
Abstract
L. donovani is an intracellular protozoan parasite, that causes visceral leishmaniasis (VL), and consequently, post-kala azar dermal leishmaniasis (PKDL). Diagnosis and treatment of leishmaniasis is crucial for decreasing its transmission. Various diagnostic techniques like microscopy, enzyme-linked immunosorbent assays (ELISA) and PCR-based methods are used to detect leishmaniasis infection. More recently, loop-mediated isothermal amplification (LAMP) assay has emerged as an ideal diagnostic measure for leishmaniasis, primarily due to its accuracy, speed and simplicity. However, point-of-care diagnosis is still not been tested with the LAMP assay. We have developed a portable LAMP device for the monitoring of Leishmania infection. The LAMP assay performed using our device can detect and amplify as little as 100 femtograms of L. donovani DNA. In a preliminary study, we have shown that the device can also amplify L. donovani DNA present in VL and PKDL patient samples with high sensitivity (100%), specificity (98%) and accuracy (99%), and can be used both for diagnostic and prognostic analysis. To our knowledge, this is the first report to describe the development and application of a portable LAMP device which has the potential to evolve as a point-of-care diagnostic and prognostic tool for Leishmania infections in future.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Leishmaniasis is caused by parasitic protozoans of the genus Leishmania which are transmitted by the bite of the female sandfly belonging to the family Phlebotominae (WHO 2013). The disease manifests clinically as (a) cutaneous leishmaniasis (CL) characterized by the presence of self-healing skin ulcers, (b) mucocutaneous leishmaniasis (MCL) which causes skin and mucosal ulcers and (c) visceral leishmaniasis (VL; kala-azar) which affects the liver, spleen, bone marrow and lymph nodes (WHO 2015). VL, caused by Leishmania donovani, is endemic in East Africa and South Asia, while autochthonous cases of VL in South America are caused by L. infantum chagasi (WHO 2019). India alone accounts for more than 80% of the total VL cases in the Indian subcontinent (WHO 2015). In India, VL is endemic in the eastern states of Bihar, West Bengal, Jharkhand and Uttar Pradesh, where around 130 million people are at risk (Mandal et al. 2018). Another manifestation, post-kala-azar dermal leishmaniasis (PKDL) is a dermal sequel of apparently cured VL (WHO 2019). PKDL is endemic in East Africa and South Asia (WHO 2019). In the Indian subcontinent, up to 15% of apparently cured VL patients develop PKDL (Ramesh et al. 2015). As PKDL is the proposed disease reservoir for VL, early detection and treatment of cases of PKDL is the need of the hour. Currently, there are no accessible diagnostic tests for this disease, creating a challenge for the physician.
In order to manage patients and decrease disease transmission, accurate and rapid laboratory diagnosis succeded by proper treatment is essential. The gold standard for VL diagnosis is the parasitological confirmation of biopsy specimens by microscopy. Additionally, serological diagnostics include rapid diagnostic tests (RDTs) and enzyme-linked immunosorbent assays (ELISAs) based on the rK39 antigen (Boeleart et al. 2014). Although the most definitive diagnostic approach in VL and PKDL would be parasite detection, it has an unacceptably low detection rate ranging from 4–58%. Furthermore, in PKDL, especially in the macular variant, a major challenge is their hypopigmented lesions are indistinguishable from pitryasis versicolor and is further confounded by the inability to detect LD bodies. In such cases, detection of parasite DNA remains the best option. Moreover, in PKDL cases, positivity of RDTs can be attributed to a past episode of VL (Adams et al. 2013). Another promising approach is PCR-based assays used to diagnose leishmaniasis, viz. restriction fragment length polymorphism analysis, nested PCR, triplex PCR, multiplex PCR and quantitative PCR, all of which offer high specificity and sensitivity (Koltas et al. 2016; Schönian et al. 2003; Reithinger et al. 2007; da Cunha Gonçalves-de-Albuquerque et al. 2014; Rodríguez-González et al. 2007; Adams et al. 2018). However, the requirement for complex and expensive equipment and infrastructure means that such diagnostic techniques are not suitable for use in the field.
An advancement to standard PCR-based diagnosis techniques came when Notomi et al. (2000) designed the loop-mediated isothermal amplification (LAMP) technique. In contrast to PCR, wherein the reaction is carried out at a series of alternating temperature steps or cycles, isothermal amplification is carried out at a constant temperature, using four sets of primers (to enable high specificity) and a Bst polymerase with high strand displacement activity in addition to a replication activity (Notomi et al. 2000). Thus, this technique obviates the need for a thermal cycler and can be performed in a dry bath, enhancing its field applicability. LAMP has been used for the diagnosis of infections caused by bacteria, viruses, fungi and parasites like Trypanosoma brucei, T. cruzi, Plasmodium falciparum and L. donovani (Qiao et al. 2007; Villari et al. 2013; Inacio et al. 2008; Wozniakowski et al. 2013; Poon et al. 2006; Nzelu et al. 2014; Takagi et al. 2009). More recently, LAMP has also been used to detect SARS CoV-2 RNA in clinical samples (Thi et al. 2020). In order to diagnose leishmaniasis, LAMP primers can target the small ribosomal subunit (18S rRNA gene), kinetoplast DNA (kDNA) and the Internal Transcribed Spacer 1 (ITS-1). Additionally, LAMP has also been used to detect parasites in their insect vectors, viz. T. cruzi and T. rangeli (Thekisoe et al. 2010).
Although LAMP has emerged to be an efficient, quick and specific technique to diagnose Leishmania infection, the requirement of a bulky dry bath and fluorescence spectrophotometer limits its applicability in the field work or point-of-care analysis. Therefore, in the present study, we describe the development and application of a portable, table-top LAMP device and fluorescence readout unit which can be used for the rapid point-of-care diagnosis of Leishmania infection. The LAMP device has enabled the development of a highly sensitive, specific and cheap diagnostic tool for patients with VL and PKDL by using as little as 100 fg of DNA. As per our knowledge, this is the first report of a portable, point-of-care LAMP device to diagnose VL and PKDL.
2 Materials and methods
2.1 Chemicals
Bst DNA polymerase large Fragment (NEB, USA), Thermo Pol Buffer (provided with the enzyme), magnesium sulphate (NEB, USA), betaine (Sigma-Aldrich, USA) and SYBR Gold nucleic acid stain, 10,000x concentration (Thermo Fischer Scientific, USA) were used in this study. All other materials used were of analytical grade and commercially available.
2.2 Parasite DNA
Genomic DNA from L. donovani Bob strain (LdBob strain/MHOM/SD/62/1SCL2D), initially obtained from Dr. Stephen Beverley (Washington University, St. Louis, MO, USA), L. donovani AG83 strain and P. falciparum 3D7 strain provided by Dr. Pawan Malhotra (International Centre for Genetic Enginnering and Biotechnology, New Delhi, India) were used in this study. Promastigotes were cultured at 26°C in M199 medium (Sigma-Aldrich, USA), supplemented with 100 units/ml penicillin (Sigma-Aldrich, USA), 100 µg/ml streptomycin (Sigma-Aldrich, USA) and 10% heat-inactivated fetal bovine serum (FBS; Biowest). DNA was isolated using phenol-chloroform extraction method.
2.3 Study population and sample management
The study was based on a convenience sampling method. Coded samples (n = 70) of peripheral blood from patients clinically diagnosed with VL (10 naïve, 5 post-treatment samples) and dermal biopsies from patients diagnosed with PKDL (40 confirmed, 20 suspected samples) respectively, were used in this study. As controls, blood from healthy volunteers from non-endemic regions of leishmaniasis (15 samples) was provided along with skin biopsies from patients with leprosy (10 samples; confirmed by a restriction length M. leprae‑specific repetitive element PCR) (Moulik et al. 2018a, b). From 2003 to date, patients clinically diagnosed with VL or PKDL were recruited by passive surveillance from the Tropical Medicine and Dermatology outpatient departments of the School of Tropical Medicine/Calcutta Medical College/ Institute of Postgraduate Medical Education and Research, Kolkata, West Bengal or by active surveillance from 2015 onwards wherein active field surveys were conducted in VL endemic districts of West Bengal (Malda, Dinajpur, Darjeeling and Birbhum) by a camp approach. An initial house-to-house survey was conducted by first-line health workers (Kala-azar Technical Supervisors) using standard case definitions and defined risk factors e.g. living in an endemic area and having an epidemiological link (past history of VL). In addition, none suffered from any co-infection or pre-existing disease and pregnant women were excluded. In patients suspected with VL, peripheral blood was collected at disease presentation and within 48h of completion of treatment. The blood was stored at −20°C until DNA isolation was performed. DNA isolation was performed from the blood according to the manufacturer’s protocol. At disease presentation, rK39 strip test was performed for initial diagnosis, and ITS-1 PCR was additionally performed for confirmation along with determination of parasite load (no of parasites/µg of genomic DNA). At 24–48 h after completion of treatment with a single dose of Liposomal Amphotericin B, heparinised blood was collected, and a repeat ITS-1 PCR and parasite load quantification were performed, prior to the patient being discharged. When no product was observed for ITS-1 PCR and the parasite load was <10, the patient was considered as negative for VL. In suspected cases of PKDL (based on clinical suspicion, a rk39 strip test positivity and a past history of VL), a 4 mm punch biopsy was collected at disease presentation and on completion of treatment. The punch biopsies were collected in sterile PBS and within 24 h transferred to RNAlater solution. The DNA isolation was performed immediately or the tissues were stored in RNAlater solution at -80°C. The suspected cases of PKDL (n = 20) tested as ITS-1 PCR negative. These cases were recruited during active field surveys from different VL endemic districts of West Bengal but did not subsequently report to the medical camps and were lost to follow up. Hence, they were considered as PKDL negative, but their final diagnosis could not be determined. The study received approval from the Institutional Ethics Committee, IPGMER, Kolkata and all patients or their legally accepted representative provided informed written consent.
2.4 DNA isolation from clinical samples and diagnosis by ITS-1 PCR
DNA extraction was performed according to manufacturer’s instructions (Qiagen, Hilden, Germany) from skin biopsies collected in phosphate buffered saline (20 mM, pH 7.4), excised into small pieces, and DNA eluted in 50 µL of DNA elution buffer. PCR was performed using Leishmania-specific primers LITSR [5′-CTGGATCATTTTCCGATG-3′] and L5.8S [5′-TGATACCACTTATCGCACTT-3′]) using Red Taq polymerase in a Master cycler (Applied Biosystems, California, USA) (Das et al. 2011). The PCR products were visualized by agarose gel electrophoresis (2.0%) and analyzed in G-BOX gel doc (Syngene, Cambridge, UK) using Gene Tools (version 4.01.04) software. DNA from L. donovani strain MHOM/IN/1983/AG83 served as the positive control, while DNA isolated from the foreskin of healthy individuals (undergoing voluntary circumcision) served as the negative control. Informed consent was obtained from the patient or in case of a minor from their legally accepted representative.
2.5 Determination of parasite load
For measurement of parasite load, a standard curve was generated as previously described (Moulik et al. 2018a, b). Briefly, blood (180 µl) from a healthy control was added to L. donovani parasites (ranging from 10 to 1 X 105). DNA was then eluted and real-time PCR was performed. A standard curve was prepared, the number of parasites was extrapolated and the final parasite load was expressed as the number of parasites/µg genomic DNA.
2.6 LAMP primer design
Four set of primers for L. donovani kinetoplast minicircle DNA (GenBank accession no. Y11401) were designed using Primer Explorer software. Primer sequences are given in table 1.
2.7 LAMP assay
Primer stocks (100 µM) were diluted to make a working stock of 10X, containing 16 µM FIP, 16 µM BIP, 2 µM F3 and 2 µM B3C primers. The LAMP reaction was set-up in a total volume of 25 µl, and contained 1X Thermo Pol buffer, 6 mM MgSO4, 1.4 mM dNTPs, 0.8 M betaine, nuclease-free water, 1X primer mix, 8 units of Bst DNA polymerase and template DNA. Serial dilutions containing 1 nanogram (ng), 100 picogram (pg), 1 pg and 100 femtogram (fg) of L. donovani genomic DNA and clinical sample DNA were prepared and used as template in the LAMP reaction. A non-template control was used as a negative control in every reaction. To ensure that there was no cross-reactivity of the LAMP primers, they were tested with 1 pg of P. falciparum 3D7 genomic DNA. The reaction was performed at 65°C for 1 h in the LAMP device. A schematic description of LAMP assay performed in the device is illustrated in (figure 1).

Schematic representation of the workflow for LAMP amplification and readout measurement. Diagnostic test workflow: the samples were processed to isolate the total DNA. The isolated DNA was added into the vials containing reagents. The vials were placed in the LAMP device and amplification was performed at 65°C for 1 h. SYBR Green gold dye was then added to the amplification products, followed by measuring the fluorescence in the portable readout unit, which was connected to a laptop. The data was analyzed by comparing the ADC values of control and test sample, and the report was generated.
2.8 Detection of LAMP amplification
LAMP amplification was detected by adding 2 µl of SYBR Gold DNA stain diluted 1:10 in nuclease-free water. Positive amplification was indicated by an orange to green color change in the sample, whereas negative controls remained orange. The color change was visualized by naked eyes, and quantified by a readout unit that detects fluorescence intensity values (hereby referred to as ADC values). A schematic description of the detection of LAMP amplification in the readout unit is illustrated in (figure 1). Further confirmation of amplification was done by electrophoresing 3 µl of the reaction mixture on a 2.5% agarose gel and detecting the characteristic ladder-like band pattern by staining the gel with ethidium bromide.
2.9 LAMP amplification device
The LAMP device's core consists of a resistive heating block and a system controller: the heating block heats and holds samples at specific temperatures, typically in the range of 40–100°C, while the system controller maintains the particular temperature and time duration set by the user. These components are placed in a 3D printed casing shown in (figure 2A). The amplification reaction temperature ranges between 50–100°C. For point-of-care applications, it is necessary to provide the test results immediately after completion of the test. Hence, a portable LAMP readout module has been developed to help obtain the LAMP reaction test result in point-of-care settings.

Components of the portable LAMP device and readout unit. (A) The LAMP device consists of 16 sample wells, a heating thermal block and the lid of the device which features a heating element to prevent condensation during the LAMP amplification process. There are three indicator lights in the front panel: red to show the operation status of system power, orange to show the Bluetooth connection, and green to display the amplification reaction process. The reaction settings are set using a mobile application developed for this device, that connects the LAMP device to a smartphone. The app has a simple graphical user interface to set the temperature and time for the reaction process and shows the real-time temperature of the block. (B) The fluorescence readout unit has an integrated LED light source, optical filters (excitation and emission filters suitable for SYBR Gold), a sample vial holder, a photodiode, and the electronic driving circuit. The LAMP samples (control/test) are mixed with SYBR Gold and placed in the sample holder for fluorescence readout. When the measurement is initiated the LED and photodiode are synchronized to read the fluorescence emission from the sample. The system measures the fluorescence levels at an interval of 3 seconds (1 second on and 2 seconds off). The data obtained from the unit is logged into a file where it can be used for further comparative analysis between control and test samples.
The LAMP device consists of an aluminum thermal block with 16 sample vials, with an integrated heater lid to prevent condensation in the sample vial during the amplification process. The reaction settings are set using a mobile application developed for this device, that connects the LAMP device to a smartphone. The app has a simple graphical user interface to set the temperature and time for the reaction process and shows the real-time temperature of the block. There are three indicator lights in the front panel, red, orange and green, to show the operation status of system power, Bluetooth connection, and amplification reaction process, respectively (figure 2A).
The LAMP device has integrated custom-designed hardware powered by Nordic semiconductor nrf52832 microcontroller IC with Bluetooth system-on-chip (SoC). The system consists of a control board for controlling input/output and Bluetooth operation, a carrier board for driving the heaters in the thermal block and the lid, and monitoring and controlling the temperature through the integrated sensors. The thermal block is made of aluminum, designed and machined to hold 16 standard PCR tubes with a reaction volume range between 10–50 µL. The system has 25-W resistors attached to two sides of the thermal block walls for heating and a thermistor to measure the block temperature; the maximum operating temperature of the block is 100°C with +1°C accuracy. The lid is integrated with a 25-W resistive heater, and the temperature is by default set to 10°C above the set block temperature to prevent condensation. The system requires 12V, 5A DC power source for operation and can also be powered using a portable power bank.
2.10 LAMP fluorescent readout unit
The components of the LAMP readout unit are illustrated in (figure 2B). The unit is designed to analyse the test samples and controls using nucleic acid intercalating dyes, such as the SYBR Gold nucleic acid stain. The SYBR Gold stain is a flurophore that has a peak excitation wavelength of 497 nm and the emission when bound to DNA is centred at 520 nm. The best source for excitation is a laser, but complexity and cost hinder its use in point-of-care devices. Therefore, a high power LED with wider spectrum has been used in the readout system (LXML-PE01-0070-High Power LEDs - Single Colour Cyan 70lm, 350mA wavelength range of 490–515nm). Because the LED has a wider spectrum which overlaps with the emission wavelength ranges, an excitation filter between the LED and sample (CWL 490nm, Dia 12.5mm, BW: 15nm for excitation) has been introduced. However, the light emitted by LEDs is unfocused, and to overcome this problem a 15 mm focus lens has been used to focus light from the excitation filter to a smaller spot on the sample. The emitted light from the sample is separated from the LED using an emission filter (CWL 520 nm, Dia 12.5 mm, BW: 15 nm) and a collector lens is used to focus the emitted light to the photodiode (BPW21R) which has peak sensitivity at 565 nm. The current produced by the photodiode is then sent to LTC 1051 op-amp. The signal from op-amp is read by the ADC pin of microcontroller (Atmega328P) and can be stored in a laptop using the serial data logger software. The emitted fluorescence values are read at a 3 second interval wherein the LED is on for 1 second and off for the remaining 2 seconds.
2.11 Statistical analysis
LAMP assay ADC value data were analyzed by GraphPad prism and represented as mean ±standard error of the mean (S.E.M.). The percentage sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV) and accuracy with 95% confidence intervals (CI) were calculated as follows using Medcalc software: sensitivity = [true positive / true positive + false negative)], specificity = [true negative / (true negative + false positive)], PPV = [true positive / (true positive + false positive)], NPV = [true negative / (true negative + false negative)] and accuracy = (true positive + true negative) / (true positive + false negative + false positive + true negative). The Spearman’s correlation coefficient (with 95% CI) between LAMP assay ADC values and parasite load was also calculated by GraphPad prism. Student’s unpaired 2-tailed t-test was used to calculate significance. P <0.05 was considered statistically significant.
3 Results
3.1 Inclusivity and exclusivity of the LAMP assay performed in the device
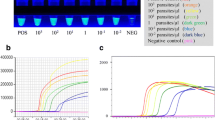
The foremost parameters used to evaluate the LAMP assay performed in the device were the inclusivity and exclusivity of the reactions. One ng of genomic DNA from L. donovani Bob and AG83 strains was successfully amplified in the device, as evident by an orange to green color change in the positive samples upon the addition of SYBR Gold stain (figure 3A and B) and the characteristic ladder-like pattern observed when the samples were electrophoresed on an agarose gel (figure 3C). Therefore, the LAMP assay performed in the device appears to be inclusive when amplifying L. donovani DNAs. The assay was also observed to be exclusive, as the L. donovani kinetoplast minicircle DNA primers were unable to amplify P. falciparum genomic DNA, as apparent by the lack of color change or ladder-like band pattern (supplementary figure 1A-C). In order to determine the ideal time for LAMP amplification of L. donovani DNA, a time-course analysis was performed. One ng of L. donovani Bob genomic DNA was subjected to LAMP amplification in the device for 15, 30, 45 and 60 min. While some amplification as evident by orange to green color change was observed at 15, 30 and 45 min (supplementary figure 2A and B), strong bands were observed after 60 min (supplementary figure 2C), thereby implying that 60 min is the ideal time for the LAMP amplification of L. donovani DNA in the device.

Inclusivity of the LAMP reaction performed in the device. The inclusivity of the LAMP reaction performed in the device was determined. One ng of genomic DNA from each of L. donovani Bob and AG83 strains was used in a LAMP reaction for 60 min, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control, Ld Bob: genomic DNA from L. donovani Bob strain, Ld AG83: genomic DNA from L. donovani AG83 strain. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder.
3.2 Anticipated range of L. donovani DNA amplified in the LAMP device
Another parameter to evaluate the analytical performance of the LAMP device was to ascertain the range of L. donovani DNA that can be amplified. Several concentrations of L. donovani Bob genomic DNA were tested for amplification, and it was observed that 1 ng, 100 pg, 1 pg and 100 fg could be amplified in the LAMP device (figure 4A-C). Therefore, 100 fg of L. donovani DNA is the limit of detection of the LAMP device. The range of detection was also determined for L. donovani DNA present in clinical samples. For this, 1 ng, 100 pg, 1 pg and 100 fg of DNA from a positive and negative sample were used in a LAMP reaction, and while all concentrations of the positive sample showed amplification, 1 ng of the negative sample showed a false-positive result and lower concentrations showed no amplification (figure 5A-C). Therefore, 100 pg was chosen as the ideal concentration at which the positive clinical sample showed positive result and the negative clinical sample showed negative result in the LAMP reaction.

Anticipated range of L. donovani genomic DNA amplified in the LAMP device. One ng, 100 pg, 1 pg and 100 fg of L. donovani Bob genomic DNA were used in a LAMP reaction for 60 min, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder.

Anticipated range of L. donovani DNA in clinical samples amplified in the LAMP device. 1 ng, 100 pg, 1 pg and 100 fg of DNA each from a positive and a negative clinical sample were used in a LAMP reaction for 60 min, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder.
3.3 Detection of L. donovani DNA in samples from VL and PKDL patients
In order to test the ability of the LAMP device to detect and amplify L. donovani DNA in clinical samples, DNA from 10 naïve VL cases was used in a LAMP assay using this device. ITS-1 PCR was performed as a standard diagnostic test and the parasite load was determined in all the samples and all the 10 samples tested positive in the LAMP assay (figure 6A-C, table 2). DNA from 60 PKDL patients (40 confirmed and 20 suspected cases) was also tested in the LAMP device. ITS-1 PCR was performed as a standard diagnostic test and the parasite load was determined in all the samples (table 3). All the 40 confirmed samples tested positive whereas the 20 suspected samples tested negative in both ITS-1 PCR and LAMP assay. A representative image of LAMP assay data from 9 PKDL patient samples is shown in (figure 7A-C). Therefore, an extremely significant correlation between parasite load obtained by ITS-1 PCR and ADC values obtained in the LAMP assay was observed in VL and PKDL patient samples (n = 50, 95% CI = 0.9134 to 0.9725; Spearman r = 0.9510; P< 0.0001) (figure 8).

LAMP amplification of L. donovani DNA from VL patient samples. One hundred pg of DNA from 10 naïve VL patient samples were used in a LAMP reaction for 60 min, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder.

LAMP amplification of L. donovani DNA from PKDL patient samples. One hundred pg of DNA from 60 (40 confirmed, 20 suspected) PKDL patient samples were used in a LAMP reaction for 60 min, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder. A representative image of data from 9 PKDL patient samples is shown.

Scatterplot of VL and PKDL patient parasite load and LAMP ADC value. One hundred pg of DNA from 10 naïve VL and 40 confirmed PKDL patient samples were used in a LAMP reaction for 60 min, as described in Materials and Methods. The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The scatter graph shows the parasite load in the samples vs. the ADC values obtained in the LAMP assay.
3.4 Detection of L. donovani DNA in non-endemic control (NEC) and leprosy patient samples
In order to test the specificity of the L. donovani LAMP assay, 25 negative controls were used. ITS-1 PCR was performed as a standard diagnostic test and the parasite load was determined in all the samples. Fifteen samples from NEC (supplementary table 1) and 10 samples from leprosy patients (supplementary table 2) were used in a LAMP assay performed in the device. All the NEC and 9 out of 10 leprosy patient samples tested negative in the LAMP assay. LAMP assay data from all 15 NEC samples (supplementary figure 3A-C) and 10 leprosy patient samples (supplementary figure 4A-C) is shown.
3.5 Detection of L. donovani DNA in post-treatment VL patient samples
The above experiments have unequivocally proven the efficacy of the LAMP device in the diagnosis of leishmaniasis. We wanted to determine if the device can also be used in testing the prognosis of treatment. Therefore, 5 post-treatment VL patient samples were subjected to LAMP assay, after testing them in ITS-1 PCR (supplementary table 3). In concordance with ITS-1 PCR data, none of the samples showed amplification in the LAMP assay (supplementary figure 5A-C), thereby implying that the LAMP device can function as an effective prognostic tool.
3.6 Comparison of diagnostic test evaluation parameters of ITS-1 PCR and LAMP assay
An evaluation of the analytical performance of the LAMP assay, in comparison to that of ITS-1 PCR was performed (table 4). Out of the total 50 true positive (10 naïve VL and 40 confirmed PKDL) and 50 true negative (20 suspected PKDL, 15 NEC, 10 leprosy and 5 post-treatment VL) samples tested in the LAMP device, no false negative and 1 false positive results were obtained. The LAMP assay performed in the device was highly sensitive (100.00%; 95% CI= 92.89-100.00), specific (98.00%; 95% CI= 89.35-99.95) and accurate (99.00%; 95% CI= 94.55-99.97).
4 Discussion
Leishmaniasis is a neglected tropical disease which impacts the poor in endemic regions, with no or limited access to a correct diagnosis due to a restricted health service infrastructure. Prompt diagnosis and treatment is imperative to reduce the morbidity of those affected by leishmaniasis (WHO 2010). The widely used rK39 strip test as a diagnostic test is appropriate for VL but not for PKDL as a positive result may occur because of persisting antibodies from a past episode of VL. Hence, the identification of PKDL patients is crucial for the success of the VL Elimination Program as they serve as durable reservoirs during inter-epidemic episodes (WHO 2010). Since its development by Notomi et al., the Loop-mediated isothermal amplification (LAMP) test has been used in the diagnosis of several infectious and hereditary diseases (Notomi et al. 2000, 2015). Recently, a LAMP test developed by Eiken Chemical Co. has been given the green signal by the WHO to be used in the diagnosis of tuberculosis (WHO 2016). LAMP has also been shown to detect Leishmania infection in sand flies, in the diagnosis of canine leishmaniasis, as well as human VL, CL and PKDL with high sensitivity and specificity either using direct examination with the naked eye, or fluorimetry or turbidimetry (Nzelu et al. 2014, 2019, 2016; Takagi et al. 2009; Kothalawala and Karunaweera 2016; Verma et al. 2013, 2017; Khan et al. 2012; Chaouch et al. 2013; Avelar et al. 2019).
Despite its numerous advantages, the requirement of bulky equipment like water baths, PCR machines and fluorescent detectors limits the applicability of LAMP in field work or point-of-care analysis. Our present study attempts to solve this problem with the introduction of a handy, portable LAMP device and a fluorescent detection device which are easy-to-operate and yield accurate and quantitative results. The development of less invasive methods of sample collection like finger-prick blood sampling and direct LAMP assays avoiding DNA isolation steps are warranted for faster and convenient field-based diagnosis.
In the present study, we have developed a portable, table-top LAMP device and fluorescence read-out unit which can be used for the rapid point-of-care diagnosis of Leishmania infection. We have shown that the LAMP device can amplify minute quantitites of L. donovani DNA, ranging from 1 ng to 100 fg in one hour. In a pilot study, we have also successfully amplified L. donovani DNA from VL and PKDL patients using the LAMP device with excellent sensitivity, specificity and accuracy. Previous studies evaluating LAMP for the diagnosis of VL using kDNA as the target have also shown high sensitivity, ranging from 93-100%, and specificity, ranging from 90.7-100% (Khan et al. 2012; Verma et al. 2013, 2017; Ghasemian et al. 2014; Mukhtar et al. 2018). Our LAMP assay analysis is concordant with these previous studies, with a sensitivity of 100% and specificity of 98%. We have performed ITS-1 PCR and kDNA real time PCR as reference tests on all samples, and our LAMP results show an exteremely significant correlation with the reference test results. In order to further test the specificity of our LAMP assay, we included samples from non-endemic controls and leprosy patients in our study. LAMP assay performed on non-endemic controls did not detect the presence of L. donovani, which is in agreement with the results of the reference tests (n=15/15). Similarly, in leprosy patients with no exposure to Leishmania, both the reference tests and the LAMP assay showed no presence of L. donovani (n=10/10). Therefore, our LAMP assay system can be used as an efficient tool for the diagnosis of VL and PKDL as it is simpler and faster.
Several read-out methods for LAMP assay have been developed, which include detection by agarose gel electrophoresis of LAMP products, detection by turbidity, colorimetric dyes like hydroxy naphthol blue and malachite green or fluorescent dyes like SYBR Green. Visual inspection of tubidity is not always very accurate. Recent advances have been made in colorimetric detection methods and real time fluorimetry with the development of Genie III real time fluorometer and ES Quant TS2.2 real time fluorimeters (Ibarra-Meneses et al. 2018; Dixit et al. 2021). These devices are portable, battery-operated and provide opportunities for diagnostic connectivity using Wifi/Bluetooth. The fluorescent detection unit of our LAMP assay system is also a handy, table-top device that can quantify the fluorescent intensity of LAMP products (in terms of ADC values).
A recent study has reported the development and clinical evaluation of LAMP assay for the diagnosis of VL in Brazil, targeting the K26 antigen-coding gene of L. donovani complex (Avelar et al. 2019). The authors have reported a limit of detection as low as 1fg of parasite DNA, which is slightly lower than that obtained by our kinetoplast minicircle DNA-based LAMP assay system (100 fg). However, our LAMP assay is clinically more sensitive (100%) as compared to that of Avelar et al. (2019) (98%), which could be attributed to the large copy number (approximately 10,000 copies) of kinetoplast minicircle DNA. Moreover, in our system, the mode of assay read-out is by measurement of fluorescence (in terms of ADC values) using our fluorescence detection device, which is more accurate and quantitative as compared to visual inspection of turbidity as reported in the study by Avelar et al. (2019). Another critical difference between the two studies is that our LAMP system has also been validated for the detection of PKDL, which has not been reported by Avelar et al. (2019).
LAMP assay has also been utilized in the assessment of cure at VL and PKDL post-treatment stages (Verma et al. 2017). We have also demonstrated that our LAMP device can be used not just as a diagnostic tool, but also for the monitoring of treatment. LAMP assay performed in our device did not detect parasite DNA in the post-treatment VL (n = 5/5) cases, and similar results were obtained by ITS-1 PCR and kDNA real time PCR. This concordance in the results of LAMP and reference assays at the post-treatment stage emphasizes the utility of LAMP assay as a prognostic assay for the point-of-care test for assessment of cure of VL cases. However, a detailed evaluation of the assay in more patients is required to evaluate its prognostic efficacy.
Eiken Chemical Co. and partners have recently developed the Loopamp™ Leishmania Detection Kit for the diagnosis of leishmaniasis, which has been evaluated for the diagnosis of VL in Sudan and Bangladesh (Mukhtar et al. 2018; Hossain et al. 2021). Although the kit showed excellent sensitivity in VL detection, further evaluation studies of this kit are necessary in India.
Our LAMP detection system has various novel features. The foremost advantage of our system is that we have designed a compact fluorescent detection device which can be used to quantify the fluorescence intensity of LAMP products (as measured by ADC values). Our data depicts a significant correlation between fluorescence intensity of LAMP products and parasite load present in the sample. Another salient feature of our system is that our LAMP device is Bluetooth enabled, and can be operated through a dedicated mobile app, which makes the operation highly user-friendly. Additionally, our LAMP system can be used to distinguish between PKDL and leprosy infections, which will aid the physician in differential diagnosis. Our system is also low-cost: one test in our device costs less than 1.5 USD. This is extremely beneficial for under developed and developing countries, which carry the highest burden of disease for neglected tropical diseases like leishmaniasis. With disruptive cost advantage of more than 10x compared to imported devices with similar capabilities, our LAMP assay system has the potential to serve as a platform for affordable molecular diagnosis for leishmaniasis and have a significant impact in the healthcare diagnostics sector in India.
References
Adams ER, Versteeg E and Leeflang MMG 2013 Systematic review into diagnostics for post-kala-azar dermal leishmaniasis (PKDL). J. Trop. Med. https://doi.org/10.1155/2013/150746
Adams ER, Schoone G, Versteeg I, et al. 2018 Development and Evaluation of a novel loop-mediated isothermal amplification assay for diagnosis of cutaneous and visceral Leishmaniasis. J. Clin. Microbiol. 56 e00386-e418
Avelar DMD, Carvalho DM and Rabello A 2019 Development and clinical evaluation of loop-mediated isothermal amplification (LAMP) assay for the diagnosis of human visceral Leishmaniasis in Brazil. Biomed. Res. Int
Boelaert M, Verdonk K, Menten J, Sunyoto T, van Griensven J, Chappuis F and Rijal S 2014 Rapid tests for the diagnosis of visceral leishmaniasis in patients with suspected disease. Cochrane Database Syst. Rev.
Chaouch M, Mhadhbi M, Adams ER, Schoone GJ, Limam S, Gharbi Z, Darghouth MA, Guizani I and BenAbderrazak S 2013 Development and evaluation of a loop-mediated isothermal amplification assay for rapid detection of Leishmania infantum in canine leishmaniasis based on cysteine protease B genes. Vet. Parasitol. 198 78–84
da Cunha Gonçalves-de-Albuquerque S, e Silva RP, de Morais RC, Trajano-Silva LA, Régis-da-Silva CG, Brandão-Filho SP and Paiva-Cavalvanti M 2014 Tracking false-negative results in molecular diagnosis: proposal of a triplex-PCR based method for leishmaniasis diagnosis. J. Venom Anim. Toxins Incl. Trop. Dis. 20 16
Das NK, Singh SK, Ghosh S, Sarkar A, Mukhopadhyay D, Roy S, Ganguly DN, Barbhuiya JN, Saha B and Chatterjee M 2011 Case series of misdiagnosis with rK39 strip test in Indian leishmaniasis. Am. J. Trop. Med. Hyg. 84 688–691
Dixit KK, Ramesh V, Gupta R, Negi NS, Singh R and Salotra P 2021 Real-time fluorimetry loop-mediated isothermal amplification for diagnosis of leishmaniasis and as a tool for assessment of cure for post–kala-azar dermal leishmaniasis. Am. J. Trop. Med. Hyg. 104 2097–2107
Ghasemian M, Gharavi MJ, Akhlaghi L, Mohebali M, Meamar AR, Aryan E and Oormazdi H 2014 Development and assessment of loop-mediated isothermal amplification (LAMP) assay for the diagnosis of human visceral leishmaniasis in Iran. Iran. J. Parasitol. 9 50–59
Hossain F, Picado A, Owen SI, et al. 2021 Evaluation of LoopampTM Leishmania detection kit and Leishmania antigen ELISA for post-elimination detection and management of visceral leishmaniasis in Bangladesh. Front. Cell Infect. Microbiol. 11 670759
Ibarra-Menesses AV, Cruz I, Chicharro C, Sánchez C, Biéler S, Broger T, Moreno J and Carrillo E 2018 Evaluation of fluorimetry and direct visualization to interpret results of a loop-mediated isothermal amplification kit to detect Leishmania DNA. Parasit. Vectors 11 250
Inacio J, Flores O and Spencer-Martins I 2008 Efficient identification of clinically relevant Candida yeast species by use of an assay combining panfungal loop-mediated isothermal DNA amplification with hybridization to species-specific oligonucleotideprobes. J. Clin. Microbiol. 46 713–720
Khan MG, Bhaskar KR, Salam MA, Akther T, Pluschke G and Mondal D 2012 Diagnostic accuracy of loop-mediated isothermal amplification (LAMP) for detection of Leishmania DNA in buffy coat from visceral leishmaniasis patients. Parasit. Vectors 5 280
Koltas IS, Eroglu F, Uzun S and Alabaz D 2016 A comparative analysis of different molecular targets using PCR for diagnosis of old world leishmaniasis. Exp. Parasitol. 164 43–48
Kothalawala HS and Karunaweera ND 2016 Loop-mediated isothermal amplification assay as a sensitive diagnostic tool for Leishmania donovani infections in Sri Lanka. Ceylon Med. J. 61 68–70
Mandal R, Kesari S, Kumar V and Das P 2018 Trends in spatio-temporal dynamics of visceral leishmaniasis cases in a highly-endemic focus of Bihar, India: an investigation based on GIS tools. Parasit. Vectors 11 220
Mercaldo ND, Lau KF and Zhou XH 2007 Confidence intervals for predictive values with an emphasis to case–control studies. Stat. Med. 26 2170–2183
Moulik S, Sengupta R, Dighal A, Sardar B, Saha B, Das NK and Chatterjee M 2018 Identification of atypical dermal leishmaniasis resolved by restriction fragment lengthpolymorphism. Indian J. Dermatol. Venereol Leprol. https://doi.org/10.4103/ijdvl.IJDVL_14_18
Moulik S, Chaudhuri SJ, Sardar B, Ghosh M, Saha B, Das NK and Chatterjee M 2018b Monitoring of parasite kinetics in Indian post kala azar dermal leishmaniasis. Clin. Infect. Dis. 66 404–410
Mukhtar M, Ali SS, Boshara SA, et al. 2018 Sensitive and less invasive confirmatory diagnosis of visceral leishmaniasis in Sudan using loop-mediated isothermal amplification (LAMP). PLoS Negl. Trop. Dis. 12 e0006264
Notomi T, Okayama H, Masubuchi H, Yonekawa T, Watanabe K, Amino N and Hase T 2000 Loop-mediated isothermal amplification of DNA. Nucleic Acids Res. 28 E63
Notomi T, Mori Y, Tomita N and Kanda H 2015 Loop-mediated isothermal amplification (LAMP): principle, features, and future prospects. J. Microbiol. 53 1–5
Nzelu CO, Gomez EA, Caceres AG, et al. 2014 Development of a loop-mediated isothermal amplificationmethod for rapid mass-screening of sand flies for Leishmania infection. Acta Trop. 132 1–6
Nzelu CO, Cáceres AG, Guerrero-Quincho S, et al. 2016 A rapid molecular diagnosis of cutaneous leishmaniasis by colorimetric malachite green-loop-mediated isothermal amplification (LAMP) combined with an FTA card as a direct sampling tool. Acta Trop. 153 116–119
Nzelu CO, Kato H and Peters NC 2019 Loop-mediated isothermal amplification (LAMP): An advanced molecular point-of-care technique for the detection of Leishmania infection. PLoS Negl. Trop. Dis. 13 e0007698
Poon LL, Wong BW, Ma EH, Chan KH, Chow LM, Abeyewickreme W, Tangpukdee N, Yuen KY, Guan Y, Looareesuwan S and Peiris JSM 2006 Sensitive and inexpensive molecular test for Falciparum malaria: detecting Plasmodium falciparum DNA directly from heat-treated blood by loop-mediated isothermal amplification. Clin. Chem. 52 303–306
Qiao YM, Guo YC, Zhang XE, Zhou YF, Zhang ZP, Wei HP, Yang RF and Wang DB 2007 Loop-mediated isothermal amplification for rapid detection of Bacillus anthracis spores. Biotechnol. Lett. 29 1939–1946
Ramesh V, Kaushal H, Mishra AK, Singh R and Salotra P 2015 Clinico-epidemiological analysis of Post kala-azar dermal leishmaniasis (PKDL) cases in India over last two decades: a hospital based retrospective study. BMC Public Health 15 1092
Reithinger R and Dujardin JC 2007 Molecular diagnosis of leishmaniasis: current status and future applications. J. Clin. Microbiol. 45 21–25
Rodríguez-González I, Marín C, Longoni SS, et al. 2007 Identification of New World Leishmania species from Peru by biochemical techniques and multiplex PCR assay. FEMS Microbiol. Lett. 267 9–16
Schönian G, Nasereddin A, Dinse N, Schweynoch C, Schallig HD, Presber W and Jaffe CL 2003 PCR diagnosis and characterization of Leishmania in local and imported clinical samples. Diagn. Microbiol. Infect. Dis. 47 349–358
Takagi H, Itoh M, Islam MZ, Razzaque A, Ekram AR, Hashighuchi Y, Noiri E and Kimura E 2009 Sensitive, specific, and rapid detection of Leishmania donovani DNA by loop-mediated isothermal amplification. Am. J. Trop. Med. Hyg. 81 578–582
Thekisoe OM, Rodriguez CV, Rivas F, Coronel-Servian AM, Fukumoto S, Sugimoto C, Kawazu SI and Inoue N 2010 Detection of Trypanosoma cruzi and T. rangeli infections from Rhodniuspallescens bugs by loop-mediated isothermal amplification (LAMP). Am. J. Trop. Med. Hyg. 82 855–860
Thi VLD, Herbst K, Boerner K, Meurer M, Kremer LPM, Kirrmaier D, Freistaedter A, et al. 2020 A colorimetric RT-LAMP assay and LAMP-sequencing for detecting SARS-CoV-2 RNA in clinical samples. Sci. Transl. Med. 12 556
Verma S, Avishek K, Sharma V, Negi NS, Ramesh V and Salotra P 2013 Application of loop-mediated isothermal amplification assay for the sensitive and rapid diagnosis of visceral leishmaniasis and post-kala-azar dermal leishmaniasis. Diagn. Microbiol. Infect. Dis. 75 390–395
Verma S, Singh R, Sharma V, Bumb RA, Negi NS, Ramesh V and Salotra P 2017 Development of a rapid loop-mediated isothermal amplification assay for diagnosis and assessment of cure of Leishmania infection. BMC Infect. Dis. 17 223
Villari C, Tomlinson JA, Battisti A, Boonham N, Capretti P and Faccoli M 2013 Use of loop-mediated isothermal amplification fordetection of Ophiostomaclavatum, the primary blue stain fungus associated with Ipsacuminatus. Appl. Environ. Microbiol. 79 2527–2533
World Health Organization 2013 Post-kala-azar dermal leishmaniasis: a manual for case management and control: report of a WHO consultative meeting, Kolkata, India, 2–3 July 2012 World Health Organization. https://apps.who.int/iris/handle/10665/78608
World Health Organization 2016 The use of loop-mediated isothermal amplification (TB-LAMP) for the diagnosis of pulmonary tuberculosis: policy guidance. WHO/HTM/TB/2016.07
WHO 2010 World Health Organization Control of the leishmaniasis: report of a meeting of the WHO Expert Committee on the Control of Leishmaniases, Geneva 22–26 March 2010. WHO technical report series No. 949. Geneva:WHO
World Health Organization 2015 Kala-azar elimination programme: Report of a WHO consultation of partners Geneva, Switzerland
World Health Organization 2019 Fact sheet on leishmaniasis
Wozniakowski G, Samorek-Salamonowicz E and Kozdrun W 2013 Comparison of loop-mediated isothermal amplification and PCR for the detection and differentiation of Marek’s disease virus serotypes 1, 2, and 3. Avian Dis. 57 539–543
Acknowledgements
We thank Dr. Pawan Malhotra (International Centre for Genetic Enginnering and Biotechnology, New Delhi, India) for kindly providing us with the P. falciparum 3D7 genomic DNA.
Funding
RM is an A. S. Paintal Distinguished Scientist Chair of ICMR. MP and EM are University Grants Commission D. S. Kothari Post-Doctoral Fellows. HKB is a recipient of funding from the Council of Scientific & Industrial Research, India. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors have declared that no competing interests exist.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Sudha Bhattacharya.
Corresponding editor: Sudha Bhattacharya
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary figure 1.
Exclusivity of the LAMP reaction performed in the device. The exclusivity of the LAMP reaction performed in the device was checked. One ng of P.falciparum 3D7 genomic DNA was used in a LAMP reaction for 60 minutes, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control, NC Pf: negative control with P. falciparum 3D7 genomic DNA and L. donovani primers. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder. (TIF 271 KB)
Supplementary figure 2.
Time-course analysis of LAMP reaction. A time-course analysis of the LAMP reaction performed in the device was carried out. One ng of L. Donovani Bob genomic DNA was used in a LAMP reaction for 15, 30, 45 and 60 minutes, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder. (TIF 376 KB)
Supplementary figure 3.
LAMP assay to detect L. donovani DNA in non-endemic control (NEC) samples. One hundred pg of DNA from 15 NEC samples were used in a LAMP reaction for 60 minutes, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder. (TIF 146 KB)
Supplementary figure 4.
LAMP assay to detect L. donovani DNA in leprosy patient samples. One hundred pg of DNA from 10 leprosy patient samples were used in a LAMP reaction for 60 minutes, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder. (TIF 146 KB)
Supplementary figure 5.
LAMP assay to detect L. donovani DNA in post-treatment VL patient samples. One hundred pg of DNA from 5 post-treatment VL samples were used in a LAMP reaction for 60 minutes, as described in Materials and Methods. (A) The confirmation of LAMP amplification was done by SYBR Gold detection. Diluted SYBR Gold nucleic acid stain was added to the LAMP products, and the color change was detected by visual examination. --: non-template control. (B) The quantification of SYBR Gold fluorescence was done by measuring the fluorescence intensity of the samples by a detection device, designated as ADC values. The mean + SEM of 10 ADC values is plotted for each sample. (C) Electrophoresis of LAMP products on a 2.5% agarose gel. M: 100 bp DNA ladder. (TIF 317 KB)
Rights and permissions
About this article

Cite this article
Puri, M., Brar, H.K., Mittal, N. et al. Rapid diagnosis of Leishmania infection with a portable loop-mediated isothermal amplification device. J Biosci 46, 92 (2021). https://doi.org/10.1007/s12038-021-00211-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s12038-021-00211-0