comp. https://doi.org/10.1080/10406638.2022.2097281
Koparir P, Omar R A, Sarac K, Ahmed L O, Karatepe A, Taskin-Tok T and Safin D A 2022 Synthesis, Characterization and Computational Analysis of Thiophene-2,5-Diylbis((3-Mesityl-3-Methylcyclobutyl)Methanone) Polycycl. Aromat. Comp. https://doi.org/10.1080/10406638.2022.2112712
Burkhanova T M, Krysantieva A I, Babashkina M G, Konyaeva I A, Monina L N, Goncharenko A N and Safin D A 2022 In silico analyses of Betulin: DFT studies, corrosion inhibition properties, ADMET prediction and molecular docking with a series of SARS-CoV-2 and monkeypox proteins Struct. Chem. https://doi.org/10.1007/s11224-022-02079-8
Dennington R, Keith T A and Millam J M 2016 GaussView, Version 6.0, Semichem Inc., Shawnee Mission, KS
Frisch M J, Trucks G W, Schlegel H B, Scuseria G E, Robb M A, Cheeseman J R, Scalmani G, Barone V, Mennucci B, Petersson G A, Nakatsuji H, Caricato M, Li X, Hratchian H P, Izmaylov A F, Bloino J, Zheng G, Sonnenberg J L, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Montgomery Jr. J A, Peralta J E, Ogliaro F, Bearpark M, Heyd J J, Brothers E, Kudin K N, Staroverov V N, Keith T, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant J C, Iyengar S S, Tomasi J, Cossi M, Rega N, Millam J M, Klene M, Knox J E, Cross J B, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann R E, Yazyev O, Austin A J, Cammi R, Pomelli C, Ochterski J W, Martin R L, Morokuma K, Zakrzewski V G, Voth G A, Salvador P, Dannenberg J J, Dapprich S, Daniels A D, Farkas O, Foresman J B, Ortiz J V, Cioslowski J and Fox D J 2013 Gaussian 09, Revision D.01
Krishnan R, Binkley J S, Seeger R and Pople J A 1980 Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions J. Chem. Phys. 72 650
Becke A D 1993 Density-functional thermochemistry. III. The role of exact exchange J. Chem. Phys. 98 5648
Frisch M J, Pople J A and Binkley J S 1984 Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets J. Chem. Phys. 80 3265
Trott O and Olson A J 2010 AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading J. Comput. Chem. 31 455
Eberhardt J, Santos-Martins D, Tillack A F and Forli S 2021 AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings J. Chem. Inf. Model. 61 3891
Rose Y, Duarte J M, Lowe R, Segura J, Bi C, Bhikadiya C, et al. 2021 RCSB protein data bank: Architectural advances towards integrated searching and efficient access to macromolecular structure data from the PDB archive J. Mol. Biol. 433 166704
BIOVIA, Dassault Systèmes, BIOVIA Discovery Studio, 2020, San Diego: Dassault Systèmes
Daina A, Michielin O and Zoete V 2017 SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules Sci. Rep. 7 42717
WebGRO for Macromolecular Simulations, https://simlab.uams.edu/.
Schüttelkopf A W and van Aalten D M F 2004 PRODRG: a tool for high-throughput crystallography of protein-ligand complexes Acta Crystallogr. 60 1355
Berendsen H J C, Grigera J R and Straatsma T P 1987 The missing term in effective pair potentials J. Phys. Chem. 91 6269
Diana A and Zoete V 2016 A BOILED-Egg To Predict Gastrointestinal Absorption and Brain Penetration of Small Molecules ChemMedChem 11 1117
Banerjee P, Eckert A O, Schrey A K and Preissner R 2018 ProTox-II: a webserver for the prediction of toxicity of chemicals Nucleic Acids Res. 46 w257
Pérez P, Domingo L R, Aizman A and Contreras R 2007 Chapter 9 The electrophilicity index in organic chemistry. In Theoretical and Computational Chemistry A Toro-Labbé (Ed.) (Elsevier B.V.) Vol. 19 pp. 139–291
Li H, Wang H-Y, Kang S, Silverman R B and Poulos T L 2016 Electrostatic Control of Isoform Selective Inhibitor Binding in Nitric Oxide Synthase Biochemistry 55 3702
Tok T T and Tatar G 2017 Structures and functions of coronavirus proteins: molecular modeling of viral nucleoprotein Int. J. Virol. Infect. Dis. 2 001
Tok T T and Gowder S J T 2020 An Updated Review on Covid-19 with Special Reference to Structural Elucidation and Functional Properties Biomed. J. Sci. Tech. Res. 31 24345
Shamsi A, Mohammad T, Anwar S, Amani S, Khan M S, Husain F M, et al. 2021 Potential drug targets of SARS-CoV-2: From genomics to therapeutics Int. J. Biol. Macromol. 177 1
Ding X-C, He J, Zhang X, Jiang C, Sun Y, Zhang Y, et al. 2021 Crucial Mutations of Spike Protein on SARS-CoV-2 Evolved to Variant Strains Escaping Neutralization of Convalescent Plasmas and RBD-Specific Monoclonal Antibodies Front. Immunol. 12 693775
https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON385
Author information
Authors and Affiliations
Corresponding authors
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Omar, R.A., Koparir, P., Sarac, K. et al. A novel coumarin-triazole-thiophene hybrid: synthesis, characterization, ADMET prediction, molecular docking and molecular dynamics studies with a series of SARS-CoV-2 proteins. J Chem Sci 135, 6 (2023). https://doi.org/10.1007/s12039-022-02127-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12039-022-02127-0