
Abstract
Parkinson’s disease (PD) represents the most common neurodegenerative movement disorder. We recently identified 16 novel genes associated with PD. In this study, we focused the attention on the common and rare variants identified in the lysosomal K+ channel TMEM175. The study includes a detailed clinical and genetic analysis of 400 cases and 300 controls. Molecular studies were performed on patient-derived fibroblasts. The functional properties of the mutant channels were assessed by patch-clamp technique and co-immunoprecipitation. We have found that TMEM175 was highly expressed in dopaminergic neurons of the substantia nigra pars compacta and in microglia of the cerebral cortex of the human brain. Four common variants were associated with PD, including two novel variants rs2290402 (c.-10C > T) and rs80114247 (c.T1022C, p.M341T), located in the Kozak consensus sequence and TM3II domain, respectively. We also disclosed 13 novel highly penetrant detrimental mutations in the TMEM175 gene associated with PD. At least nine of these mutations (p.R35C, p. R183X, p.A270T, p.P308L, p.S348L, p. L405V, p.R414W, p.P427fs, p.R481W) may be sufficient to cause the disease, and the presence of mutations of other genes correlated with an earlier disease onset. In vitro functional analysis of the ion channel encoded by the mutated TMEM175 gene revealed a loss of the K+ conductance and a reduced channel affinity for Akt. Moreover, we observed an impaired autophagic/lysosomal proteolytic flux and an increase expression of unfolded protein response markers in patient-derived fibroblasts. These data suggest that mutations in TMEM175 gene may contribute to the pathophysiology of PD.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is a common neurodegenerative disorder characterized by a wide spectrum of motor symptoms (bradykinesia, muscular rigidity, resting tremor, and postural instability) which are caused by the progressive loss of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc) [1, 2]. The pathological hallmark of PD is the presence of Lewy bodies containing alpha-synuclein aggregates [3] in some surviving SNpc neurons [4].
In the last years, genome-wide association studies (GWAS) and next-generation sequencing (NGS) analysis of large cohorts of familiar and sporadic cases identified a number of genes/variants associated with PD; however, for the majority of them, the causal role in the pathogenesis of PD has not yet been identified [5,6,7].
In a recent study, we identified 16 novel candidate genes for PD in Italian families. These genes are expressed in the mesencephalon and are involved in pathways, controlling mitochondrial metabolism and oxidative stress, vesicular trafficking, microtubule dynamics, and autophagy, all pathways potentially deregulated in PD [8]. We also demonstrated that polygenic inheritance of rare variants in PD Mendelian genes might predict the disease occurrence in both familial and sporadic cases highlighting the importance of rare variants also in the genetics of sporadic PD [8].
TMEM175, one of the novel genes identified in our cohort of PD families, is located in a highly significant PD GWAS peak at chromosome 4 [9, 10]. Recently, two common coding variants, rs34884217 (c.A194C, p.Q65P) and rs34311866 (c.T1178C, p.M393T), have been associated with PD risk in several populations [11,12,13,14]. This gene encodes for an endolysosomal K+ channel that is involved in K+ conductance, pH stability, and α-synuclein clearance [15,16,17,18]. Deficiency in TMEM175 leads to the loss of dopaminergic neurons and impairment in motor function in mice [19].
TMEM175 is a protein of great interest for PD etiopathogenesis because it might be potentially modulated by pharmacological agents. However, a detailed genetic analysis is still missing.
In this study, we performed a comprehensive molecular and genetic analysis in PD Italian patients. We identified novel common and rare variants associated with PD and examined their potential role in the pathophysiology of the disease.
Patients and Methods
Study Participants
Four hundred PD patients (243 males and 157 females; 178 familiar and 222 sporadic cases) were recruited at the Istituto di Ricovero e Cura a Carattere Scientifico (IRCCS, Scientific Institute for Research, Hospitalization and Healthcare) Mediterranean Neurological Institute (MNI) Neuromed in Pozzilli, here called MNI cohort. The project was approved by the ethical committees of IRCCS Neuromed, and written informed consent was signed by all participants. All subjects were of European ancestry and were evaluated by qualified neurologist of the Parkinson Centre from June 2015 to December 2017, with a thorough protocol comprising neurological examination and evaluation of non-motor domains. Information about family history, demographic characteristics, anamnesis, and pharmacological therapy was also collected [8, 20, 21].
The Movement Disorder Society revised version of the Unified Parkinson’s Disease Rating Scale Part III (18 items, maximum score 72; hereafter called UPDRS) [22] was used for the assessment of clinical motor symptoms. These included language, facial expressions, tremor, rigidity, agility in movements, stability, gait, and bradykinesia. Cognitive abilities were tested through an Italian validated version of the Montreal Cognitive Assessment (MoCA) [23]. The assessed cognitive domains include short-term memory (5 points); visuospatial abilities via clock drawing (3 points) and a cube copy task (1 point); executive functioning via an adaptation of Trail Making Test Part B (1 point), phonemic fluency (1 point), and verbal abstraction (2 points); attention, concentration, and working memory via target detection (1 point), serial subtraction (2 points), digits forward and backward (1 point each); language via confrontation naming with low-familiarity animals (3 points), and repetition of complex sentences (2 points); and orientation to time and place (6 points). The total score was given by the sum of these domains and then divided by the maximum score that could be obtained (30 points). Where one or more domains could not be tested (e.g., visuospatial tasks, due to unavailability of optical devices), the corresponding score was subtracted from the total score obtainable.
Non-motor symptoms were assessed through an Italian-validated version of Non-motor Symptoms Scale (NMMS) for Parkinson’s disease [24]. This scale tests 9 items, including cardiovascular domain, sleep/fatigue, mood/cognition, perceptual problems/hallucinations, attention/memory, gastrointestinal, urinary, sexual function, and ability to taste or smell. For each item, both severity and frequency of symptoms are measured, so that the scale accounts for both aspects. Here, the sleep domain was slightly modified by adding a further question on the occurrence of vivid dreams. This question was treated like all the others, i.e., the severity of impairment was scored from 0 (no symptoms) to 3 (severe impairment), and the frequency of impairment was scored from 0 (less than once a week) to 4 (daily impairment); then the total score of the sub-item was computed as the product of severity by frequency and added to the scores of the other sub-items.
A subset of participants carrying deleterious variants in TMEM175 gene underwent a standardized magnetic resonance imaging (MRI) protocol with a 3 T SIGNA HD platform (United States General Electric Medical Group, GE Healthcare) including the following sequences: high-resolution 3-dimensional T1-weighted (T1-3D) magnetization-prepared rapid acquisition with gradient echo sequence (repetition time [TR] = 1900 ms, echo time [TE] = 2.93 ms, flip angle = 9, field of view [FOV] = 260 mm, matrix = 256 × 256, 176 contiguous sagittal 1-mm thick slices) and dual turbo spin-echo proton density and T2-weighted images (TR = 3320 ms, TE = 10/103 ms, FOV = 220 mm, matrix = 384 × 384, 25 axial 4-mm thick slices, 30% gap).
Whole-exome sequencing (WES) data were available for 112 PD cases. WES was performed by using the SureSelect All Exome kit v6 (Agilent® Technologies, Santa Clara, CA, USA) for DNA fragmentation and capture. Exomes were barcoded and sequenced at Helmholtz Zentrum, München, Germany, using the Illumina® HiSeq2000 platform.
Next-generation sequencing-targeted resequencing (NGS-TR) was performed on 288 PD cases. Probes specific for 100 genes, as described previously [8], were designed with Nimble Design software (Roche Diagnostics, Mannheim Germany). Targeting regions were enriched using the SeqCap kit based on DNA fragmentation (Kapa Hyper plus kit) and capture (Roche Diagnostics, Mannheim Germany). Targeted regions were barcoded and sequenced on MiSeq platform (Illumina, San Diego, CA, USA).
Italian control cohort included whole genome data of 107 samples of the Tuscan Italians (TSI) population of the 1000 Genome Project (phase 3 release) and NGS-TR data of 192 healthy subjects from Neuromed biobank [8].
Human Dermal Fibroblast (hDF) Cell Lines
The hDF cells were generated in our laboratory. Skin samples of PD patients and unrelated healthy individuals were obtained by skin punch biopsy mostly at the inner side of the upper arm. Tissue pieces were enzymatically digested for 5 h at 37 °C by using Collagenase/Dispase kit (Roche Diagnostics, Mannheim Germany), followed by mechanically disaggregation with knife. Cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4.5 g/L D-glucose, 0.11 g/L sodium pyruvate, 20% FBS, 1% NEAA, 1% L-glutamine, and 1% Pen-Strep at 37 °C in a humidified CO2 5% air for about 4–5 weeks. hDF cell lines were maintained and were used in all the experiments in sub-confluent monolayers. Starvation was performed for 1 h in HBSS. In bafilomycin treatment, cells were starved for 2 h in HBSS with/without 100 mM bafilomycin.
Plasmid Preparation, Cell Culture, and Transfection
The human wild-type (wt) TMEM175 construct was purchased at Source Bioscience and was used as template for PCR reaction to obtain the full-length cDNA fragment that was introduced into the pCagMCSEGFPiresNEO expression vector to generate a GFP-tagged TMEM175 construct. The mutagenesis of the wt cDNA to obtain mutant forms (c.C103T, p.R35C; c.G808A, p.A270T; c.C923T, p.P308L; c.T1022C, p.M341T; c.C1213G, p.L405V) was carried out by using the Quick Change II XL site-directed mutagenesis technique according to the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA, USA). The coding region of wt and mutant plasmids was sequenced to confirm the mutagenesis and to exclude undesired mutations.
The mouse dopaminergic neuronal cell line, MN9D, was cultured in high-glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA) and 2% L-glutamine in a humidified atmosphere of 5% CO2 at 37 °C.
Ten micrograms of the constructs were linearized and electroporated in MN9D cells. Stable transfected clones were grown and selected by puromycin. The mutant and wt clones were analyzed by WB and immunoprecipitation (IP) assay.
Human embryonic kidney 293 (HEK293) cells and HeLa cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with 4.5 g/L D-glucose, 0.11 g/L sodium pyruvate, 10% FBS (Thermo Fisher Scientific, Waltham, MA, USA), 1% L-glutamine, and 1% Pen-Strep at 37 °C in a humidified atmosphere of 5% CO2. Ten micrograms of the constructs were electroporated in HEK293 cells for transient transfection.
Patch-Clamp Experiments
HEK293 cells transfected with the GFP-tagged wt or mutant proteins were plated (80,000 cells) on 35-mm tissue culture dishes 24/48 h before the experimental day. Currents were recorded with a Multiclamp 700B amplifier and Clampex 10.5 software (Molecular Devices, San Jose, CA, USA) in the perforated patch-clamp configuration to reduce the perturbation of the intracellular milieu, by using escin (50 μM). Patch-clamp recordings were obtained using borosilicate glass electrodes (4–6 MΩ) filled with intracellular solution contained (in mM) 140 CsMetSO3, 5 BAPTA-AM, 2 MgCl2, 2 MgATP, 10 HEPES buffered with CsOH, and pH = 7.4. During recordings, cells were continuously perfused using normal external solutions (NES), containing (in mM) 140 Na-gluconate (Na-Glu), 2.8 KCl, 2 MgCl2, 2 CaCl2, 10 glucose, 10 HEPES buffered with NaOH, and pH 7.4. Cells were held at − 70 mV, and currents were elicited with voltage ramps (from − 100 to + 100 mV, 200 ms) every 2 s. The current density at + 30 mV was determined as the current amplitude/cell capacitance (pA/pF) [15]. Cell capacitance was continuously monitored.
Data analysis was performed using SigmaPlot 14.0 (Systat). All data are expressed as means ± standard error mean and analyzed using Student’s t-test. Significance for all tests was set at P < 0.05.
Western Blot and Immunoprecipitation (IP) Assay
hDF cells were collected and washed with cold PBS (2.7 mM KCl, 1.2 mM KH2PO4, 8.1 mM Na2HPO4, 138 mM NaCl [pH 7.4]). Total proteins were solubilized in lysis buffer (20 mM Tris–HCl [pH 7.4], 1 mM ethylenediaminetetraacetic acid (EDTA), 0.4 mM NaF, 0.04 mM Na3VO, 1% NP40, protease inhibitors) and sonicated in ice. Extraction of separate cytoplasmic and nuclear protein fractions was performed with NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Fisher Scientific, Waltham, MA, USA). Proteins were separated by SDS-PAGE and transferred on PVDF membrane (Millipore, Bedford, Massachusetts, USA). The level of expression of TMEM175, LC3I/II, p62, LAMP1, TFEB, GAPDH, and PARP1 was determined by immunoblot analysis using anti-LAMP1 (ab25630, Abcam, Cambridge, UK, 1:1000), anti-LC3B (2775, Cell Signaling, Danvers, Massachusetts, USA, 1:1000/), anti-TMEM175 (19,925–1-AP, Proteintech, Manchester, UK, 1:1000) and anti-p62/SQSTM1 (P0067, Sigma-Aldrich, St. Louis, MO, USA, 1:15,000), anti-TFEB (A303-673A, Bethyl Laboratories (Waltham, MA, USA), 1:1000), anti-PARP1 (66,520-1Ig, Proteintech, Manchester, UK, 1:20,000), and anti-GAPDH (sc-32233, Santa Cruz Biotechnology (Dallas, Texas USA), 1:1000). Cytoplasmic proteins were normalized to GAPDH, whereas nuclear proteins were normalized to PARP1.
Immunoprecipitation (1 mg total lysate) was performed using the anti-Akt antibodies (Immunological Sciences, Cat. N. MAB-94320) complexed to protein G-Sepharose (Invitrogen, Cat. N. 101,243). The immunoprecipitated proteins were resolved on 10% SDS–PAGE, transferred for 2 h at room temperature on PVDF membrane, and detected by immunoblotting with GFP (1:1000) (Synaptic System, Cat. N. 132 002) and Akt (1:1000). HRP-conjugated secondary antibodies (Immunological Sciences, Cat. N. IS20402) were used at 1:5000 dilution. Protein bands were detected by ECL and visualized by Quantity One software (Bio-Rad Laboratories).
The mean standard deviations of 3 independent experiments were analyzed as multiple datasets with ANOVA test for multiple comparisons or with Student’s t-test. Data were plotted as histogram representation. A value of p < 0.05 was considered statistically significant.
Expression Studies
Data of the human brain (cortex and substantia nigra) single-cell expression profile were from Webber laboratory [25]. Total RNA from human adult tissues was from Stratagene (La Jolla, CA, USA); total RNA from hDF was isolated from 106 cells; midbrain, cortex, striatum, and hippocampus RNAs were extracted from three adult mice (P45). RNA was isolated by using EuroGold Trifast kit (Euroclone, IT) protocol. Five micrograms of total RNA was digested with TURBO™ DNase (RNase free) kit (Thermo Fisher Scientific, Waltham, MA, USA) to eliminate genomic DNA contamination. Two micrograms of DNA-free RNA were reverse transcribed with the Superscript III-First strand kit (Thermo Fisher Scientific, Waltham, MA, USA). Quantitative PCR (qPCR) reactions were performed in triplicate, using gene specific primers (Table S1) and ITaq Universal Sybr Green Supermix (Bio-Rad, Hercules, CA, USA) following the manufacturer’s directions. Results were normalized to the expression of the glyceraldehydes-3-phosphate dehydrogenase (GAPDH) gene. Standard deviation was calculated by using data of three different experiments.
Immunofluorescence Staining of hDF Cell Lines
hDF cells (20,000 cells in 24-well plates on coverslip) were grown in complete medium for 24 h. Starvation was performed for 1 h in Hanks’ Balanced Salt solution (HBSS) before fixing with paraformaldehyde (PFA) 4% and staining with anti-LAMP1, anti-LAMP2, anti-p62, anti-LC3B, anti-TMEM175, anti-GC, and anti-GM antibodies. Briefly, incubation with blocking buffer (1 × PBS/0.1% triton/5% serum) was performed for 15 min in a humidified chamber. Primary and then secondary antibody incubations (1 × PBS/0.1% triton/antibody) were performed at 4 °C overnight and at RT for 1 h, respectively. Anti-LAMP1 (ab25630, Abcam, Cambridge, UK, 1:20), anti-LAMP2 (ab25631, Abcam, Cambridge, UK, 1:100), anti-LC3B (2775, Cell Signaling, Danvers, MA, USA, 1:200), anti-TMEM175 (19,925–1-AP, Proteintech, Manchester, UK, 1:100) and anti-p62/SQSTM1 (P0067, Sigma-Aldrich, St. Louis, MO, USA, 1:500), anti-GlcCer (RAS0010, Glycobiotech, Kuekels, Germany, 1:50) primary antibodies and anti-mouse IgG-Cy3 (C2181, Sigma-Aldrich, St. Louis, MO, USA, 1:200), anti-rabbit IgG-Cy3 (C2306, Sigma-Aldrich, St. Louis, MO, USA, 1:200), Donkey anti-Mouse IgG (H + L), Alexa Fluor™ 488 (A-21202, Thermo Fisher Scientific CA USA, 1:400), and Donkey anti-Rabbit IgG (H + L) Alexa Fluor™ 488 (A-21206, Thermo Fisher Scientific CA USA, 1:400) secondary antibodies were used. GM1 was detected through 2-h incubation with Cholera toxin subunit B (CT-B) conjugated with Alexa fluor 488 (c34775, Molecular Probes, Eugene, OR, USA, 1:50). Hoechst H3570 (Thermo Fisher Scientific, CA, USA, 1:1000) was used to visualize the nuclei. Slides were visualized with Nikon Confocal Microscope A1R and with Nikon Eclipse Ni-E Microscope.
Immunohistochemistry Assays
Brain tissues were obtained from control animals of 3 months of age, previously authorized in the project 545/2019-PR. Immunohistochemistry was performed on paraformaldehyde-fixed, wax-included brains as described previously [26]. In brief, slides were xylene deparaffinized and rehydrated through a descending series of alcohol to water before being boiled in citrate buffer (pH 6.0) for antigen retrieval. Sections were then incubated with a blocking solution containing 0.5% milk, 10% FBS, and 1% BSA and hybridized with the primary and Alexa Fluor secondary antibodies (Molecular probes, Eugene, Oregon, USA). Primary antibodies were rabbit anti-Tmem175 (1:200, bs-18922R Bioss Inc, Boston, MA, USA), mouse anti-GFAP (1:200, MAB3402 Chemicon, Temecula, CA, USA), mouse anti-NeuN (1:100, MAB377 Chemicon, Temecula, CA, USA), and Chicken anti-Iba1 (1:2000, 234,009 Synaptic Systems, Goettingen, Germany). The immunohistochemistry experiment to visualize Tmem175, Iba1, and NeuN was performed with three compatible antibodies; fluorescence was excited at 488 nm (Tmem175, green), 555 nm (NeuN, red), and 647 nm (Iba1, red) for secondary antibodies. All immunostaining images were captured with a Nikon eclipse NI microscope.
Statistical Analysis
Categorical variables were compared with χ2-test or Fisher’s exact test, as appropriate. Genotype and allele frequencies were computed, and their distribution in cases and controls was analyzed by χ2-test with 2 DF and 1 DF. Concordance to the frequency predicted by the Hardy–Weinberg equilibrium (HWE) was assessed by χ2-test with 1 DF.
SPSS statistical software (SPSS Inc., Chicago, IL, USA, version 12.0) was used for most of the statistical analysis. Tests for deviation from Hardy–Weinberg equilibrium and tests for association were performed with DeFinetti (http://ihg.gsf.de/cgi-bin/hw/hwa1.pl). The difference between the observed means was calculated with MedCalc (https://www.medcalc.org/calc/comparison_of_means.php). The power of our case–control sample was calculated by the QUANTO statistical software. This study had the ability to detect, assuming 80% power and a two-sided 0.05 significance level with a minor allele frequency of 20% and an odds ratio > 1.51 (https://www.stat.ubc.ca/~rollin/stats/ssize/caco.html). All calculations were considered significant for P < 0.05. Bonferroni correction for the number of SNP was tested (α = 0.05/7 = 0.007).
In the endophenotype analysis, we used UPDRS score < 30 (corresponding to a mild phenotype: 1–2 of Hoehn and Yahr (HY) scale) and ≥ 30 (corresponding to a severe phenotype: 3–5 HY scale) [27], while in non-motor symptoms, we used as cutoff ≤ 54 and > 54 (9 items × 2 (mild impairment) × 3 (weekly impairment)). Statistical significance was set to α = 0.01, correcting for five phenotypes tested.
Results
Expression Profile of TMEM175 Gene in Human and Mouse Tissues
Quantitative PCR analysis showed that the transcript of TMEM175 gene was ubiquitously expressed in both adult human and mouse brain tissue (Fig. 1a, c). A higher expression was observed in the brain, pancreas, and skeletal muscle in human (Fig. 1a) and in midbrain and forebrain in the mouse brain (Fig. 1c). Analysis of human single-cell RNA sequencing data [25] showed the highest TMEM175 expression in dopaminergic neurons (DaN_1 and DaN_2) of the SNpc and in microglia of the cerebral cortex (Fig. 1b). In mouse brain, the highest expression of Tmem175 protein was observed in all DA neurons (TH + cells) of the SNpc (Fig. 2a, b) and in microglia (Iba1 + cells) of the dentate gyrus, hippocampus, and cortex (Fig. 2d, e, f). A lower expression was also detected in cortical neurons (NeuN + cells) (Fig. 2f) and in astrocytes (GFAP + cells) in the hippocampus (Fig. 2i). Overall, these data suggest a role for TMEM175 in the etiopathogenesis of PD and neuroinflammation.

Expression analysis of TMEM175 in human and mouse tissues. a, b qPCR experiments showed that TMEM175 mRNA is highly expressed in the human brain (brown bar) and mouse midbrain and forebrain (blue and yellow bar) at P45. Results were normalized by GAPDH. Data are expressed as absolute values with SD calculated from three biological replicates. c Single-cell RNA sequencing analysis of human substantia nigra (SN) and cerebral cortex showed the highest expression of TMEM175 mRNA in dopaminergic neurons of SN and in microglia of the cerebral cortex. Abbreviations: SN, substantia nigra; ODC, oligodendrocytes; OPC, oligodendrocyte progenitor cell

TMEM175 expression in the substantia nigra and dorsal telencephalon of the mouse adult brain. a–j Immunohistochemistry assays performed on mouse adult brain sections with Tmem175 and TH (a, b); NeuN (c); Tmem175, IBA1, and NeuN (d–f); GFAP (g); and Tmem175 and GFAP (h–j) shows that in the substantia nigra Tmem175 protein is co-expressed with TH in all dopaminergic neurons (a, b); in the dorsal telencephalon, Tmem175 exhibits high expression level in IBA1+ microglia cells (arrows in d–f) and a lower expression level in virtually all NeuN+ cortical neurons (f) and in a small fraction of GFAP+ putative astrocytes prevalently distributed in a territory including the oriens layer of the hippocampus, the alveus, and the white matter of corpus callosum (arrows in i). The area demarcated in (a) corresponds to sections shown in (b), and those demarcated in (c) and (g) correspond to sections shown in (d–f) and (h–j), respectively. Abbreviations: SN, substantia nigra; Cx, cortex; Hi, hippocampus; DG, dentate gyrus. Scale bars are indicated
Comprehensive Molecular Analysis of TMEM175 Gene in Italian Late-Onset PD (LOPD) Patients
The TMEM175 gene is located on chromosome 4 in the TMEM175/GAK/DGKQ GWAS locus associated with PD [9]. Recently, the TMEM175 p.M393T common variant was described as a risk factor for PD in several populations [11, 14], and rare deleterious variants were associated with PD in Italian families [8].
To assess the genetic contribution of TMEM175 gene in the Italian population, we performed a retrospective observational monocentric study of 400 PD patients (178 familiar and 222 sporadic cases; mean age at diagnosis 58.26 years (standard deviation (SD) 9.74)) [8, 20] and 300 unrelated healthy subjects (mean age 77 years; SD 5.4). We found 66 variants including 34 exonic (12 synonymous, 20 non-synonymous, 1 frameshift deletion, and 1 stopgain) variants, 9 variants in untranslated (3′ and 5′) regions, and 23 variants in intronic regions adjacent to the exon/intron boundaries. The complete list of identified variants is reported in Table 1.
To perform a case–control association study, we selected 7 variants that were located throughout the gene (Fig. 3a), were annotated both in WES and NGS-TR dataset, and showed a minor allele frequency (MAF) > 0.01 in multiple genetic databases. Results are reported in Table 2. Four variants showed significant association with PD: rs2290402 (nucleotide position c.-10C > T; odds ratio (OR) (confidence interval (CI)) = 1.5 [1.12–2.04], p value (p) = 0.003), rs34884217 (c.A194C, amino acid position p.Q65P; OR [CI] = 0.67 [0.44–1.01], p = 0.03), rs80114247 (c.T1022C, p.M341T; OR [CI] = 1.84 [0.93–3.63], p = 0.05], and rs34311866 (c.T1178C, p.M393T; OR [CI] = 1.38 [1.02–1.87], p = 0.02). Interestingly, two of these variants (rs34884217 and rs34311866) have been already described as associated with PD in other populations [11, 12, 28], while rs2290402 and rs80114247 are novel variants never described before. The rs2290402 variant (transcript number NM_032326; c.-10C > T), which remained statistically significant after correction for multiple testing, is located upstream (in the 5′-direction) from the ATG start codon in the Kozak consensus sequence (Fig. 3a), suggesting a functional role in modulating protein translation. The rs80114247 variant (c.T1022C, p.M341T) affected a highly conserved residue located in the TM3-II transmembrane domain, suggesting a role in the channel activity (Fig. 3b, c).

Graphical distribution of the TMEM175 gene variants identified in Italian PD patients. a The 7 common variants identified in the TMEM175 gene are reported on the exon/intron structure of the gene. Exons are shown as blue rectangles and introns as yellow bars. The start codon site (ATG) and the stop codon (TAG) are indicated. The human Kozak consensus sequence is reported in the upper part of the figure. b The 13 rare variants identified in the Italian PD patients are indicated on the graphical representation of the protein. TMEM175 consists in 6 trans-membrane motifs (TM1-TM6), each repeated twice. Five variants are exposed to the cytoplasm (p.P14L, p.T105A, p.R335H, p.L405V, and p.R481W), 8 variants are in the transmembrane motifs (p.R35C, p.R183X, p.A270T, p.P308L, p.M341T, p.S348L, p.R414W, and p.P427fs), and 1 is exposed to the lumen (p.R370H). c Eleven variants, including 10 rare variants and the p.M341T common variant, affect highly conserved residues among vertebrates (highlighted in red and yellow)
The assessment of motor and non-motor PD symptoms revealed a significant association with Unified Parkinson’s Disease Rating and Non-motor Symptoms (UPDRS/NMS) scales for the rs2290402 variant (UPDRS: OR [CI] = 1.63 [0.99–2.63], p = 0.03; NMS: OR [CI] = 1.46 [0.98–2.20], p = 0.03). Overall data showed that the rs2290402 variant might influence the occurrence and the severity of the PD phenotype, although the contrast would not survive correction for multiple testing of five phenotypes (Table 3).
Analysis of Rare Detrimental Variants in TMEM175 Gene
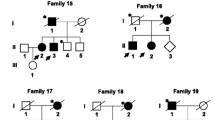
Thirteen rare detrimental variants were identified in 22 Italian patients. All affected residues identified in PD patients occupied functionally important amino acid positions, and seven of them were highly conserved among vertebrates (Fig. 3b, c). Considering the polygenic inheritance associated with PD, disclosed in our previous study [8], we identified 4 patients carrying one mutation in TMEM175 and one pathogenic mutation in GBA (p.N409S or H294Q); three patients carrying one mutation in TMEM175 and one pathogenic mutation in PARK2, PARK7, and PINK1 respectively; and 8 patients carrying one mutation in TMEM175 and one rare deleterious variant in other PD candidate genes such as AIMP2, KIF24, MAN2C1, SNCAIP, SPTBN1, and TVP23A. Eight PD patients carried a single deleterious variant in TMEM175 alone (Table 4, Fig. 4). The segregation analysis was performed only in 7 families (PD2, PD7, PD9, PD10, PD12, PD14, PD21) in which more PD patients, in the same family, were available for the genetic test (Fig. 4).

Graphical representation of the 22 Italian families carrying TMEM175 mutations. Affected individuals are indicated with dark symbols. Patients who underwent WES analysis are shown with dark arrows. Additional family members from whom DNA was available are indicated with an asterisk. Only affected subjects were included in the segregation analysis. The rare deleterious variants reported in Table 4 were carried by the affected family members
A complete description of all clinical data of the 22 patients is reported in Table 4; pedigrees are reported in Fig. 4. Mean age (± SD) at first symptoms was 59.5 ± 8.2 years (range 44–79 years). A higher age at onset (AAO) was found in patients carrying only the mutation in the TMEM175 gene compared to patients also presenting mutations of other genes (62.5 ± 6.5 years vs 56.3 ± 7 years; p = 0.05). No difference in motor, non-motor, and cognitive (Montreal Cognitive Assessment (MoCA) score) symptoms were identified. Considering the high expression of TMEM175 gene in microglia of human and mouse cerebral cortex, we investigated the presence of areas of cortical atrophy in our cohort of patients carrying detrimental mutations. Indeed, previous positron emission tomography (PET)/magnetic resonance imaging (MRI) studies have already largely investigated the relationship between neuroinflammation and gray matter alterations in Alzheimer’s disease [29,30,31]. However, more recently a negative correlation between an increased microglia activation and gray matter volume reduction has been demonstrated also in alpha-synucleinopathies such as PD [32]. Accordingly, in vitro evidences have suggested that neuronal release of aggregated alpha-synuclein in the central nervous system may activate microglia with production of proinflammatory mediators increasing the rate of neurodegeneration [32].
Six patients underwent MRI in our Parkinson Centre. In five patients, MRI was performed in the first 2 years of the disease; in one patient carrying the p.P14L variant, MRI performed at 5-year follow-up showed widespread cortical atrophy (Fig. 5).

MRI analysis of PD patient. Three-dimensional T1-weighted images showing widespread cortical atrophy in a PD patient carrying the TMEM175 p.P14L mutation (coronal slice, left; axial slice, right)
Overall, these data did not permit us to conclude that the mutations in the TMEM175 gene might be sufficient to cause the disease; however, the co-inheritance of mutations in other PD genes anticipated the clinical onset of the disease of about 6 years.
Functional Analysis of TMEM175 Deleterious Variants
TMEM175 is an endolysosomal K+ channel that regulates lysosome pH stability and K+ conductance [15]. We evaluated the impact of the most detrimental variants identified in the TMEM175 gene in Italian PD patients on mRNA and protein stability and sub-cellular localization in dermal fibroblasts (hDF) derived from PD patients and healthy subjects (Fig. 6a–c). We observed a nonuniform gene expression pattern between the two groups. In particular, a significant increase in the protein amount was observed in PD5 carrying the p.P427fs mutation (Fig. 6b), suggesting that this mutated protein might accumulate into the cell. Interestingly, reduced amount of TMEM175 protein was observed in hDFs derived from PD14 carrying the p.R35C mutation co-inherited with the SNCAIP_S2N mutation, compared to the expression observed in PD16 carrying the p.R35C mutation alone, suggesting that the expression of TMEM175 might be perturbed by the polygenic variant burden (Fig. 6b). A co-immunostaining with the lysosomal marker, LAMP1, was observed in patient and control cells, suggesting that these mutations did not affect protein folding and localization (Fig. 6c, Fig. S1).

Expression analyses of TMEM175 in hDF of PD patients and healthy subjects. a qPCR and b western blot experiments. A nonuniform gene expression pattern was observed between the two groups (PD patients, black bar in the graph; healthy subjects, red bar in the graph). The mutations p.S348L, p.P427fs, p.R35Ca, p.R35Cb, p.R414W, and p.R481W correspond to the dermal fibroblasts of the patients PD3, PD5, PD14, PD16, PD17, and PD22, respectively (Table 4). Data were normalized to GAPDH and expressed as absolute values with SD calculated from three biological replicates. A significant increase in protein levels was observed in PD5 carrying the p.P427fs mutation. c and d Immunofluorescence experiments to visualize the sub-cellular localization of the TMEM175 protein. c Human dermal fibroblasts (hDF) of the healthy subject (CNT3) and of the PD11 patient carrying the p.L405 mutation. d HeLa cells transfected with the wild-type (WT) or the mutant (L405V) TMEM175 protein GFP-tagged. Hoechst H3570 (blue) was used to detect nuclei. c In hDF the endogenous TMEM175 protein was stained with the anti-TMEM175/CY3 (red), and the lysosomal markers LAMP1 was stained using anti-LAMP1/488 (green). d In HeLa cells, the exogenous GFP-tagged TMEM175 protein was in green, and the lysosomal marker LAMP2 was stained using anti-LAMP2/CY3 (red). Both endogenous TMEM175 protein and exogenous GFP-tagged TMEM175 protein are co-localized with lysosomal markers. White arrows in d indicate the localization of the GFP-tagged TMEM175 protein into the plasma membrane. Images were acquired by Nikon Confocal Microscope A1R, and scale bars correspond to c 50 μm and d 5 μm
However, as the analyzed patients were heterozygous for TMEM175 mutations, we were not able to clearly discriminate between reference and mutant alleles. We also generated a panel of GFP-tagged wt and mutant proteins (Fig. 3c) and demonstrated that mutant proteins were able to translocate to lysosomal membranes in HeLa cells (Fig. 6d, Fig. S2). However, we also observed intense staining signal into the cytoplasm and faint staining signal on plasma membrane, suggesting that the excess protein, produced by the construct, remained free in the cytoplasm or translocated into plasma membrane (Fig. 6d, Fig. S2).
Next, we investigated the functional properties of the mutant channels in HEK293 cells. In transfected HEK293 cells, the GFP-tagged TMEM175 protein was able to translocate into the plasma membrane (data not shown), as also demonstrated in a previous study [15]. Electrophysiological measurements were performed in the perforated patch-clamp configuration, avoiding cell dialysis [33]. Under this condition, during a stimulation protocol consisting of voltage ramps from − 100 to + 100 mV applied every 2 s, overexpression of the wt channel induced a clear increase in transmembrane current, as exemplified in Fig. 7a. Current density measured at + 30 mV changed from a mean value of 2.15 ± 0.34 pA/pF in non-transfected cells to a fivefold increased value of 11.08 ± 3.38 pA/pF (p = 0.027) in cells overexpressing the wt protein (Fig. 7b). The mutant channels (p.R35C, p.P308L, p.A270T, p.L405V) showed no significant current increase compared to non-transfected cells (Fig. 7b; 3.25 ± 0.84 pA/pF n = 5; 3.11 ± 0.94 pA/pF, 2.75 ± 0.44 pA/pF, and 2.54 ± 0.41 pA/pF, respectively), suggesting that these variants might affect channel activity. Interestingly, the polymorphic variant p.M341T, associated with PD in the case–control analysis, showed an increase of the K+ current across the plasma membrane (6.63 ± 1.67 pA/pF, p = 0.043), which was intermediate between that detected with the wt and mutant proteins (Fig. 7b), indicating that this change might partially affect the functional properties of the channel.

Mutations in TMEM175 alter channel activity. a Typical I-V relation obtained by applying a single voltage ramp (from − 100 to + 100 mV, 200 ms) on a representative HEK293 cell transfected with wt TMEM175-GFP-fused construct (red line), TMEM175-M341T polymorphic variant (gray line), and TMEM175 R35C, P308L, A270T, and L405V rare deleterious variants (black line). Note the increase in the current in cell overexpressing the wt plasmid. b Histogram representing the mean current density measured at + 30 mV in different experimental conditions, as indicated. Note the increase in current density in cells overexpressing the wt TMEM175 protein compared to non-transfected cells (n = 8 and n = 13 biological replicates, respectively). Current density measured in cells transfected with the mutant proteins TMEM175 R35C, P308L, A270T, and L405V (n = 5, n = 6, n = 6, n = 7 biological replicates, respectively) was similar to that measured in non-transfected cells, while the polymorphic variant TMEM175 M341T showed an increase of the K + current across the plasma membrane that was intermediate between that detected with the wt and mutant protein (n = 6 biological replicates). Data (mean ± SEM) were analyzed with Student’s t-test and plotted as histogram representation. (*) indicates a p value < 0.05. c On the top representative immunoblotting of anti-GFP in immunoprecipitated protein with anti-Akt in MN9D cells expressing GFP-conjugated TMEM175 wt and mutant proteins. In the left, total proteins blotted for input control. On the bottom, representative histogram of the fold of wt densitometric ratio between the anti-GFP and anti-Akt blots (n = 3 biological replicates). Note the loss of binding between Akt and the mutant TMEM175 alleles P308L, L405V, and M341T expressed in stable cell line. The mean ± SEM of three biological replicates were analyzed with Student’s t-test. Data were plotted as histogram representation. (**) indicates a p value < 0.01, (***) indicates a p value < 0.001
Furthermore, we investigated if some of the identified changes affected the binding with the regulatory factor protein kinase B (Akt) [19]. The interaction was investigated in MN9D cells with a stable expression of GFP-tagged wt or mutant proteins. A significant reduction in Akt-binding affinity was observed for the p.P308L, p.L405V, and p.M341T mutations (p < 0.001, p = 0.007, p = 0.006, respectively) (Fig. 7c), located across the TM2II-TM4II domains of TMEM175 (Fig. 3c).
TMEM175 Mutations Affect the Expression of Markers Involved in the Autophagy-Lysosomal Pathway and Unfolded Protein Response (UPR)
Quantitative PCR analysis showed, under stress conditions (1-h serum starvation), a significant increase in the expression of the mRNA of the autophagy-lysosomal markers, TFEB, mTOR, and SQSTM1, and the UPR markers, BIP, ATF6, and CALR, in patient-derived fibroblasts (PD3, PD5, PD14, PD16, PD17, PD22) compared to fibroblasts of healthy-unrelated subjects (CNT1-CNT5) (Fig. 8). A significant increase in the mRNA expression of TFEB and BIP was also observed under basal conditions (Fig. 8). A reduced expression of LC3 II and LAMP1 proteins was observed in PD3, PD5, PD17, and PD22 (Fig. 9a), and a significant increase in the expression of TFEB protein was observed in the cell nucleus in patients PD14, PD17, and PD22 (Fig. 9c). These data were suggestive of a defective autophagy in PD patients, either mutated in TMEM175 alone (PD17) or carrying two pathogenic mutations, one in TMEM175 and one in another PD gene (PD3, PD5, PD14, and PD22) (Table 4). A significant reduction of p62, LC3 II, and LAMP1 was also observed in PD17 cells (p.R414W-TMEM175), under bafilomycin treatment, further supporting autophagy impairment and a causal role for this mutation (Fig. 9b). p62 regulates the formation of protein aggregates and is removed by autophagy. Accumulation of p62-positive aggregates in cells and tissues are among the best-known characteristics of autophagy-deficiency; in fact, an increase or decrease in the amounts of p62 protein and aggregates can reflect a change in autophagic activity [34]. In patient-derived cells carrying TMEM175 mutations we observed, under stress conditions, a larger number of p62 punctate signals that were suggestive of impaired autophagy, compared to healthy subjects (Fig. 10, Fig. S3).

TMEM175 mutations affect the expression of markers involved in the autophagy-lysosomal pathway and unfolded protein response (UPR). qPCR experiments were performed in human dermal fibroblasts, under basal conditions (HBSS −) and under serum starvation (1 h) (HBSS +) to examine the expression of genes involved in the surveillance of autophagy-lysosomal and URP pathways. In the graph, dermal fibroblasts of healthy subjects (mean value of 5 healthy subject-derived cells: CNT1-CNT5) are indicated as red bars and dermal fibroblasts of PD patients (mean value of 6 patient-derived cells: PD3, PD5, PD14, PD16, PD17, and PD22 carrying the mutations p.S348L, p.P427fs, p.R35Ca, p.R35C.b, p.R414W, and p.R481W respectively (Table 4) are indicated as black bars. The mean ± SD of three biological replicates was analyzed with Student’s t-test. Data were plotted as histogram representation. (*) indicates a p value < 0.05, (**) indicates a p value < 0.01, (***) indicates a p value < 0.001

Autophagy-lysosomal pathway was defective in patient-derived fibroblasts. a Cells were harvested for 1 h, and the transformation of LC3 I into LC3 II and the expression of p62 and LAMP1 were detected by WB. Patient-derived cells show a reduced expression of LC3 II and LAMP1 compared to healthy subjects. The mean ± SD of three biological replicates was analyzed with Student’s t-test. Data were plotted as histogram representation: healthy subjects (mean value of 3 healthy subject-derived cells) are indicated as red bars, and patients are indicated as black bars. The mutations p.S348L, p.P427fs, p.R35Ca, p.R35Cb, p.R414W, and p.R481W correspond to the dermal fibroblasts of the patients PD3, PD5, PD14, PD16, PD17, and PD22, respectively (Table 4). (*) indicates a p value < 0.05, (**) indicates a p value < 0.01, and (***) indicates a p value < 0.001. b Cells were harvested for 2 h in HBSS w/wo 100 mM bafilomycin. Expression of LC3 I/LC3 II, p62, and LAMP1 was detected by WB. PD17-derived cells showed a reduced expression of p62, LC3 II, and LAMP1. Note that PD17 is a heterozygous carrier of TMEM175-R414W alone. c The translocation of TFEB into the nucleus was detected by WB. Patient-derived cells show a significant increase of expression of TFEB protein in the nucleus compared to healthy subjects

Immuno-localization of LAMP1 and p62 in patient-derived fibroblasts. The lysosomal marker, LAMP1, is in green, whereas the autophagic marker, p62, is in red. Under stress conditions, patient-derived fibroblasts displayed an increased number, size, and intensity of p62 puncta (white arrow in the lower panel) compared to healthy subject-derived cells. Hoechst H3570 (blue) was used to detect nuclei. Slides were visualized with Nikon Eclipse Ni-E 60 × magnification, and scale bars correspond to 50 μm
Finally, we investigated whether TMEM175 mutations might affect the amount and distribution of glucosylceramide (GlcCer), which is cleaved into glucose and ceramide by the lysosomal enzyme glucocerebrosidase (GBA), as well as the expression of ganglioside GM1, which is reduced in PD patients [35]. Immunofluorescent experiments in hDF showed staining of GlcCer and GM1 in the cytoplasm and lysosomes. Higher levels of GlcCer, together with reduced amount of GM1, were observed in PD patients compared to healthy controls (Fig. 11). These changes were prominent in patients PD21 and PD14 carrying three mutations (TMEM175 L405V/PARK2 R402C/GBA H294Q) and two mutations (TMEM175 R35C/SNCAIP S2N), respectively (Fig. 11). Interestingly for PD11, which carried the TMEM175 L405V mutation alone, no difference in GM1, compared to healthy subjects, was observed (Fig. 11).

Immuno-localization of glucosylceramide (GlcCer) and ganglioside GM1 in patient-derived fibroblasts. LAMP1 is shown green when associated with Glc-Cer (in red) and is shown in red when associated with GM1 that was detected with Cholera toxin subunit B (CT-B)/488. Nuclei were stained with Hoechst. Slides were visualized with Nikon Eclipse Ni-E 60 × magnification. A strong intense signal was observed for GlcCer in patient-derived cells. GM1 was reduced in patients compared to control cells
Discussion
To date, the genetic diagnosis of PD is still challenging due to the high genetic heterogeneity associated with the disease and the difficulty in interpreting results of genetic testing. Genetic characterization may pave the way to tailored treatments targeting different pathophysiological mechanisms in individual PD patients [36].
In the current study, we examined the role of common and rare variants of the TMEM175 gene in Italian PD patients through the analysis of next-generation sequencing data followed by association tests.
We found a statistically significant association between PD and the common TMEM175 variant rs2290402 (c.-10C > T; p = 0.003) and nominal associations (p < 0.05) between PD and the other variants, rs34884217 (c.A194C, p.Q65P), rs80114247 (c.T1022C, p.M341T), and rs34311866 (c.T1178C, p.M393T) (Table 2). Interestingly, the most associated variant, in our cohort of patients, was observed in the Kozak consensus sequence, 10 nucleotides upstream of the ATG start codon. The presence of the T allele predicted the ATG start site with a score 8% lower than the C allele, by using the ATGpr software (https://atgpr.dbcls.jp). This suggests the association of PD with a putative regulatory variant, affecting the level of the translated protein. Further association studies, performed on a higher number of subjects and targeting both intronic regions and other potentially regulatory regions outside the 5′ and 3′ UTRs will be necessary to characterize any association between PD and other potential additional TMEM175 variants.
To establish whether rare and highly penetrant variants of TMEM175 were causal mutations, we analyzed the segregation of the selected changes in PD families in which multiple patients were available. We identified 13 rare variants in 22 PD patients. These included 1 frameshift, 1 stop codon, and 11 missense amino acid changes, 8 of them affecting highly conserved amino acid residues (Fig. 3c).
In several patients, the same TMEM175 variant was either present as single causal mutation or co-inherited with mutations in other PD genes, and the presence of multiple mutations in PD genes is associated with earlier onset of the disease.
From a survey of the literature, we observed that some of the identified changes were in important domains of the protein. In particular, the p.R35C change is in the RxxxFSD motif, which is important for channel folding [15], p.A270T is in the TM1-II domain, important for the selectivity of K+ permeability [15], p.P308L is in the TM1II-TM2II linker, which protrudes outwards from the main body of the TM domain and is the entrance space of the channel pore [15], and p.M341T and p.S348L are in the TM3-II domain.
In the light of that, we investigated whether some of the identified mutations might have any functional consequences. To this regard, the electrophysiological analysis revealed a significant effect on channel opening probability for all selected mutations (p.R35C, p.A270T, p.P308L, p.L405V), while the effect, mediated by the common variant p.M341T, was less dramatic.
A recent study demonstrated that TMEM175 channel is activated by growth factors and gated by Akt [19]. The authors showed that Akt binds the TM2-TM3 domains of repeat II, and this complex is required for the channel to remain open after activation. Our results are coherent with these observations and revealed a significant reduction of Akt-TMEM175 binding affinity for the mutant proteins p.P308L, p.L405V, and p.M341T, which are in the TM2-TM3 domains of repeat II. These findings confirmed the key role of these amino acidic residues in the TMEM175/Akt interaction [19].
Considering the important role of the lysosomal-autophagic pathway in the pathogenesis of PD [37,38,39], we analyzed this pathway in patient-derived fibroblasts heterozygous for the mutant proteins p.R35C, p.S348L, p.R414W, p.P427fs, and p.R481W. We revealed a defective expression of different proteins involved in this pathway, including p62, LC3I/II, LAMP1, and TFEB in both basal and upon stress conditions. In particular, we found increased expression of TFEB at both mRNA and protein level and a massive TFEB translocation from a cytosolic location to the nucleus. This observation provided evidence for endogenous activation of TFEB as an adaptive response to lysosomal stress [37]. These findings were in line with other studies which demonstrated the requirement of TMEM175 for the maintenance of the physiological functions of lysosomes [15, 16, 19]. In fact, it is well demonstrated that TMEM175 depletion alters lysosome pH and leads to abnormal organelle fusion during autophagy [16, 19].
Moreover, we observed in these patients the activation of the UPR markers BIP, ATF6, and CALR, upon stress condition. This data are consistent with the results of other studies showing that sustained endoplasmic reticulum (ER) stress and dysfunction of autophagy are closely related in PD pathology [40].
Additional evidence of a possible impairment in lysosomal activity emerged by the analysis of glycosphingolipids in human fibroblast from PD patients.
A number of studies indicate that TMEM175 deficiency is associated with a decreased GBA activity and with the accumulation of GlcCer within lysosomes. This leads to impaired lysosomal protein degradation and increased exosomal release of alpha-synuclein [17, 41,42,43]. On the other hand, reduced levels of GM1, observed in both brain and peripheral tissues of PD patients, has been reported to contribute to the pathogenesis of the disease [29, 44]. Our data are in line with these observations and suggest an impairment of glycosphingolipid metabolic pathways in PD subjects, carrying TMEM175 mutations, possibly due to defective endomembrane systems.
Taken together, these data suggest that the studied variants (p.R35C, p.A270T, p.P308L, p.M341T, p.S348L, p. L405V, p.R414W, p.P427fs, p.R481W) might affect important domains of the protein and alter lysosome functioning, suggesting a pathogenic relevance. Among the other identified variants, p.R183X is a truncating mutation that might affect protein function. We also performed in silico predictions of the variants p.T105A, p.R335H, and p.R370H with Dynamut software that allows assessing the impact of mutations on protein stability and dynamics [45]. We showed an alteration of the interatomic interactions and of the vibrational entropy energy between wt and mutant proteins, suggesting a possible functional role for these changes (Fig. S4).
Interestingly, the nine highly penetrant pathogenic mutations (p.R35C, p. R183X, p.A270T, p.P308L, p.S348L, p. L405V, p.R414W, p.P427fs, p.R481W) identified in TMEM175 gene were present in 14 unrelated PD patients who represent the 3.5% of the entire MNI cohort. This percentage increases at 5.5% if we also consider those patients carrying the three mutations p.T105A, p.R335H, and p.R370H, predicted as deleterious by in silico analysis. This data might be very promising for the genetic diagnosis of PD if we consider that the major causative event of late onset PD is the p.G2019S mutation in LRRK2 [46], which is present in our cohort in 8 out of 400 PD patients (2%) [8]. Therefore, in our opinion, inserting the genetic analysis of TMEM175 gene in the diagnostic protocols of late-onset PD should be recommended; this could allow to diagnose the disease in a larger number of patients.
Another interesting finding emerging from our data is the demonstration that, in human and mouse brain, TMEM175 is highly expressed in mdDA neurons of the SNpc and in microglia in the cerebral cortex. MRI carried out in one patient carrying the p.P14L mutation located in the signal peptide of TMEM175 showed intense areas of widespread cortical atrophy. This evidence paves the way to an in-depth investigation of a larger cohort of patients to understand if mutations in TMEM175 may be responsible for a more severe phenotype characterized by cortical atrophy, as a result of neuroinflammation with ensuing neurodegeneration.
In recent years, inflammatory processes have emerged as prominent contributors to the pathogenesis of PD. A large body of evidence suggests that microglial cells may either protect or damage surrounding neurons depending on their phenotype. A proinflammatory, damaging, microglial phenotype is consistently observed in the PD brain [47]. It is well demonstrated that PD genes, including between others, SNCA and LRRK2, are expressed in microglial cells [48], and dominant mutations in these genes affect microglial function by promoting neuroinflammation via activation of microglia and inflammatory signaling pathways [49,50,51]. Moreover, it is well described that autophagy is involved in the regulation of the inflammatory status of microglia [52, 53] and may have a role in the pathophysiology of inflammation [54].
Conclusions
Our results highlight the importance of performing fine mapping of common and rare variants of PD genes to obtain a comprehensive genetic contribution on pathogenesis of the disease and to ameliorate the genetic tests. Furthermore, the study identifies functionally important amino acid residues in TMEM175 that cause the disease. These findings will pave the way to facilitate and accelerate the identification of molecular targets capable of modulating the channel’s activity.
Data Availability
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Change history
12 March 2023
Missing Open Access funding information has been added in the Funding Note.
Abbreviations
- PD:
-
Parkinson’s disease
- LOPD:
-
Late-onset Parkinson’s disease
- GWAS:
-
Genome-wide association studies
- mdDA:
-
Mesencephalic-diencephalic dopaminergic
- SNpc:
-
Substantia nigra pars compacta
- WES:
-
Whole exome sequencing
- MNI:
-
Mediterranean Neurological Institute
- NGS:
-
Next-generation sequencing
- TR:
-
Targeted resequencing
- SNVs:
-
Single-nucleotide variants
- MAF:
-
Minor allele frequency
- CADD:
-
Combined annotation-dependent depletion
- TSI:
-
Tuscan Italians
- qPCR:
-
Quantitative PCR
- FPD:
-
Familial PD
- SPD:
-
Sporadic PD
- ER:
-
Endoplasmic reticulum
References
Bloem BR, Okun MS, Klein C (2021) Parkinson’s disease. Lancet 397:2284–2303. https://doi.org/10.1016/S0140-6736(21)00218-X
Chen APF, Chen L, Kim TA, Xiong Q (2021) Integrating the roles of midbrain dopamine circuits in behavior and neuropsychiatric disease. Biomedicines 9:647. https://doi.org/10.3390/biomedicines9060647
Stefanis L (2012) α-Synuclein in Parkinson’s disease. Cold Spring Harb Perspect Med 2:a009399. https://doi.org/10.1101/cshperspect.a009399
Langston W, Schüle B, Rees L, Nichols RJ, Barlow C (2015) Multisystem Lewy body disease and the other parkinsonian disorders. Nat Genet 47:1378–1384. https://doi.org/10.1038/ng0329-473b
Verstraeten A, Theuns J, Van Broeckhoven C (2015) Progress in unraveling the genetic etiology of Parkinson disease in a genomic era. Trends Genet 31:140–149. https://doi.org/10.1016/j.tig.2015.01.004
Fernández-Santiago R, Sharma M (2022) What have we learned from genome-wide association studies (GWAS) in Parkinson disease? Ageing Res Rev 17:101648. https://doi.org/10.1016/j.arr.2022.101648
Singleton A, Hardy J (2019) Progress in the genetic analysis of Parkinson’s disease. Hum Mol Genet 28:R215–R218. https://doi.org/10.1093/hmg/ddz183
Gialluisi A, Reccia MG, Modugno N, Nutile T, Lombardi A, Di Giovannantonio LG et al (2021) Identification of sixteen novel candidate genes for late onset Parkinson’s disease. Mol Neurodegener 16:35. https://doi.org/10.1186/s13024-021-00455-2
Nalls MA, Pankratz N, Lill CM, Do CB, Hernandez DG, Saad M et al (2014) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 056:1–7. https://doi.org/10.1038/ng.3043
Chang D, Nalls MA, Hallgrímsdóttir IB, Hunkapiller J, van der Brug M, Cai F et al (2017) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49:1511–1516. https://doi.org/10.1038/ng.3955
Jinn S, Blauwendraat C, Toolan D, Gretzula CA, Drolet RE, Smith S et al (2019) Functionalization of the TMEM175 p. M393T variant as a risk factor for Parkinson disease. Hum Mol Genet 28:3244–3254. https://doi.org/10.1093/hmg/ddz136
Iwaki H, Blauwendraat C, Leonard HL, Liu G, Maple-Grødem J, Corvol JC et al (2019) Genetic risk of Parkinson disease and progression: an analysis of 13 longitudinal cohorts. Neurol Genet 5:e348. https://doi.org/10.1212/NXG.0000000000000348
Postuma RB, Berg D, Stern M, Poewe W, Olanow CW, Oertel W et al (2015) MDS clinical diagnostic criteria for Parkinson’s disease. Mov Disord 30:1591–1601. https://doi.org/10.1002/mds.26424
Grover S, Ashwin AKS, Pihlstrom L, Domenighetti C, Schulte C, Sugier P-E et al (2022) Genome-wide association and meta-analysis of age-at-onset in Parkinson disease: evidence from COURAGE-PD Consortium. Neurology 10:1212. https://doi.org/10.1212/WNL.0000000000200699
Lee C, Guo J, Zeng W, Kim S, She J, Cang C et al (2017) The lysosomal potassium channel TMEM175 adopts a novel tetrameric architecture. Nature 547:472–475. https://doi.org/10.1038/nature23269
Cang C, Aranda K, Seo YJ, Gasnier B, Ren D (2015) TMEM175 is an organelle K+ channel regulating lysosomal function. Cell 162:1101–1112. https://doi.org/10.1016/j.cell.2015.08.002
Jinn S, Drolet RE, Cramer PE, Wong AH-K, Toolan DM, Gretzula CA et al (2017) TMEM175 deficiency impairs lysosomal and mitochondrial function and increases α-synuclein aggregation. Proc Natl Acad Sci USA 114:2389–2394. https://doi.org/10.1073/pnas.1616332114
Brunner JD, Jakob RP, Schulze T, Neldner Y, Moroni A, Thiel G et al (2020) Structural basis for ion selectivity in tmem175 k+ channels. Elife 9:e53683. https://doi.org/10.7554/eLife.53683
Wie J, Liu Z, Song H, Tropea TF, Yang L, Wang H et al (2021) A growth-factor-activated lysosomal K+ channel regulates Parkinson’s pathology. Nature 591:431–437. https://doi.org/10.1038/s41586-021-03185-z
Gialluisi A, Reccia MG, Tirozzi A, Nutile T, Lombardi A, De Sanctis C et al (2020) Whole exome sequencing study of Parkinson’s disease and related endophenotypes in the Italian population. Front Neurol 10:1362. https://doi.org/10.3389/fneur.2019.01362
Tirozzi A, Modugno N, Palomba NP, Ferese R, Lombardi A, Olivola E et al (2021) Analysis of genetic and non-genetic predictors of levodopa induced dyskinesia in Parkinson’s disease. Front Pharmacol 12:640603. https://doi.org/10.3389/fphar.2021.640603
Hentz JG, Mehta SH, Shill HA, Driver-Dunckley E, Beach TG, Adler CH (2015) Simplified conversion method for unified Parkinson’s disease rating scale motor examinations. Mov Disord 30:1967–1970. https://doi.org/10.1002/mds.26435
Conti S, Bonazzi S, Laiacona M, Masina M, Coralli MV (2015) Montreal Cognitive Assessment (MoCA)-Italian version: regression based norms and equivalent scores. Neurol Sci 36:209–214. https://doi.org/10.1007/s10072-014-1921-3
Cova I, Di Battista ME, Vanacore N, Papi CP, Alampi G, Rubino A et al (2017) Validation of the Italian version of the Non Motor Symptoms Scale for Parkinson’s disease. Park Relat Disord 34:38–42. https://doi.org/10.1016/j.parkreldis.2016.10.020
Agarwal D, Sandor C, Volpato V, Caffrey TM, Monzón-Sandoval J, Bowden R et al (2020) A single-cell atlas of the human substantia nigra reveals cell-specific pathways associated with neurological disorders. Nat Commun 11(1):4183. https://doi.org/10.1038/s41467-020-17876-0
Di Giovannantonio LG, Acampora D, Omodei D, Nigro V, Barba P, Barbieri E, Chambers I, Simeone A (2021) Direct repression of Nanog and Oct4 by OTX2 modulates the contribution of epiblast-derived cells to germline and somatic lineage. Development 148(10):dev199166. https://doi.org/10.1242/dev.199166
Shulman LMM, Gruber-Baldini AL, Anderson KE, Vaughan CG, Reich SG, Fishman PS, Weiner WJ (2008) The evolution of disability in Parkinson disease. Mov Disord 23:790–796. https://doi.org/10.1002/mds.21879
Krohn L, Öztürk TN, Vanderperre B, Ouled Amar Bencheikh B, Ruskey JA, Laurent SB et al (2020) Genetic, structural, and functional evidence link TMEM175 to synucleinopathies. Ann Neurol 87:139–153. https://doi.org/10.1002/ana.25629
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE et al (2008) Microglia, amyloid, and cognition in Alzheimer’s disease: an [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis 32:412–419. https://doi.org/10.1016/j.nbd.2008.08.001
Femminella GD, Ninan S, Atkinson R, Fan Z, Brooks DJ, Edison P (2016) Does microglial activation influence hippocampal volume and neuronal function in Alzheimer’s disease and Parkinson’s disease dementia? J Alzheimers Dis 51:1275–1289. https://doi.org/10.3233/JAD-150827
Nicastro N, Malpetti M, Mak E, Williams GB, Bevan-Jones WR, Carter SF (2020) Gray matter changes related to microglial activation in Alzheimer’s disease. Neurobiol Aging 94:236–242. https://doi.org/10.1016/j.neurobiolaging.2020.06.010
Stefanova N (2022) Microglia in Parkinson’s disease. J Parkinsons Dis 12(s1):S105–S112. https://doi.org/10.3233/JPD-223237
Palomba NP, Martinello K, Cocozza G, Casciato S, Mascia A, Di Gennaro G et al (2021) ATP-evoked intracellular Ca2+ transients shape the ionic permeability of human microglia from epileptic temporal cortex. J Neuroinflammation 18:44. https://doi.org/10.1186/s12974-021-02096-0
Komatsu M, Waguri S, Koike M, Sou YS, Ueno T, Hara T et al (2007) Homeostatic levels of p62 control cytoplasmic inclusion body formation in autophagy-deficient mice. Cell 131:1149–1163. https://doi.org/10.1016/j.cell.2007.10.035
Chowdhury S, Ledeen R (2022) The key role of GM1 ganglioside in Parkinson’s disease. Biomolecules 12:173. https://doi.org/10.3390/biom12020173
Schneider SA, Alcalay RN (2020) Precision medicine in Parkinson’s disease: emerging treatments for genetic Parkinson’s disease. J Neurol 267:860–869. https://doi.org/10.1007/s00415-020-09705-7
Ballabio A, Bonifacino JS (2020) Lysosomes as dynamic regulators of cell and organismal homeostasis. Nat Rev Mol Cell Biol 21:101–108. https://doi.org/10.1038/s41580-019-0185-4
Lie PPY, Nixon RA (2019) Lysosome trafficking and signaling in health and neurodegenerative diseases. Neurobiol Dis 122:94–105. https://doi.org/10.1016/j.nbd.2018.05.015
Abeliovich A, Gitler AD (2016) Defects in trafficking bridge Parkinson’s disease pathology and genetics. Nature 539:207–216. https://doi.org/10.1038/nature20414
Ren H, Zhai W, Lu X, Wang G (2021) The cross-links of endoplasmic reticulum stress, autophagy, and neurodegeneration in Parkinson’s disease. Front Aging Neurosci 13:691881. https://doi.org/10.3389/fnagi.2021.691881
Sidransky E, Nalls MA, Aasly JO, Aharon-Peretz J, Annesi G, Barbosa ER et al (2009) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361:1651–1661. https://doi.org/10.1056/NEJMoa0901281
Magalhaes JM, Gegg ME, Migdalska-Richards A, Doherty MK, Whitfield PD, Schapira AHV (2016) Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet 25:3432–3445. https://doi.org/10.1093/hmg/ddw185
Papadopoulos VE, Nikolopoulou G, Antoniadou I, Karachaliou A, Arianoglou G, Emmanouilidou E et al (2018) Modulation of β-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. Hum Mol Genet 27:1696–1710. https://doi.org/10.1093/hmg/ddy075
Alselehdar SK, Chakraborty M, Chowdhury S, Alcalay RN, Surface M, Ledeen R (2021) Subnormal GM1 in PBMCs: promise for early diagnosis of Parkinson’s disease? Int J Mol Sci 22:11522. https://doi.org/10.3390/ijms222111522
Rodrigues CH, Pires DE, Ascher DB (2018) DynaMut: predicting the impact of mutations on protein conformation, flexibility and stability. Nucleic Acids Res 46(W1):W350–W355. https://doi.org/10.1093/nar/gky300
Healy DG, Falchi M, O’Sullivan SS, Bonifati V, Durr A, Bressman S et al (2008) Phenotype, genotype, and worldwide genetic penetrance of LRRK2-associated Parkinson’s disease: a case-control study. Lancet Neurol 7(7):583–590. https://doi.org/10.1016/S1474-4422(08)70117-0
Badanjak K, Fixemer S, Smajić S, Skupin A, Grünewald A (2021) The contribution of microglia to neuroinflammation in Parkinson’s disease. Int J Mol Sci 22:4676. https://doi.org/10.3390/ijms22094676
MacMahon Copas AN, McComish SF, Fletcher JM, Caldwell MA (2021) The pathogenesis of Parkinson’s disease: a complex interplay between astrocytes, microglia, and T lymphocytes? Front Neurol 12:666737. https://doi.org/10.3389/fneur.2021.666737
Kam TI, Hinkle JT, Dawson TM, Dawson VL (2020) Microglia and astrocyte dysfunction in Parkinson’s disease. Neurobiol Dis 144:105028. https://doi.org/10.1016/j.nbd.2020.105028
Miki YM, Shimoyama S, Kon T, Ueno T, Hayakari R, Tanji K et al (2018) Alteration of autophagy-related proteins in peripheral blood mononuclear cells of patients with Parkinson’s disease. Neurobiol Aging 63:33–43. https://doi.org/10.1016/j.neurobiolaging.2017.11.006
Lee HJ, Cho ED, Lee KW, Kim JH, Cho SG, Lee SJ (2013) Autophagic failure promotes the exocytosis and intercellular transfer of α-synuclein. Exp Mol Med 45:e22. https://doi.org/10.1038/emm.2013.45
Hou X, Watzlawik JO, Fiesel FC, Springer W (2020) Autophagy in Parkinson’s disease. J Mol Biol 432:2651–2672. https://doi.org/10.1016/j.jmb.2020.01.037
Qin Y, Qiu J, Wang P, Liu J, Zhao Y, Jiang F, Lou H (2021) Impaired autophagy in microglia aggravates dopaminergic neurodegeneration by regulating NLRP3 inflammasome activation in experimental models of Parkinson’s disease. Brain Behav Immun 91:324–338. https://doi.org/10.1016/j.bbi.2020.10.010
Cheng J, Liao Y, Dong Y, Hu H, Yang N, Kong X et al (2020) Microglial autophagy defect causes Parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 16:2193–2205. https://doi.org/10.1080/15548627.2020.1719723
Acknowledgements
We thank the participating patients and their families. The authors also thank the Microscopy Facility at IGB and William Delli Quadri for graphical assistance. This work was supported by Italian Ministry of Health grant n. RF 2019–12370224 to TE and Current Research to TE.
Funding
Open access funding provided by Consiglio Nazionale Delle Ricerche (CNR) within the CRUI-CARE Agreement. This work was supported by the Italian Ministry of Health grant no. RF 2019–12370224 to TE and Current Research to TE. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
TE, NPP, and GF designed the study. NM and SP recruited the patients and performed clinical study. NM, SP, NPP, and TE collected the data. RC and VD performed skin biopsies. MS performed the analysis of HCV, HBV, and HIV before skin biopsy. NPP and TE performed sample management and biobanking. NPP and TE realized the clinical database. TE performed the analysis of WES and NGS-TR data. LI performed the mutation analysis of the patients. GF generated dermal fibroblast cells. NPP, GF, GP, ID, LI, GM, and FC performed qPCR, WB and immunohistochemistry experiments, plasmid preparation, cell culture, and transfection. LGDG and AS performed immunohistochemistry experiments on mouse brain. GP performed immunoprecipitation assay. NPP, KM, and SF performed patch-clamp experiments. VV and CW performed single-cell analysis. NPP, GF, VM, FN, SF, and TE performed the analysis of data. NPP and GF wrote the manuscript. All authors revised the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval
The ethical board of the IRCCS Neuromed approved the study protocol (N°19 of the November 16, 2020).
Consent to Participate
Written informed consent was obtained from all participants.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Palomba, N.P., Fortunato, G., Pepe, G. et al. Common and Rare Variants in TMEM175 Gene Concur to the Pathogenesis of Parkinson’s Disease in Italian Patients. Mol Neurobiol 60, 2150–2173 (2023). https://doi.org/10.1007/s12035-022-03203-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03203-9