
Abstract
Fibroblast growth factors (FGFs) act as key signalling molecules in brain development, maintenance, and repair. They influence the intricate relationship between myelinating cells and axons as well as the association of astrocytic and microglial processes with neuronal perikarya and synapses. Advances in molecular genetics and imaging techniques have allowed novel insights into FGF signalling in recent years. Conditional mouse mutants have revealed the functional significance of neuronal and glial FGF receptors, not only in tissue protection, axon regeneration, and glial proliferation but also in instant behavioural changes. This review provides a summary of recent findings regarding the role of FGFs and their receptors in the nervous system and in the pathogenesis of major neurological and psychiatric disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The FGF and FGF Receptor Families
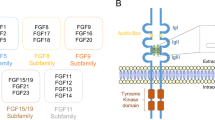
Fibroblast growth factors (FGFs) comprise a large family of polypeptides. They are expressed in nearly all organisms, ranging from nematodes to vertebrates. The 22 members of the FGF family are highly conserved in gene structure and amino acid sequence. Several of these factors are secreted and implicated in differentiation and migration during organ development. Ten of them are expressed in the brain [1]. FGF3, FGF8, FGF15, FGF17, and FGF18 play key roles in early development by imparting positional information and regulation of gene expression involved in brain patterning [2]. At the adult stage, FGFs primarily act as homeostatic factors in tissue repair and cellular proliferation. Several FGFs and their receptors (listed in Table 1) have been demonstrated to be involved in the pathogenesis of neurological disorders including Parkinson’s and Alzheimer’s [3, 4] and will be the focus of this review. The experimental data presented here were mainly obtained in rats and mice, the leading model organisms used in biomedical research. Very few aspects were validated in humans, and those are included as well.
FGF1 and FGF2 (acidic and basic FGF) are the most widely studied members of the FGF family expressed in neurons and glial cells [5]. An unusual feature of those ligands is the lack of a conventional signal sequence for export out of the cell and their exit via non-canonical mechanisms. Other FGFs like FGF11-14 remain intracellular and exert intracrine functions [6]. Some translocate from early endosomes into the cytosol and enter the nucleus [7]. This complexity is further enhanced by the expression of isoforms. For example, human FGF2 is expressed in five different isoforms derived from a single mRNA species [8]. This is the result of N-terminal sequence extensions in higher molecular weight isoforms as compared with low molecular weight FGF2 in some species [9, 10]. While the intranuclear functions of FGF1 and FGF2 are not fully understood [11], signalling through membrane-bound tyrosine kinase receptors has been described in detail [12]. Different FGF subfamilies exhibit preferences for one of the FGF receptors (FGFRs). FGF1 is the only member that can activate all four FGFR variants.
FGFRs share 46% amino acid identity and code for receptors of 125-160 kDa molecular weight. A fifth FGFR, FGFRL1, lacks the tyrosine kinase domain and is a putative co-receptor for FGFR1 [13]. FGFR1-3 are characterised by three extracellular immunoglobulin (Ig)-like domains, a heparin-binding region, and an acidic box domain. Ligand binding to D2 and D3 domains results in a 2:2:2 ternary complex of FGF, FGFR, and heparan sulphate [14]. The Ig-like D1 domain and the acidic box, located between D1 and D2, inhibit ligand binding by electrostatic interactions [15]. Alternative splicing in the D3 domain generates the IIIb and IIIc isoforms of FGFR1-3 with different ligand-binding properties (FGFR1b, -1c, -2b, -2c, -3b, and -3c). The b and c isoforms are restricted mainly to epithelial and mesenchymal tissues, respectively [16]. The extracellular D2 domain interacts with heparan sulphate proteoglycans (HSPGs) that facilitate the dimerisation and stabilisation of ligand interactions. The intracellular region of FGFRs beneath the transmembrane segment harbours a split tyrosine kinase domain. The binding of FGFs at HSPGs allows the formation of defined ligand gradients required for paracrine signalling, in particular during development.
Nuclear FGFR Signalling
In addition to its canonical role as membrane-bound tyrosine kinase receptor, FGFR1 has been described as a nuclear protein [17]. It translocates to the nucleus via an importin-β-dependent mechanism [18]. On the functional level, the nuclear receptor is a major signalling hub (designated as nuclear FGFR1 signalling, INFS) regulating neuronal growth and differentiation, amongst others [11, 19, 20]. Nuclear FGFR1 colocalises with transcriptionally active chromatin, binds to CREB-binding protein (CBP) or ribosomal S6 kinase isoform 1 (RSK1), and forms complexes with retinoid and Nurr receptors. Developmental signals are thereby directly forwarded to CBP and RSK1. RSK1 binding promotes FGFR1 release from the pre-Golgi to the cytosol, increases the mobile population of FGFR1, and facilitates nuclear accumulation. Novel interactive features of FGFR1 allow the newly synthesised 90 kDa protein to be released from pre-Golgi membranes and translocate into the cell nucleus along with the nuclear localisation signal (NLS)-containing FGF2 ligand [21]. Granzyme B-dependent cleavage of the C-terminal part of FGFR1 may also play a role [22]. The mRNAs for FGF2 and tyrosine hydroxylase are up-regulated in response to the nuclear shuttling of the receptor [23]. Importantly, nerve growth factor (NGF) utilises INFS for its neurodevelopmental and gene-activating functions [24]. NGF induces process outgrowth and transcriptional programming in a neuronal cell line (PC12) via nuclear translocation of FGFR1. FGFR1 interacts with the orphan nuclear receptor Nurr1, and this complex regulates tyrosine hydroxylase (TH) expression by binding to the TH gene promoter [25]. Furthermore, INFS appears to be necessary for dendritic outgrowth of sympathetic neurons in response to bone morphogenetic protein, BMP7 [26]. Recent evidence suggests a broader function of nuclear FGFR1 on several genes relevant in nuclear development [21, 27].
Distribution of FGF Receptors in the Nervous System
FGFR1-3 are widely distributed in the brain and bind to FGF ligands with different affinities and specificities [28, 29]. FGFR4 plays a role in early brain development but is absent from the adult brain (apart from one small nucleus). FGFR1 is most abundant in the nervous system with a predominant expression in neurons, astrocytes, and radial glia [30]. FGFR2 and FGFR3 are preferentially expressed in astrocytes and oligodendrocytes [1]. Studies of mice carrying null mutations in each of the FGFR genes revealed that FGFR1 and FGFR2 are essential for early embryonic development, which reflects their key roles in neuralisation and precursor proliferation. In contrast, animals lacking FGFR3 survive and exhibit no obvious telencephalic defects. However, FGFR3 plays an important role in cortex development [3]. Mutations in FGFR2 lead to either Apert or Crouzon syndrome, resulting in prominent changes of several brain structures [31].
The overlapping pattern of FGF ligand binding to similar FGF receptors implies a certain level of redundancy. In fact, different FGF family members activate FGFR subtypes to different degrees, depending on their ability to bind with high or low affinity to each receptor subtype [32]. Moreover, receptor specificity is modulated by the expression of other receptors and by the specific lipid composition of the plasma membrane. For example, in oligodendrocytes, a fraction of FGFR2 resides within the cholesterol/glycosphingolipid-enriched membrane microdomains (lipid rafts) [33]. Lipid rafts concentrate and segregate surface receptors together with their signalling molecules, and this compartmentalises and enhances intracellular signal transduction [34].
The complexity of FGFR activation is further increased by membrane molecules that may directly bind to and activate FGFRs in the absence of canonical ligands. Some of them play a pivotal role in the nervous system. For example, neural cell adhesion molecule (N-CAM), neuronal cadherin (N-cadherin), Eph receptor A4 (EphA4), and Anosmin-1 use FGFR as a signalling mediator and interfere with intracellular FGFR transport [35,36,37]. Hence, FGFRs are not specific to FGFs, and the phenotype of mice deficient in one or more FGFRs may be due to the lack of FGFR activation by ligands unrelated to FGFs or other receptors.
Signal Transduction of FGF Receptors
Ligand binding in cooperation with accessory HSPG triggers dimerisation of receptor monomers. This results in their mutual activation [38]. All possible FGFR combinations formed amongst FGFR1-3 suggest that FGFR heterodimers are as functionally important as homodimers [39]. FGFR dimers may also form in the absence of ligands at their physiological concentrations (Fig. 1). The ligand-independent dimers are stabilised through contacts below the transmembrane domains. These receptors are auto-phosphorylated, which explains why FGFR overexpression can lead to certain forms of cancer. The primary effect of ligand binding lies in a structural change in the pre-formed dimers and thereby enhanced receptor phosphorylation. Diverse ligands change receptor kinase activities in different ways. For example, FGF2-bound dimers show the smallest separation between the transmembrane domains but the highest possible phosphorylation [40].

The neuronal FGFR signalling network, from the binding of ligands to downstream events. FGFR1-3 monomers (1) form homodimers (2) and heterodimers (3) either in a ligand-dependent (2, 3) or independent (4) mode. The latter may undergo autophosphorylation. FGF ligand binding leads to enhanced receptor phosphorylation. The ordered and cooperative post-translational modifications are depicted as an activation code with sequentially phosphorylated tyrosine residues (boxed inset, 4). Certain downstream pathways like PLCγ and STAT3 require specific phosphorylation of additional tyrosine residues. Phosphorylation activates downstream pathways such as PLCγ, AKT, ERK, and STAT3. These are regulated by a number of proteins that provide an inhibitory feedback, thereby limiting activation (SPRY, Sef, and DUSP6; depicted in red). The lipid-anchored fibroblast growth factor receptor substrate 2 (FRS2) undergoes phosphorylation by FGFR kinase activity and recruits downstream factors PI3K and Ras/ERK as signalling hubs. However, this mechanism is FGFR-specific, with FGFR1 showing higher activity than the other FGFRs (boxed inset). Moreover, FGFR subtypes differentially activate downstream ERK and PLCγ with FGFR1 showing stronger activation than FGFR2 or FGFR4, respectively (boxed inset). For references, see text
Phosphorylated FGFRs activate canonical scr-homology 2 (SH2)-linked signalling proteins (PLCγ, CRKL) and recruit adapter proteins for connecting the receptor to PI3K/MAPK pathways. The analysis of chimeric receptors composed of cytoplasmic FGFRs and extracellular PDGF receptors has revealed that all FGFR subtypes stimulate the same pathways but with different magnitudes [38]. Prominent differences are found between FGFR1/FGFR2 and FGFR3/FGFR4 signalling. This may be due to differences in ligand activation and/or intracellular receptor transport following internalisation. For example, FGFR1 activates ERK and PLCγ more strongly than FGFR4, and higher ERK activation is caused by FGFR1 rather than by FGFR2 [16].
Key signalling hubs such as PI3K and Ras/ERK are recruited by the lipid-anchored 80 kDa docking protein FRS2 (also referred to as SNT1). FRS2 constitutively binds the juxtamembrane region of FGFR1 and is phosphorylated most efficiently by FGFR1 as compared with other receptor isoforms [41]. Activated FRS2α associates with the Grb2/SOS complex to relay activation of Ras and downstream MAPK signalling [42]. Additionally, FRS2α recruits the tyrosine phosphatase SHP2 [43]. FRS2α is also involved in neurotrophin receptor (Trk) signalling and appears to act as a ‘conning centre’ responsible for differential pathway activation. Furthermore, FRS2 is crucial for FGFR ubiquitination and trafficking due to its ability to constitute local signalling platforms and to recruit feedback inhibitors [44]. The latter initiate a cascade of negative signalling events that decrease the amplitude of positive signals and modulate the level of stimulation.
Six tyrosine (Y) residues in the split kinase domain need to be sequentially phosphorylated for the full activation of at least four major signalling pathways (Y653 → Y583 → Y463, Y766, and Y585 → Y654) [12]. Y653 increases tyrosine kinase activity by fifty- to one hundred-fold, and Y654 by a further ten-fold. Additional tyrosines are required for the activation of phospholipase Cγ (Y766) and STAT3 (Y677). The STAT pathway changes nuclear gene expression, whereas activation of PLCγ at the plasma membrane produces inositol trisphosphate (IP3) and diacylglycerol (DAG), thereby releasing calcium from endoplasmic reticulum stores and causing the activation of protein kinase C (PKC), respectively. DAG also produces ligands that can activate the endocannabinoid receptor CB1 in the brain [45]. Importantly, cannabinoid receptors transactivate FGFR1 in lipid rafts [46].
Inhibitory feedback mechanisms are induced by FGFR stimulation, which is essential to limit excessive signalling. Their de-regulation may result in brain tumours. They involve the coordinated action of ubiquitin ligase (c-Cbl), adapters (Grb2) and proteins such as Sef, phosphatases (DUSP), and Sprouty proteins [47, 48]. The latter function as crucial FGFR antagonists during brain development and in the adult, mainly by interfering with processes upstream of ERK [49]. Interestingly, FGFRs themselves are the subject of negative feedback mechanisms, because the prevention of ERK-dependent phosphorylation at serine 777 of FGFR1 (or the mutation of this serine to alanine) promotes receptor tyrosine phosphorylation and, consequently, cellular proliferation, migration, and axon growth [50].
Intracellular FGF Receptor Transport
FGFR activation is followed by rapid endocytosis and degradation of the receptor and the ligand. Ligand binding induces receptor mono-ubiquitination by the ubiquitin ligase c-Cbl, which functions as a signal for the sorting of the receptor into intraluminal vesicles of multivesicular endosomes and its subsequent delivery to lysosomes [51]. Receptor tyrosine kinases, such as FGFR, EGFR, and PDGFR, are mono-ubiquitinylated at multiple sites, while cytoplasmatic phosphorylated protein tyrosine kinases are poly-ubiquitinylated and degraded in the proteasome [52]. In the case of FGFR signalling, c-Cbl does not directly bind to the receptor but catalyses the ubiquitination of the receptor via interaction with FRS2 and Grb2. Hence, the competition of c-Cbl with SOS for Grb2 abrogates MAPK signalling [53].
The transport of FGFRs from the cell surface to different subcellular compartments influences the biological response to receptor activation. This has recently been confirmed by optogenetics [54]. Overexpressed FGFR1-eGFP fusion proteins bind FGF2 and activate signalling hubs at various locations. FGFRs internalise and shuttle to the recycling and degradation compartments in neurons and glial cells. This has consequences for the strength and duration of signalling pathway activation [55, 56].
Plasma membrane levels of neuronal FGFRs at the adult stage appear to be significantly lower than those of neurotrophin receptors, because the effects of neurotrophins on neuronal survival and neurite outgrowth are significantly stronger when compared with FGFs. However, overexpression of FGFR1 stimulates axon growth [57]. This effect is further enhanced by protease inhibitors such as leupeptin, which inhibits lysosomal protein degradation and promotes receptor recycling [58].
RTK recycling depends on the number of intracellular lysine residues that are required for receptor ubiquitination. For example, FGFR1 comprises 29 lysine residues, while the intracellular part of FGFR4 harbours 16 lysine residues only. Accordingly, lysine mutants of FGFR1 that are deficient in ubiquitination will be sorted to the recycling pathway rather than to degradation in lysosomes [59]. Overexpression of FGFR1 mutants exhibiting reduced numbers of lysines modifies axon outgrowth [60]. FGFR1-15R (with 14 instead of 29 lysine residues) preferentially recycles back to the plasma membrane similarly to FGFR4 and strongly promotes elongative axon growth without stimulating axon branching. Interestingly, the ERK inhibitor PD98059 does not reduce elongative axon growth induced by FGFR1-15R overexpression. This raises the possibility that ERK has independent effects on the axonal cytoskeleton through enhanced receptor recycling of FGFR1 which probably shows increased interaction with other growth promoting membrane receptors (e.g., NCAM [61]). The functional significance of the Ras/RAF/ERK pathway for adult axon regeneration remains a subject of controversy because in some studies ERK inhibitors did not interfere with axon outgrowth of adult primary neurons in culture [62].
FGFs in Neurological Disorders
Neuronal Degeneration and Repair
As master regulators of brain organogenesis and homeostasis, FGFs play an important role in the regeneration and repair of the nervous system. In fact, FGFR1 and FGFR2 stimulation induces complete neural tissue regeneration in planarians and vertebrate embryos [63]. Moreover, FGFs often synergise with other growth factors and cytokines in the generation of multipotent progenitors, for example, in the zebrafish retina [64]. In adult mammalian species, however, FGFs cannot replace damaged tissue, although stimulation of FGFR signalling assists in adult neurogenesis [65] and promotes neuronal survival after injury [1]. Conversely, expression of dominant-negative FGFR results in increased neuronal vulnerability [66]. The neuroprotective functions of FGFs are at least partially mediated by direct stimulation of neuronal FGFRs and are related to the inhibition of autophagy/protein clearance in a PI3K/AKT/mTOR-dependent manner [67].
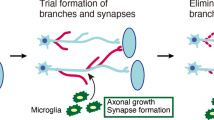
In addition to preventing or delaying neuronal cell death, FGFs are involved in the repair of synaptic connections. The formation of new excitatory synaptic contacts is regulated by FGF22, which is expressed in spinal interneurons and long propriospinal neurons [68]. In fact, a lack of FGF22 or targeted deletion of FGFR1 and FGFR2 in the motor cortex reduces synapse formation between corticospinal collaterals and relay neurons and attenuates functional recovery in response to spinal cord injury [69]. FGFR1b and 2b are required for excitatory and inhibitory presynaptic differentiation in response to FGF22 and FGF7, respectively [70, 71]. Both receptors mediate the excitatory presynaptic response to FGF22, whereas only FGFR2b elicits the inhibitory presynaptic response to FGF7.
FGF7-deficient mice exhibit epileptogenic changes in the hippocampus. This indicates that inhibitory synapse formation may be impaired, resulting in mossy fibre sprouting and enhanced neurogenesis during development [72]. Blocking FGF22 while activating FGF7 signalling may help to alleviate epileptogenesis. In general, it is assumed that FGFs are implicated in both seizure susceptibility and seizure-induced plasticity. It has been suggested that FGF2 favours acute seizures but reduces seizure-induced cell death [73, 74]. In the amyotrophic lateral sclerosis (ALS) model of mutant SOD1 mice, FGF deficiency causes a significant delay in disease onset, less impaired motor function, and prolonged survival when compared with mice with normal FGF2 levels, probably due to an up-regulation of neurotrophic factors such as CNTF and GDNF [75].
FGF2 and FGF20 synergise to increase dopaminergic neuron numbers in stem cell models [3]. FGF20 has been found to be preferentially expressed in the substantia nigra, pars compacta. It stimulates survival of dopaminergic neurons via activation of FGFR1IIIc [76]. In addition, FGF2 facilitates the formation of functional dopaminergic neurons from stem cells [77, 78]. In the 6-hydroxydopamine lesion model, infusion of FGF20 into the substantia nigra protects against cell death in both the substantia nigra and striatum, and this is accompanied by improved motor function [79]. Moreover, intrastriatal expression of FGF2 results in dopaminergic neuron recovery following chemically induced lesions [80].
With regard to Alzheimer’s disease, overexpression of FGF2 restores spatial learning, long-term potentiation, and neurogenesis. These effects are probably mediated by FGFR1-activated increases in CD200, the OX-2 membrane glycoprotein that regulates microglial activity and promotes neurite outgrowth and neuronal survival [81]. Furthermore, exogenous FGF2 ameliorates tau pathology and spatial memory deficits by down-regulating the amyloid precursor protein-cleaving enzyme (BACE1) that is involved in the production of amyloid β [82]. In primary hippocampal neuron culture, protection against amyloid β-induced neurotoxicity has been demonstrated to be dependent on the AKT but not the ERK signalling pathway. Interestingly, high molecular weight isoforms of FGF2 are more efficient than those of low molecular weight in this paradigm [83].
Endogenous FGF2 is secreted by neurons upon damage by glutamate or oligomeric amyloid β. This is followed by enhanced microglial migration and neuroprotection because of increased phagocytosis of neuronal debris via FGFR3 activation involving ERK and Wnt signalling [84]. However, loss of all three FGFRs in astrocytes results in microglia hypertrophy and proliferation [85]. These findings indicate a key role for FGF2 and FGFRs in orchestrating the crosstalk between degenerating neurons, microglia, and astrocytes. They also show that cellular activation and proliferation are distinct and that FGF-dependent processes are induced at different points after injury (Fig. 2).

Key mechanisms of FGF/FGFRs in the nervous system. The central nervous system comprises a large number of functionally and structurally diverse neuronal and glial cell types. The figure depicts model neurons forming synaptic connections as well as oligodendrocytes, astrocytes, and microglia. Modulation of synaptic connections by FGFs (boxed inset): FGF22 regulates the formation of excitatory synapses together with FGFR1b and R2b. Although both are involved in excitatory synapse regulation, inhibitory synapses are regulated by FGF7 via FGFR2b only. FGF2 and 20 synergise to regulate differentiation of dopaminergic neurons by using FGFR1IIIc as the receptor (FGF20 also binds to FGFR1IIIc in other neurons). When secreted by neurons, FGF2 enhances microglia activation, leading to increased removal of neuronal debris in case of neuronal damage. Moreover, FGF2 restores spatial learning, long-term potentiation, and neurogenesis in Alzheimer´s disease. Mechanistically, FGFR1 regulates CD200, which in turn mediates microglia responses and neurite outgrowth. This factor also feeds back by activation of FGFR1. Axonal growth and regeneration is stimulated mainly by FGF1 and FGF2. FGF2 secreted by neurons stimulates astrocytes via FGFR1-3 activation. Signalling from astrocytes to oligodendrocytes is accomplished by FGF2 influencing the survival and proliferation of oligodendrocyte precursor cells (OPCs). FGFR1 and R2 regulate myelin thickness and gene expression. For references, see text
Glutamate-mediated neuronal damage is observed in the hippocampus following temporal lobe epilepsy. This brain structure is highly dependent on FGFR1 signalling during development via FGF-mediated stimulation of hippocampal progenitor and stem cells [86]. Hippocampal deficits observed in patients with neurodegeneration, trauma, Alzheimer’s disease, and in normal ageing may therefore be counteracted by the incorporation of newly born neurons into existing networks. In fact, hippocampal neurogenesis has been observed to facilitate learning and memory in rodents. Adult neurogenesis in the human hippocampus is, however, a heavily debated issue. Recently, neurogenesis has been demonstrated to be limited to early development and childhood [87]. Other groups have observed ongoing neurogenesis in the hippocampus and a modest decline with age by applying improved strategies for the visualisation of neuronal precursor cells [88].
Stimulating neurogenesis through enhanced FGF signalling in the adult hippocampus may therefore be beneficial, particularly since neurogenesis decreases progressively in the brains of Alzheimer’s disease-affected patients. It is not yet clear, however, whether intrinsic precursor cell activity or changes in their environment determines such decline. Although FGFs promote the proliferation of cultured adult hippocampal precursor cells, their requirement for in vivo hippocampal neurogenesis in the adult and ageing brain still needs to be demonstrated. Elegant studies including conditional expression of mutated FGFRs have revealed that FGF signalling is clearly required for stem cell maintenance and increased neuron production [89,90,91]. Moreover, activated FGFR restores age-related decline in neurogenesis to a level found in young adult animals [92].
Neuronal degeneration is often associated with oedema formation and vascular pathology. These are particularly common in ageing and regularly affect hippocampal formation [73]. FGFs exert beneficial effects in some of these conditions, for example, in retinal cell swelling [93] and ischemic-reperfusion or hypoxic injury [94, 95]. FGF1 mixed into fibrin glue as a slow-release carrier reduces ischemia-induced focal brain infarction and attenuates functional deficits. Hippocampal and cortical neuron loss as well as microglial infiltration are also reduced. In addition, FGFs induce up-regulation of tight junction proteins via RhoA inhibition, thereby mitigating blood-brain barrier (BBB) breakdown and secondary brain injury [96].
Trauma in the CNS
Adult axon regeneration across spinal cord injuries and into intact spinal cord tissue generally fails in all higher vertebrates. Only a few growth factors, amongst them FGF1 and FGF2, were shown to promote axonal growth and functional recovery in spinal cord injury (SCI) models [97, 98]. The observed beneficial effects have been attributed generally to an attenuation of astrogliosis, increased numbers of neuronal progenitors, and/or stimulation of bipolar astrocyte morphology, which result in glial bridge formation guiding regenerating axons across the lesion site [99]. In fact, astrocytes use FGF2 as an auto- and intracrine signal to promote proliferation and structural changes in glial cells via FGFR1 or FGFR2 signalling. This effect can be enhanced by exogenous FGF2 [100]. FGF2 treatment shortly after spinal cord hemisection results in a significant reduction of TNFα expression at the lesion site, gliosis, and monocyte/macrophage infiltration 2 weeks later. Levels of astrocyte-derived chondroitin sulphate proteoglycans (CSPGs) are also markedly decreased, and functional recovery significantly improved [101]. Interestingly, similar effects were observed in the spinal cord of injured mice lacking Spry4, an endogenous feedback inhibitor of FGF signalling [102].
Recent evidence suggests, however, that FGF signalling is also required for the re-establishment of the non-reactive state of astrocytes following the initial phase after CNS injury. After applying conditional genetic approaches to manipulate FGFRs specifically in adult astrocytes, strong activation was observed in the lesioned neocortex of FGFR1-3 triple knock-out mice [85]. Both FGF1 and FGF2 inhibit GFAP expression via FGFR3 signalling [103, 104]. Since the formation of astrocytic scars clearly inhibits axon regeneration in the CNS, the reversal of the active state in astrocytes (probably involving changes in heparin sulphatation [105]) is likely to contribute to the positive effects of FGF1 and FGF2 on functional recovery after axotomy of intrinsic spinal cord or peripheral axons projecting into the CNS [91, 106,107,108].
A recent study [109] provided convincing evidence for a reversal of the dogma that adult CNS axons do not regenerate over long distances, by overcoming three obstacles simultaneously. The authors demonstrated that enhancing the intrinsic neuronal growth machinery and providing a supportive extracellular matrix via stimulation of FGF signalling, together with the application of chemoattractive cues, results in robust and successful axon regrowth across at least one spinal segment after complete spinal cord injury in rodents. In this model, osteopontin, IGF1, and CNTF were applied before injury. Growth-supportive substrates were induced by a combination of EGF and FGF2, which increased astrocytic proliferation as well as laminin, collagen, and fibronectin production inside the lesions. Then, propriospinal axons were attracted with GDNF delivered from biomaterial depots. All of these steps must be performed in combination to stimulate axon regeneration significantly, i.e. by a factor of around 100. Treatment with FGFs alone did not support axonal growth through astrocyte scars and across the lesion core into spared neural tissue. The combined approach resulted in the formation of terminal synapses and the re-establishment of electrophysiological conduction capacity [109].
Other studies confirm that stimulation of FGFR1 signalling alone is not sufficient to promote axon regeneration in the CNS. Up-regulation of FGFR1 in neurons projecting into the corticospinal tract (CST) does not enhance axon outgrowth. Injection of AAV serotype 1 overexpressing FGFR1 in a rat model of unilateral pyramidotomy did not increase sprouting of intact contralateral CST axons with overexpressed FGFR1, nor was it accompanied by functional improvements over control AAV injected animals [100]. Overexpression of FGFR1 in cultured cerebellar granule neurons even resulted in decreased neurite outgrowth. It is possible that key adaptor proteins, such as FRS2, are sequestered away from neurotrophic receptors promoting strong axon outgrowth, such as TrkA [110]. It has been shown previously that FGF2 exerts inhibitory effects on neurite outgrowth of cerebellar neurons plated on cortical astrocytes [111]. In addition, FGF1 and FGF2 may even interact with inhibitory receptors, such as the Nogo-66 receptor 1 (NgR1) [112].
NgR1 is a member of the Nogo receptor family implicated in the binding of myelin inhibitors and chondroitin sulphate proteoglycans [113]. It is part of a multi-component receptor complex comprising Lingo-1, p75, or TROY, which induces activation of the small GTPase RhoA, a well-known pathway involved in growth cone collapse and neurite outgrowth inhibition [114]. Interestingly, FGF2-dependent neurotrophic effects such as neuronal differentiation of PC12 cells and axonal branching in cortical neuron cultures are fully blocked by the ectopic overexpression of NgR1. Direct interaction between the two receptors could not be demonstrated; however, high-affinity binding of FGF2 to NgR1 was observed, suggesting that FGF family members also act as ligands at completely unrelated receptors [112].
Nerve Injury in the PNS
In the peripheral nervous system (PNS), axons do regenerate in the absence of exogenous growth factor support, provided that the proximal nerve stump containing the severed axons is connected to its distal counterpart. FGF1, FGF2, FGF7, and FGFR3 are all rapidly up-regulated in the lesioned nerve and in corresponding ganglia after axotomy [115, 116]. FGF2 prevents apoptosis of sensory neurons when applied directly to the transected sciatic nerve [117]. FGF1 and FGF2 have both been shown to improve nerve regeneration across a collagen-filled nerve conduit [118, 119]. In fact, FGF2 is one of the most promising growth factor with regard to clinically relevant muscle re-innervation, because it induces neurite elongation of motor axons similarly to GDNF [120].
Channels filled with Schwann cells overexpressing the high molecular weight (HMW) isoforms of FGF2 are particularly useful in promoting nerve regeneration [121, 122]. Low molecular weight (18kD) FGF2 released from transduced Schwann cells also accelerates regeneration and functional recovery when it is used to repair the transected sciatic nerve [123]. Because of their effect on the mitogenesis of mesoderm- and neuroectoderm-derived cells, it is assumed that FGFs support axonal regeneration mainly via increased proliferation of Schwann cells and enhanced angiogenesis [118]. Yet, direct trophic effects of FGF2 isoforms on primary neurons are observed as well. Nevertheless, adult sensory neurons must be sensitised before FGF treatment by prior axotomy in vivo. In response to such a ‘pre-conditioning’ lesion, i.e. a sciatic nerve transection 1 week before extraction of the lumbar ganglia, FGF2 isoforms stimulate axonal elongation preferably [124]. This effect can be completely blocked by SU5402, a specific FGFR antagonist, and it is mediated by ERK and PI3K activation. Naïve, untreated sensory neurons exhibit only a little FGFR1 and FGFR2 at their surface, suggesting that axotomy-induced receptor upregulation may be involved in the regenerative response.
Hence, FGF2 does not exert prominent effects on peripheral axon outgrowth if neurons have not been pre-lesioned. However, increasing FGFR1 levels by overexpression, inhibition of degradation, or promotion of receptor recycling all stimulate peripheral axon regeneration [55, 125]. Additional treatment with the protease inhibitor leupeptin further increases outgrowth [57, 58]. FGFR3 stimulation induces the opposite response, since FGFR3-deficient mice reveal reduced neuronal apoptosis in response to nerve transection [126]. Transgenic mice expressing high levels of FGF2 reveal faster axon regeneration, probably as a result of combined effects on Schwann cell proliferation, delayed myelination, and on axons directly [127]. Moreover, intramuscular injections of FGF2 increase the amplitude of compound muscle action potentials, wet muscle weight, and motor endplate density [128].
Enhanced FGF signalling has also been shown to be beneficial in response to facial nerve injury [129]. The already poor recovery of regenerating facial axons is further compromised in FGF2 knock-out mice. However, FGF2-deficient animals exhibit no difference in the number of regenerating axons in the sciatic nerve. In fact, faster recovery of mechanosensory (but not of motor) function following sciatic nerve crush was observed, suggesting compensatory mechanisms in the lesioned peripheral nervous system of global FGF2 knock-outs [130].
Our own investigations on FGFR signalling antagonists like Sprouty proteins corroborate the positive effects of FGFs on axon regeneration. Primary sensory or hippocampal neurons dissociated from Sprouty2 knock-out mice or transfected with shRNAs against Sprouty2 and Sprouty4 reveal significantly enhanced axon outgrowth [131,132,133]. Following sciatic nerve crush, more myelinated axons regenerate in heterozygous Sprouty2 knock-out mice, and this is accompanied by faster recovery of sensorimotor performance and increased expression of the regeneration-associated GAP-43 protein [133].
As stated above, FGFRs interact with other membrane components that may affect neuronal survival and even axon regeneration. For example, FGFR1 is required for neurite outgrowth stimulated by CAMs (PSA-NCAM, α7 integrin, and N-cadherin) in neurons and in neuron-like cell lines [61, 134,135,136]. NCAM/FGFR1 receptor complexes could be particularly relevant for motor axon regeneration in trans: FGFR1 is found at the plasma membrane of Schwann cells, and polysialylated (PSA)-NCAM (but not FGFR1) is localised at the membrane of elongating motor axons during the early phase of regeneration. This hypothesis is further supported by the demonstration of an increased interaction of FGFR1 and PSA-NCAM following FGF2 treatment [120]. Moreover, FGFR and neurotrophin receptor (Trk) signalling is co-dependent as well. Rat pheochromocytoma (PC12) cells expressing dominant negative FGFR exhibit reduced NGF-induced process formation and autophosphorylation of FGFRs. Selective FGFR inhibitors or oligonucleotides that interfere with receptor binding completely block neurite outgrowth induced by NGF in this cell line [137].
With regard to the possible interaction with inhibitory signalling pathways, previous studies by the Schwab group have suggested that the Nogo/NgR system is not only relevant for the CNS but for the PNS as well. Axonal regeneration and functional recovery are impaired following sciatic nerve crush in transgenic mice overexpressing Nogo-A in Schwann cells [138]. By contrast, sciatic nerve regeneration is enhanced in NgR1 knock-out mice (authors’ observation). Importantly, NgR1 is expressed in adult DRG sensory neurons and in motoneurons [139, 140]. However, there is no evidence of a potential cross-talk between NgR1/p75/RhoA and FGF signalling in the PNS yet, although FGF2 has been demonstrated to act as potent inhibitor of RhoA in primary neuron cultures [141].
With regard to lesion-induced neuropathic pain, FGF7 may play a role in injury-induced nociception. It is localised in the large dense-core vesicles (LDCVs) of small-diameter primary sensory neurons and may be transported to the dorsal spinal cord [142]. FGF7 increases the amplitude of excitatory post-synaptic current evoked by stimulating the sensory afferent fibres in spinal cord slices. Intrathecally applied FGF7 potentiates a formalin-induced acute nociceptive response, while it is diminished in FGF7 knock-out mice. Mice deficient in FGF2 or FGFR1/FGFR2 exhibit decreased thermal pain sensitivity accompanied by neuropathy of unmyelinated axons in the dorsal spinal cord [143, 144]. Furthermore, continuous intrathecal infusion of FGFR1 inhibitors reduces neuropathic pain-related behaviour in the partial sciatic nerve lesion model via inhibition of p38 MAPK [145].
Demyelination in the CNS
In the CNS, oligodendrocytes are required for myelination during development and in demyelinating disease. FGFR1 expression in these cells increases as the lineage progresses from oligodendrocyte precursor cells (OPCs) to mature oligodendrocytes. FGF2 and FGFR2 are also found in terminally differentiated oligodendrocytes [146]. Astrocyte-derived FGF2 positively influences the survival and proliferation of OPCs [147]. Conditional ablation of FGFR1 and R2 leads to the down-regulation of myelin gene expression, reduced myelin thickness, and axonal degeneration as knock-out mice age [148, 149]. FGFR3 is expressed in OPCs as well; however, FGFR3-deficient mice exhibit no changes in OPC proliferation rates [150].
Demyelination causes severe neurological deficits that are partially reversed by the spontaneous remyelination of axons by oligodendrocytes. Although the CNS is isolated from the peripheral milieu by the blood-brain barrier, remyelination can be triggered by peripheral factors that leak into the CNS after injury, including FGFs. Various FGFs are elevated in autoimmune diseases of the brain, such as multiple sclerosis: FGF1 in remyelinated lesions, FGF2 in active lesions and in the cerebrospinal fluid, FGF9 in active demyelinated lesions, and FGF21 in activated microglia or macrophages [151,152,153]. Intraventricular delivery of FGF2 induces severe disruption of mature oligodendrocytes, a marked loss of myelin, and aberrant accumulation of immature oligodendrocytes with a premyelinating phenotype [154]. Interestingly, the relative concentrations of the extracellular matrix protein Anosmin-1 and FGF2 in human MS lesions appears to be important for OPC migration through their interactions with FGFR1. This may have consequences for remyelination of lesioned axons because Anosmin-1 inhibits the effects of FGF2 on cellular migration [155,156,157].
FGF9 (also termed glia activating factor) is expressed by neurons and glia [158]. Like FGF2, FGF9 suppresses myelin protein synthesis by differentiating OPCs [159]. In multiple sclerosis (MS) lesions, FGF9 has been demonstrated to act indirectly, i.e. via the initiation of a complex astrocytic response that compromises remyelination [151]. By contrast, circulating FGF21, a member of the endocrine FGF family (expressed in the pancreas), stimulates OPC proliferation through interactions with β-klotho, an essential co-receptor of endocrine FGFs, in lysophosphatidylcholine (LPC)-induced lesions [160]. OPCs express β-klotho, the inhibition of which prevents increased OPC proliferation and remyelination.
Another level of complexity is introduced by the action of FGF1 and FGF2 in acute versus chronic MS lesions. FGF2 and FGFR1 levels are higher in MS patients than in controls, and a difference between relapse patients with higher FGF2 levels and those in remission is observed as well [161]. FGFR1 and FGFR2 double mutant mice exhibit hypomyelination in the chronic cuprizone model, indicating that FGF signalling (presumably via PI3K/AKT) is necessary for remyelination [162]. However, other studies have found the opposite, i.e., increased numbers of oligodendrocytes and improved remyelination of cuprizone-induced lesions or less myelin and axonal loss in MOG35-55-induced EAE in mice lacking FGF2 or FGFR1 [163,164,165,166,167]. This discrepancy may be due to the complexity of FGF signalling in multiple responses to injury and stress.
In experimental autoimmune encephalomyelitis (EAE), a model for MS, overexpression of FGF2 leads to enhanced remyelination and reduced axonal damage [168, 169]. Conversely, in the MOG35–55-induced EAE model of MS, FGF2 deficiency results in a more severe disease course, increased infiltration of lymphocytes and macrophages, and reduced remyelination [170]. By contrast, viral-mediated FGF2 overexpression results in less disease severity via inhibition of lymphocyte/macrophage infiltration [168]. FGF2 treatment clearly interferes with inflammation by reducing macrophages, microglia, and CD8 T-cells and limiting CD44-mediated leukocyte migration [168, 171, 172].
Brain Tumour Formation
FGFRs are commonly overexpressed in many types of cancer. High levels of FGFR1 are associated with better overall survival in peripheral nerve sheath sarcomas [173], while activating mutations in the FGFR1 kinase domain have been found in a subset of glioblastoma patients with poor prognosis [174]. FGFR2 is expressed at lower levels in high-grade gliomas, which correlates with higher proliferation and lower survival rates [175]. These grade IV malignant gliomas are amongst the most lethal human cancers, because they are resistant to neurosurgery, cytotoxic chemotherapy, and radiation [176]. Most patients present with primary glioblastoma multiforme associated with irregular signalling of epidermal growth factor (EGF) receptors or mutated PTEN (phosphatase and tensin homolog). Combined RAS/AKT signalling and PTEN deficiency have been shown to act as the main drivers of these tumours [177]. Regulators of ERK signalling, such as the Sprouty proteins, also play an important role in glioma proliferation [178].
High levels of FGF1 and FGF2 have been detected in glioma tissue relative to the normal brain [179]. Whereas expression of FGFR1 is low in normal white matter, its synthesis is dramatically increased in malignant astrocytic tumours [180]. Nuclear FGFR1 contributes to increased proliferation of glioma cells [19]. The blockade of FGF signalling by various means, amongst them FGF2 antibodies, siRNAs against FGFR1, or treatment with the FGFR/VEGFR inhibitor PD173074, reveals small but significant growth inhibitory effects in glioma cell lines [181, 182]. FGF inhibitors stimulate apoptosis, inhibit glioblastoma invasion, and suppress angiogenesis [183]. Down-regulation of FGF2 potentiates the effect of temozolomide (TMZ), an oral alkylating agent, by inhibiting proliferation and migration, blocking the cell cycle in G0/G1, and promoting apoptosis [184]. However, the efficient treatment of brain tumours with antibodies, siRNAs, or pharmacological inhibitors remains a challenge (see Therapeutic Approaches below).
Glioblastoma is also one of the most highly vascularised cancers. Therefore, inhibition of FGF activity, for example, by overexpressing dominant-negative FGFR1, may constitute a therapeutic strategy for disrupting angiogenesis-dependent signals required for glioma growth and invasion [185]. FGF2 promotes angiogenesis directly by activating the proliferation and migration of endothelial cells and indirectly by upregulating urokinase-type plasminogen activator, which also leads to cell migration [186]. Furthermore, secretion of FGF2 by glioma cells enhances the blood-brain barrier function of endothelial cells, which may contribute to drug resistance [187]. Interestingly, recent data suggest that FGFR1 inhibitors also decrease resistance to radiotherapy, a widespread problem in glioblastoma [188].
FGFs in Psychiatric Disorders
Over the years, it has become clear that FGF2 may act as key factor in neuropsychiatric syndromes via activation of FGFR1 [3, 189,190,191]. The proposed functions range from memory enhancing to anti-depressant and anxiolytic functions. In fact, FGF2 is down-regulated in rats showing high spontaneous anxiety. Knockdown of hippocampal FGF2 activity increases anxiety in naïve rats, and FGF2 treatment reduces anxiety in highly anxious rats [192]. However, mice with genetic ablation of FGF2 isoforms do not show alterations of anxiety-like stress susceptibility [193]. It should be noted, however, that other growth factors (BDNF and IGF-1) exert similar neuromodulatory effects as FGF2 on anxiety-related behaviour, as do genetic and environmental factors.
Traditional anxiolytics interfere with long-term extinction of fear memories. By contrast, chronic extinction is augmented by FGF2 that reduces the likelihood of exhibiting a relapse of extinguished fear in a new environment or following stress. Interestingly, early life treatment with FGF2 may decrease anxiety-like behaviour in adulthood, which probably involves an interaction between FGF receptors and adenosine A2 receptors or dopamine D2 receptors [194]. Furthermore, epigenetic mechanisms may be involved, such as regulation of non-coding RNAs, histone modifications, or DNA methylation [3]. FGF2 promotes the association of trimethylated histone protein H3 at lysine 9 (H3K9me3) at its own promoter [195]. Commonly used bipolar disorder drugs, such as valproate and lithium chloride, have been shown to inhibit histone deacetylases (HDACs) and to increase FGF1 expression [196].
Changes in FGFR levels and genetic variations of FGFR2 have been implicated in the pathomechanisms of schizophrenia [197]. Serum levels of FGF2 are increased in medicated schizophrenia patients and in non-medicated patients exhibiting negative symptoms [198]. FGF signalling disturbances in mood disorders might be indirect, since the disruption of schizophrenia-associated proteins, such as the neuronal PAS domain protein 3 (NPAS3), correlates with a dramatic reduction in FGFR1 mRNA, and NPAS3 deficiency behaviourally resembles FGFR1 knock-out mice [199]. Moreover, the phenotype of FGFR-deficient mice is reminiscent of other animal models for schizophrenia and mood disorders, for example, of knock-outs for disruption in schizophrenia 1 (DISC1), BDNF, and NRG1 [191]. Interestingly, nuclear FGFR1 has been implicated in the disease pathogenesis of schizophrenia. Neuron-committed cells from patients overexpressing FGFR1 reveal an association of nuclear FGFR1 with a large number of genes dysregulated in schizophrenia [27].
FGF signalling is clearly disturbed in individuals with major depressive disorder (MDD). Levels of FGF1, FGF2, FGFR2, and FGFR3 decrease in cortical areas, while FGF9 and FGF12 are elevated in the anterior cingulate and dorsolateral prefrontal cortex of depressive patients [200]. Ubiquitously found NG2 glial cells (precursors for myelinating oligodendrocytes) have been shown to secrete FGF2 during chronic stress, which may prevent glutamate abnormalities and maladaptive depressive behaviour [201]. Direct relationships were suggested when the intracerebroventricular injection of FGF2 resulted in antidepressant effects, which were also observed after FGF2 infusion into the prefrontal cortex in chronic unpredictable stress models of depression [202]. Notably, FGF2 may have indirectly enhanced neuronal activity by the stimulation of astrocyte proliferation, which is reduced in rodent depression models.
Elevated FGF2 levels may also play a role in the therapeutic effects of tricyclic antidepressants and selective serotonin re-uptake inhibitors [203]. The low and high molecular weight isoforms of FGF2 increase in response to monoamine oxidase inhibitor treatment in cortical astrocytes [204], and anti-depressive treatments in rodents elevated FGF2 expression in the hippocampus and cerebral cortex [203, 205]. Inflammatory reactions such as microglia activation and proliferation, which accompany depressive-like behaviours in LPS models, are ameliorated by neuron-derived endogenous FGF2 [171] or by FGF2 infusions [206]. FGF22 is putatively also involved in regulating affective behaviour. FGF22 knock-out mice exhibit depressive symptoms such as longer duration of floating, increased immobility in the tail suspension test, and a decreased preference for sucrose [207].
In animal models of drug use and addiction, FGF2 expression is increased in reward-related infralimbic/medial prefrontal cortex areas, and its neutralisation facilitates extinction of cocaine seeking [208]. Cocaine blocks the re-uptake of dopamine, and the auto-oxidation of dopamine results in free-radicals as by-products [209], which in turn increase FGF2 expression in astrocytes [30]. FGF2 is required for amphetamine-induced sensitisation [210], dendritic growth in dopaminergic neurons, and reductions in intrinsic excitability [211]. These effects are probably mediated not only by FGF2 but also by a cocktail of growth factors that arbitrate maladaptive stimulant induced alterations in neuronal function and structure.
Therapeutic Approaches
FGFs display a poor blood-brain barrier penetration and have a short half-life. They are vulnerable to proteolytic cleavage events resulting in their inactivation in various body fluids. A recent study demonstrated that a protein delivery coacervate can be prepared that controls FGF2 release and maintains its bioactivity by binding FGF2 via charge interaction consisting of polycation-polyethylene argininylaspartatediglyceride (PEAD) and heparin [212]. Furthermore, biodegradable micro-osmotic pumps based on microelectromechanical system (MEMS) technology have been developed for long-term controlled release of FGF2 [213]. Clearly, novel approaches including small non-peptidergic FGF mimetics will be required for the treatment of central and peripheral nervous system disorders.
Stimulants of FGFR and NCAM, such as the FGF-derived dekafin peptides, will be particularly useful in promoting tissue repair [214, 215]. It will be interesting to determine if these dendrimers might also be beneficial in normalising cognitive and social behaviour by their influence on the excitation/inhibition balance in the brain. Furthermore, FGF-dendrimer-based targeted delivery of drugs through FGFR may be a useful technology to target tumour cells or other FGFR expressing cells [216]. Novel biomaterials such as nontoxic and chemically inert hydrogels provide ideal scaffolds for the ingrowth of regenerating axons. Recently, FGF2 containing HEMA-MOETACL hydrogels were demonstrated to deliver FGF to the injured spinal cord in a localised and sustained manner [217]. Two months after implantation, the hydrogel was surrounded by an acellular vascular matrix consisting of glycosaminoglycan (GAG) and elastic/collagen fibres that promoted FGF-enhanced adhesion and migration of various cells types, resulting in nervous tissue regeneration and functional recovery in the paralysed hindlimbs of rats with complete spinal cord injury.
More recent developments include genetic approaches, for example, neuronal or glial FGF/FGFR transfer via lentiviral vectors (LVs). Gene therapy provides a useful tool for the specific down-regulation or knock-out of neuronal targets, since efficient siRNA treatments and viral gene transfer of shRNAs are now available for humans. Gene replacement therapy has been demonstrated to promote survival of patients with spinal muscular atrophy (SMA) following a single intravenous infusion of adeno-associated virus 9 (AAV9) containing cDNA coding for SMN [218]. LVs are even more useful for in vivo applications than AAVs because of their efficiency in gene delivery and their excellent safety profile. Moreover, in contrast with retroviral vectors, LVs do not depend on active division of the cell to be transduced and produce only minimal alteration in cellular physiology [219]. Alternatively, organically modified silica (ORMOSIL) nanoparticles may be used as nonviral vectors for efficient in vivo gene delivery. Nucleus-targeting FGFR1 was successfully transfected with this method [220].
Conclusions
The intricate relationship between oligodendrocytes and Schwann cells and central and peripheral axons, respectively, as well as the close association of ramified astrocytic or microglial processes with neuronal perikarya, neurites, and synapses represent the morphological basis for all FGFR-dependent signalling processes in neuronal and glial cells. More than 10 members of the FGF family function in these spatio-temporal domains, including membrane and nuclear compartments, to regulate transcriptional, post-transcriptional, and post-translational molecular events underlying instant changes in behaviour, long-lasting tissue repair, and axon regeneration. The effects of FGFs are clearly different from other neuronal growth factor families that act in the developing and adult brain. First, they strongly involve glial elements, and, second, they interact with several unrelated receptors via direct physical interaction or formation of heteromeric receptor complexes.
Currently, it is not possible to conclude with certainty whether the neurochemical and morphological alterations induced by FGFR signalling represent causes, consequences, epiphenomena, or a combination of those in the various pathological processes discussed here. Therefore, it is often not clear whether modulators of FGFRs would result in successful treatment that interferes with disease defining pathomechanisms. They could also alleviate symptoms or modify secondary, perhaps even beneficial aspects of brain disorders as exemplified by the negative effects of endogenous FGF2 in experimental models of amyotrophic lateral sclerosis [75]. Hence, the specific cellular context of a given pathology involving primarily glial and/or neuronal mechanisms will decide whether therapeutic interference with FGFRs is indicated or not.
Data availability
Not applicable
References
Reuss B, BuH O (2003) Fibroblast growth factors and their receptors in the central nervous system. Cell Tiss Res 313(2):139–157
Guillemot F, Zimmer C (2011) From cradle to grave: the multiple roles of fibroblast growth factors in neural development. Neuron 71(4):574–588
Turner CA, Eren-Kocak E, Inui EG, Watson SJ, Akil H (2015) Dysregulated fibroblast growth factor (FGF) signaling in neurological and psychiatric disorders. Sem Cell Dev Biol 53:136–143
Li X, Wang C, Xiao J, McKeehan WL, Wang F (2016) Fibroblast growth factors, old kids on the new block. Sem Cell Dev Biol 53:155–167
Eckenstein FP, Shipley GD, Nishi R (1991) Acidic and basic fibroblast growth factors in the nervous system: distribution and differential alteration of levels after injury of central versus peripheral nerve. J Neurosci 11:412–419
Nickel W (2011) The unconventional secretory machinery of fibroblast growth factor 2. Traffic 12(7):799–805
Wesche J, Malécki J, Widlocha A, Skjerpen CS, Claus P, Olsnes S (2006) FGF-1 and FGF-2 require the cytosolic chaperone Hsp90 for translocation into the cytosol and the cell nucleus. J Biol Chem 281(16):11405–11412
Arnaud E, Touriol C, Boutonnet C, Gensac MC, Vagner S, Prats H, Prats AC (1999) A new 34-kilodalton Isoform of human fibroblast growth factor 2 is cap dependently synthesized by using a non-AUG start codon and behaves as a survival factor. Mol Cell Biol 19(1):505–514
Claus P, Doring F, Gringel S, Muller-Ostermeyer F, Fuhlrott J, Kraft T, Grothe C (2003) Differential intranuclear localization of fibroblast growth factor-2 isoforms and specific interaction with the survival of motoneuron protein. J Biol Chem 278(1):479–485
Prats H, Kaghad M, Prats AC, Klagsbrun M, Lélias JM, Liauzun P, Chalon P, Tauber JP et al (1989) High molecular mass forms of basic fibroblast growth factor are initiated by alternative CUG codons. Proc Natl Acad Sci 86(6):1836–1840
Förthmann B, Aletta JM, Lee YW, Terranova C, Birkaya B, Stachowiak EK, Stachowiak MK, Claus P (2015) Coalition of nuclear receptors in the nervous system. J Cell Physiol 230(12):2875–2880
Ornitz DM, Itoh N (2015) The fibroblast growth factor signaling pathway. WIREs Dev Biol 4(3):215–266
Regeenes R, Silva PN, Chang HH, Arany EJ, Shukalyuk AI, Audet J, Kilkenny DM, Rocheleau JV (2018) Fibroblast growth factor receptor 5 (FGFR5) is a co-receptor for FGFR1 that is up-regulated in beta-cells by cytokine-induced inflammation. J Biol Chem 293(44):17218–17228
Goetz R, Mohammadi M (2013) Exploring mechanisms of FGF signalling through the lens of structural biology. Nat Rev Mol Cell Biol 14(3):166–180
Kalinina J, Dutta K, Ilghari D, Beenken A, Goetz R, A-á E, Cowburn D, Mohammadi M (2012) The alternatively spliced acid box region plays a key role in FGF receptor autoinhibition. Structure 20(1):77–88
Brewer JR, Mazot P, Soriano P (2016) Genetic insights into the mechanisms of FGF signaling. Genes Dev 30(7):751–771
Maher PA (1996) Nuclear translocation of fibroblast growth factor (FGF) receptors in response to FGF-2. J Cell Biol 134(2):529–536
Reilly JF, Maher PA (2001) Importin beta-mediated nuclear import of fibroblast growth factor receptor: role in cell proliferation. J Cell Biol 152(6):1307–1312
Stachowiak EK, Maher PA, Tucholski J, Mordechai E, Joy A, Moffett J, Coons S, Stachowiak MK (1997) Nuclear accumulation of fibroblast growth factor receptors in human glial cells--association with cell proliferation. Oncogene 14(18):2201–2211
Stachowiak MK, Maher PA, Stachowiak EK (2007) Integrative nuclear signaling in cell development—a role for FGF receptor-1. DNA Cell Biol 26(12):811–826
Stachowiak MK, Birkaya B, Aletta J, Narla S, Benson CA, Decker B, Stachowiak EK (2014) Nuclear FGF receptor-1 and CREB binding protein - an integrative signaling module. J Cell Physiol 230(5):989–1002
Chioni AM, Grose R (2012) FGFR1 cleavage and nuclear translocation regulates breast cancer cell behavior. J Cell Biol 197(6):801–817
Peng H, Myers J, Fang X, Stachowiak EK, Maher PA, Martins GG, Popescu G, Berezney R et al (2002) Integrative nuclear FGFR1 signaling (INFS) pathway mediates activation of the tyrosine hydroxylase gene by angiotensin II, depolarization and protein kinase C. J Neurochem 81(3):506–524
Lee YW, Stachowiak EK, Birkaya B, Terranova C, Capacchietti M, Claus P, Aletta JM, Stachowiak MK (2013) NGF-induced cell differentiation and gene activation is mediated by integrative nuclear FGFR1 signaling (INFS). PLoS ONE 8(7):e68931
Baron O, Förthmann B, Lee Y-W, Terranova C, Ratzka A, Stachowiak EK, Grothe C, Claus P et al (2012) Cooperation of nuclear fibroblast growth factor receptor 1 and Nurr1 offers new interactive mechanism in postmitotic development of mesencephalic dopaminergic neurons. J Biol Chem 287(24):19827–19840
Horbinski C, Stachowiak EK, Chandrasekaran V, Miuzukoshi E, Higgins D, Stachowiak MK (2002) Bone morphogenetic protein-7 stimulates initial dendritic growth in sympathetic neurons through an intracellular fibroblast growth factor signaling pathway. J Neurochem 80(1):54–63
Narla ST, Lee YW, Benson CA, Sarder P, Brennand KJ, Stachowiak EK, Stachowiak MK (2017) Common developmental genome deprogramming in schizophrenia — role of Integrative Nuclear FGFR1 Signaling (INFS). Schizophr Res 185:17–32
Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M (1996) Receptor specificity of the fibroblast growth factor family. J Biol Chem 271(25):15292–15297
Zhang X, Ibrahimi OA, Olsen SK, Umemori H, Mohammadi M, Ornitz DM (2006) Receptor specificity of the fibroblast growth factor family. The complete mammalian FGF family. J Biol Chem 281(23):15694–15700
Gonzalez AM, Berry M, Maher PA, Logan A, Baird A (1995) A comprehensive analysis of the distribution of FGF-2 and FGFR1 in the rat brain. Brain Res 701(1-2):201–226
Richtsmeier JT, Flaherty K (2013) Hand in glove: brain and skull in development and dysmorphogenesis. Acta neuropathol 125(4):469–489
Coleman SJ, Bruce C, Chioni AM, Kocher HM, Grose RP (2014) The ins and outs of fibroblast growth factor receptor signalling. Clin Sci 127(4):217–231
Bryant MR, Marta CB, Kim FS, Bansal R (2009) Phosphorylation and lipid raft association of fibroblast growth factor receptor-2 in oligodendrocytes. Glia 57(9):935–946
Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1(1):31–39
Doherty P, Walsh FS (1996) CAM-FGF receptor interactions: a model for axonal growth. Mol Cell Neurosci 8(2-3):99–111
Francavilla C, Cattaneo P, Berezin V, Bock E, Ami D, de Marco A, Christofori G, Cavallaro U (2009) The binding of NCAM to FGFR1 induces a specific cellular response mediated by receptor trafficking. J Cell Biol 187(7):1101–1116
Dijaz-Balzac CA, Lazaro-Pena MI, Ramos-Ortiz GA, Bülow HE (2015) The adhesion molecule KAL-1/anosmin-1 regulates neurite branching through a SAX-7/L1CAM-EGL-15/FGFR receptor complex. Cell Reports 11(9):1377–1384
Eswarakumar VP, Lax I, Schlessinger J (2005) Cellular signaling by fibroblast growth factor receptors. Cytok Growth Fact Rev 16:139–149
Del Piccolo N, Sarabipour S, Hristova K (2016) A new method to study heterodimerization of membrane proteins and its application to fibroblast growth factor receptors. J Biol Chem. 292(4):1288–1301
Sarabipour S, Hristova K (2016) Mechanism of FGF receptor dimerization and activation. Nat Commun 7:10262
Xu H, Lee KW, Goldfarb M (1998) Novel recognition motif on fibroblast growth factor receptor mediates direct association and activation of SNT adapter proteins. J Biol Chem 273(29):17987–17990
Wang JK, Xu H, Li HC, Goldfarb M (1996) Broadly expressed SNT-like proteins link FGF receptor stimulation to activators of Ras. Oncogene 13(4):721–729
Ong SH, Lim YP, Low BC, Guy GR (1997) SHP2 associates directly with tyrosine phosphorylated p90 (SNT) protein in FGF-stimulated cells. Biochem Biophys Res Commun 238(1):261–266
Gotoh N (2008) Regulation of growth factor signaling by FRS2 family docking/scaffold adaptor proteins. Canc Sci 99(7):1319–1325
Williams EJ, Walsh FS, Doherty P (1994) The production of arachidonic acid can account for calcium channel activation in the second messenger pathway underlying neurite outgrowth stimulated by NCAM, N-Cadherin, and L1. J Neurochem 62(3):1231–1234
Asimaki O, Leondaritis G, Lois G, Sakellaridis N, Mangoura D (2011) Cannabinoid 1 receptor-dependent transactivation of fibroblast growth factor receptor 1 emanates from lipid rafts and amplifies extracellular signal-regulated kinase 1/2 activation in embryonic cortical neurons. J Neurochem 116(5):866–873
Dikic I, Giordano S (2003) Negative receptor signalling. Curr Opin Cell Biol 15(2):128–135
Mason JM, Morrison DJ, Basson MA, Licht JD (2006) Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol 16(1):45–54
Hausott B, Klimaschewski L (2019) Sprouty2—a novel therapeutic target in the nervous system? Mol Neurobiol 56:3897–3903
Zakrzewska M, Haugsten EM, Nadratowska-Wesolowska B, Oppelt A, Hausott B, Jin Y, Otlewski J, Wesche J, Wiedlocha A (2013) ERK-mediated phosphorylation of fibroblast growth factor receptor 1 on Ser777 inhibits signaling. Sci Signal 6 (262):ra11
Bache KG, Slagsvold T, Stenmark H (2004) Defective downregulation of receptor tyrosine kinases in cancer. EMBO J 23(14):2707–2712
Haglund K, Sigismund S, Polo S, Szymkiewicz I, Di Fiore PP, Dikic I (2003) Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat Cell Biol 5(5):461–466
Wong A, Lamothe B, Lee A, Schlessinger J, Lax I, Li A (2002) FRS2 alpha attenuates FGF receptor signaling by Grb2-mediated recruitment of the ubiquitin ligase Cbl. Proc Natl Acad Sci USA 99(10):6684–6689
Csanaky K, Hess MW, Klimaschewski L (2019) Membrane-associated, not cytoplasmic or nuclear, FGFR1 induces neuronal differentiation. Cells 8(3):243
Hausott B, Rietzler A, Vallant N, Auer M, Haller I, Perkhofer S, Klimaschewski L (2011) Inhibition of fibroblast growth factor receptor 1 endocytosis promotes axonal branching of adult sensory neurons. Neuroscience 188:13–22
Irschick R, Trost T, Karp G, Hausott B, Auer M, Claus P, Klimaschewski L (2013) Sorting of the FGF receptor 1 in a human glioma cell line. Histochem Cell Biol 139(1):135–148
Hausott B, Schlick B, Vallant N, Dorn R, Klimaschewski L (2008) Promotion of neurite outgrowth by fibroblast growth factor receptor 1 overexpression and lysosomal inhibition of receptor degradation in pheochromocytoma cells and adult sensory neurons. Neuroscience 153(2):461–473
Hausott B, Vallant N, Hochfilzer M, Mangger S, Irschick R, Haugsten EM, Klimaschewski L (2012) Leupeptin enhances cell surface localization of fibroblast growth factor receptor 1 in adult sensory neurons by increased recycling. Eur J Cell Biol 91(2):129–138
Haugsten EM, Malecki J, Bjorklund SM, Olsnes S, Wesche J (2008) Ubiquitination of fibroblast growth factor receptor 1 is required for its intracellular sorting but not for its endocytosis. Mol Biol Cell 19(8):3390–3403
Hausott B, Förste A, Zach F, Mangger S, Haugsten EM, Klimaschewski L (2019) Endocytosis and transport of growth factor receptors in peripheral axon regeneration: novel lessons from neurons expressing lysine-deficient FGF receptor type 1 in vitro. Anat Rec 302:1268–1275
Francavilla C, Loeffler S, Piccini D, Kren A, Christofori G, Cavallaro U (2007) Neural cell adhesion molecule regulates the cellular response to fibroblast growth factor. J Cell Sci 120(24):4388–4394
Hausott B, Klimaschewski L (2019) Promotion of peripheral nerve regeneration by stimulation of the extracellular signal-regulated kinase (ERK) pathway. Anat Rec 302:1261–1267
Maddaluno L, Urwyler C, Werner S (2017) Fibroblast growth factors: key players in regeneration and tissue repair. Development 144(22):4047–4060
Wan J, Zhao XF, Vojtek A, Goldman D (2014) Retinal injury, growth factors, and cytokines converge on β-catenin and pStat3 signaling to stimulate retina regeneration. Cell Rep 9(1):285–297
Zhao M, Li D, Shimazu K, Zhou YX, Lu B, Deng CX (2007) Fibroblast growth factor receptor-1 is required for long-term potentiation, memory consolidation, and neurogenesis. Biol Psych 62(5):381–390
Eckenstein FP, McGovern T, Kern D, Deignan J (2006) Neuronal vulnerability in transgenic mice expressing an inducible dominant-negative FGF receptor. Exp Neurol 198(2):338–349
Zhang HY, Wang ZG, Wu FZ, Kong XX, Yang J, Lin BB, Zhu SP, Lin L et al (2013) Regulation of autophagy and ubiquitinated protein accumulation by bFGF promotes functional recovery and neural protection in a rat model of spinal cord Injury. Mol Neurobiol 48(3):452–464
Umemori H, Linhoff MW, Ornitz DM, Sanes JR (2004) FGF22 and its close relatives are presynaptic organizing molecules in the mammalian brain. Cell 118(2):257–270
Jacobi A, Loy K, Schmalz AM, Hellsten M, Umemori H, Kerschensteiner M, Bareyre FM (2015) FGF22 signaling regulates synapse formation during post-injury remodeling of the spinal cord. EMBO J 34(9):1231–1243
Terauchi A, Johnson-Venkatesh EM, Toth AB, Javed D, Sutton MA, Umemori H (2010) Distinct FGFs promote differentiation of excitatory and inhibitory synapses. Nature 465(7299):783–787
Dabrowski A, Terauchi A, Strong C, Umemori H (2015) Distinct sets of FGF receptors sculpt excitatory and inhibitory synaptogenesis. Development 142(10):1818–1830
Lee CH, Javed D, Althaus AL, Parent JM, Umemori H (2012) Neurogenesis is enhanced and mossy fiber sprouting arises in FGF7-deficient mice during development. Mol Cell Neurosci 51:61–67
Zechel S, Werner S, Unsicker K, Halbach O VB u (2010) Expression and functions of fibroblast growth factor 2 (FGF-2) in hippocampal formation. Neuroscientist 16(4):357–373
Paradiso B, Zucchini S, Simonato M (2013) Implication of fibroblast growth factors in epileptogenesis-associated circuit rearrangements. Front Cell Neurosci:7
Thau N, Jungnickel J, Knippenberg S, Ratzka A, Dengler R, Petri S, Grothe C (2012) Prolonged survival and milder impairment of motor function in the SOD1 ALS mouse model devoid of fibroblast growth factor 2. Neurobiol Dis 47(2):248–257
Itoh N, Ohta H (2013) Roles of FGF20 in dopaminergic neurons and Parkinson’s disease. Front Mol Neurosci 6:15
Grothe C, Timmer M (2007) The physiological and pharmacological role of basic fibroblast growth factor in the dopaminergic nigrostriatal system. Brain Res Rev 54(1):80–91
Nandy SB, Mohanty S, Singh M, Behari M, Airan B (2014) Fibroblast growth factor-2 alone as an efficient inducer for differentiation of human bone marrow mesenchymal stem cells into dopaminergic neurons. J Biomed Sci 21(1):83
Sleeman IJ, Boshoff EL, Duty S (2012) Fibroblast growth factor-20 protects against dopamine neuron loss in vitro and provides functional protection in the 6-hydroxydopamine-lesioned rat model of Parkinson’s disease. Neuropharmacology 63(7):1268–1277
Otto D, Unsicker K (1990) Basic FGF reverses chemical and morphological deficits in the nigrostriatal system of MPTP-treated mice. J Neurosci 10(6):1912–1921
Pankratova S, Bjornsdottir H, Christensen C, Zhang L, Li S, Dmytriyeva O, Bock E, Berezin V (2014) Immunomodulator CD200 promotes neurotrophic activity by interacting with and activating the fibroblast growth factor receptor. Mol Neurobiol 53(1):584–594
Katsouri L, Ashraf A, Birch AM, Lee KKL, Mirzaei N, Sastre M (2015) Systemic administration of fibroblast growth factor-2 (FGF2) reduces BACE1 expression and amyloid pathology in APP23 mice. Neurobiol Aging 36(2):821–831
Cheng Y, Li Z, Kardami E, Loh YP (2016) Neuroprotective effects of LMW and HMW FGF2 against amyloid beta toxicity in primary cultured hippocampal neurons. Neurosci Lett 632:109–113
Noda M, Takii K, Parajuli B, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A (2014) FGF-2 released from degenerating neurons exerts microglial-induced neuroprotection via FGFR3-ERK signaling pathway. J Neuroinflam 11(1):76
Kang W, Balordi F, Su N, Chen L, Fishell G, Hebert JM (2014) Astrocyte activation is suppressed in both normal and injured brain by FGF signaling. Proc Natl Acad Sci 111(29):E2987–E2995
Ohkubo Y, Uchida AO, Shin D, Partanen J, Vaccarino FM (2004) Fibroblast growth factor receptor 1 is required for the proliferation of hippocampal progenitor cells and for hippocampal growth in mouse. J Neurosci 24(27):6057–6069
Sorrells SF, Paredes MF, Cebrian-Silla A, Sandoval K, Qi D, Kelley KW, James D, Mayer S et al (2018) Human hippocampal neurogenesis drops sharply in children to undetectable levels in adults. Nature 555(7696):377–381
Moreno-Jiménez EP, Flor-García M, Terreros-Roncal J, Rábano A, Cafini F, Pallas-Bazarra N, Ávila J, Llorens-Martín M (2019) Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat Med 25(4):554–560
Nakatomi H, Kuriu T, Okabe S, S-i Y, Hatano O, Kawahara N, Tamura A, Kirino T et al (2002) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110(4):429–441
Yoshimura S, Teramoto T, Whalen MJ, Irizarry MC, Takagi Y, Qiu J, Harada J, Waeber C et al (2003) FGF-2 regulates neurogenesis and degeneration in the dentate gyrus after traumatic brain injury in mice. J Clin Invest 112(8):1202–1210
Ohori Y, S-i Y, Nagao M, Sugimori M, Yamamoto N, Nakamura K, Nakafuku M (2006) Growth factor treatment and genetic manipulation stimulate neurogenesis and oligodendrogenesis by endogenous neural progenitors in the injured adult spinal cord. J Neurosci 26(46):11948–11960
Kang W, Hebert JM (2015) FGF signaling is necessary for neurogenesis in young mice and sufficient to reverse its decline in old mice. J Neurosci 35(28):10217–10223
Berk BA, Vogler S, Pannicke T, Kuhrt H, Garcia TB, Wiedemann P, Reichenbach A, Seeger J et al (2015) Brain-derived neurotrophic factor inhibits osmotic swelling of rat retinal glial (Müller) and bipolar cells by activation of basic fibroblast growth factor signaling. Neuroscience 295:175–186
Fagel DM, Ganat Y, Cheng E, Silbereis J, Ohkubo Y, Ment LR, Vaccarino FM (2009) FGFR1 is required for cortical regeneration and repair after perinatal hypoxia. J Neurosci 29(4):1202–1211
Tsai MJ, Tsai SK, Huang MC, Liou DY, Huang SL, Hsieh WH, Huang WC, Huang SS et al (2015) Acidic FGF promotes neurite outgrowth of cortical neurons and improves neuroprotective effect in a cerebral ischemic rat model. Neuroscience 305:238–247
Huang B, Krafft PR, Ma Q, Rolland WB, Caner B, Lekic T, Manaenko A, Le M et al (2012) Fibroblast growth factors preserve blood-brain barrier integrity through RhoA inhibition after intracerebral hemorrhage in mice. Neurobiol Dis 46(1):204–214
Lee YS, Hsiao I, Lin VW (2002) Peripheral nerve grafts and aFGF restore partial hindlimb function in adult paraplegic rats. J Neurotrauma 19(10):1203–1216
Huang WC, Kuo HS, Tsai MJ, Ma H, Chiu CW, Huang MC, Yang LH, Chang PT et al (2011) Adeno-associated virus-mediated human acidic fibroblast growth factor expression promotes functional recovery of spinal cord–contused rats. J Gene Med 13(5):283–289
Goldshmit Y, Sztal TE, Jusuf PR, Hall TE, Nguyen-Chi M, Currie PD (2012) FGF-dependent glial cell bridges facilitate spinal cord regeneration in Zebrafish. J Neurosci 32(22):7477–7492
Haenzi B, Moon LD (2017) The function of FGFR1 signalling in the spinal cord: therapeutic approaches using FGFR1 ligands after spinal cord injury. Neural Plast 2017:2740768
Goldshmit Y, Frisca F, Pinto AR, Pebay A, Tang JK, Siegel AL, Kaslin J, Currie PD (2014) FGF2 improves functional recovery - decreasing gliosis and increasing radial glia and neural progenitor cells after spinal cord injury. Brain Behav 4(2):187–200
Goldshmit Y, Frisca F, Kaslin J, Pinto AR, Tang JKKY, Pebay A, Pinkas-Kramarski R, Currie PD (2015) Decreased anti-regenerative effects after spinal cord injury in spry4-/- mice. Neuroscience 287:104–112
Lee MJ, Chen CJ, Huang WC, Huang MC, Chang WC, Kuo HS, Tsai MJ, Lin YL et al (2011) Regulation of chondroitin sulphate proteoglycan and reactive gliosis after spinal cord transection: effects of peripheral nerve graft and fibroblast growth factor 1. Neuropath Appl Neurobiol 37(6):585–599
Kang K, Lee SW, Han JE, Choi JW, Song MR (2014) The complex morphology of reactive astrocytes controlled by fibroblast growth factor signaling. Glia 62(8):1328–1344
Higginson JR, Thompson SM, Santos-Silva A, Guimond SE, Turnbull JE, Barnett SC (2012) Differential sulfation remodelling of heparan sulfate by extracellular 6-O-sulfatases regulates fibroblast growth factor-induced boundary formation by glial cells: Implications for glial cell transplantation. J Neurosci 32(45):15902–15912
Teng YD, Mocchetti I, Taveira-DaSilva AM, Gillis RA, Wrathall JR (1999) Basic fibroblast growth factor increases long-term survival of spinal motor neurons and improves respiratory function after experimental spinal cord injury. J Neurosci 19(16):7037–7047
Tsai MC, Shen LF, Kuo HS, Cheng H, Chak KF (2008) Involvement of acidic fibroblast growth factor in spinal cord injury repair processes revealed by a proteomics approach. Mol Cell Proteomics 7(9):1668–1687
Huang MC, Lo MJ, Lin YL, Chang SE, Huang WC, Kuo WC, Tsai MJ, Kuo HS et al (2009) Functional recovery after the repair of transected cervical roots in the chronic stage of injury. J Neurotrauma 26(10):1795–1804
Anderson MA, O’Shea TM, Burda JE, Ao Y, Barlatey SL, Bernstein AM, Kim JH, James ND et al (2018) Required growth facilitators propel axon regeneration across complete spinal cord injury. Nature 561(7723):396–400
Ong SH, Guy GR, Hadari YR, Laks S, Gotoh N, Schlessinger J, Lax I (2000) FRS2 proteins recruit intracellular signaling pathways by binding to diverse targets on fibroblast growth factor and nerve growth factor receptors. Mol Cell Biol 20(3):979–989
Williams EJ, Mittal B, Walsh FS, Doherty P (1995) FGF inhibits neurite outgrowth over monolayers of astrocytes and fibroblasts expressing transfected cell adhesion molecules. J Cell Sci 108(11):3523–3530
Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y, Yeh HH, Shrager P et al (2008) Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci 28(11):2753–2765
Schwab ME (2010) Functions of Nogo proteins and their receptors in the nervous system. Nat Rev Neurosci 11(12):799–811
Auer M, Hausott B, Klimaschewski L (2011) Rho GTPases as regulators of morphological neuroplasticity. Ann Anat. 193(4):259–266
Grothe C, Haastert K, Jungnickel J (2006) Physiological function and putative therapeutic impact of the FGF-2 system in peripheral nerve regeneration - lessons from in vivo studies in mice and rats. Brain Res Rev 51(2):293–299
Bryan DJ, Litchfield CR, Manchio JV, Logvinenko T, Holway AH, Austin J, Summerhayes IC, Rieger-Christ KM (2012) Spatiotemporal expression profiling of proteins in rat sciatic nerve regeneration using reverse phase protein arrays. Proteome Sci 10(1):9
Otto D, Unsicker K, Grothe C (1987) Pharmacological effects of nerve growth factor and fibroblast growth factor applied to the transectioned sciatic nerve on neuron death in adult rat dorsal root ganglia. Neurosci Lett 83(1-2):156–160
Aebischer P, Salessiotis AN, Winn SR (1989) Basic fibroblast growth factor released from synthetic guidance channels facilitates peripheral nerve regeneration across long nerve gaps. J Neurosci Res 23(3):282–289
Danielsen N, Pettmann B, Vahlsing HL, Manthorpe M, Varon S (1988) Fibroblast growth factor effects on peripheral nerve regeneration in a silicone chamber model. J Neurosci Res 20(3):320–330
Allodi I, Casals-Diaz L, Santos-Nogueira E, Gonzalez-Perez F, Navarro X, Udina E (2013) FGF-2 low molecular weight selectively promotes neuritogenesis of motor neurons in vitro. Mol Neurobiol 47(2):770–781
Timmer M, Robben S, Muller-Ostermeyer F, Nikkhah G, Grothe C (2003) Axonal regeneration across long gaps in silicone chambers filled with Schwann cells overexpressing high molecular weight FGF-2. Cell Transplant 12(3):265–277
Haastert K, Lipokatic E, Fischer M, Timmer M, Grothe C (2006) Differentially promoted peripheral nerve regeneration by grafted Schwann cells over-expressing different FGF-2 isoforms. Neurobiol Dis 21(1):138–153
Allodi I, Mecollari V, Gonzalez-Perez F, Eggers R, Hoyng S, Verhaagen J, Navarro X, Udina E (2014) Schwann cells transduced with a lentiviral vector encoding FGF-2 promote motor neuron regeneration following sciatic nerve injury. Glia 62(10):1736–1746
Klimaschewski L, Nindl W, Feurle J, Kavakebi P, Kostron H (2004) Basic fibroblast growth factor isoforms promote axonal elongation and branching of adult sensory neurons in vitro. Neuroscience 126(2):347–353
Hausott B, Klimaschewski L (2016) Membrane turnover and receptor trafficking in regenerating axons. Eur J Neurosci 43(3):309–317
Jungnickel J, Gransalke K, Timmer M, Grothe C (2004) Fibroblast growth factor receptor 3 signaling regulates injury-related effects in the peripheral nervous system. Mol Cell Neurosci 25(1):21–29
Jungnickel J, Haase K, Konitzer J, Timmer M, Grothe C (2006) Faster nerve regeneration after sciatic nerve injury in mice over-expressing basic fibroblast growth factor. J Neurobiol 66(9):940–948
Iwata Y, Ozaki N, Hirata H, Sugiura Y, Horii E, Nakao E, Tatebe M, Yazaki N et al (2006) Fibroblast growth factor-2 enhances functional recovery of reinnervated muscle. Muscle Nerve 34(5):623–630
Seitz M, Grosheva M, Skouras E, Angelova SK, Ankerne J, Jungnickel J, Grothe C, Klimaschewski L et al (2011) Poor functional recovery and muscle polyinnervation after facial nerve injury in fibroblast growth factor-2−/− mice can be improved by manual stimulation of denervated vibrissal muscles. Neuroscience 182:241–247
Jungnickel J, Haastert K, Grzybek M, Thau N, Lipokatic-Takacs E, Ratzka A, Nolle A, Claus P et al (2010) Mice lacking basic fibroblast growth factor showed faster sensory recovery. Exp Neurol 223(1):166–172
Hausott B, Vallant N, Auer M, Yang L, Dai F, Brand-Saberi B, Klimaschewski L (2009) Sprouty2 down-regulation promotes axon growth by adult sensory neurons. Mol Cell Neurosci 42(4):328–340
Hausott B, Vallant N, Schlick B, Auer M, Nimmervoll B, Obermair GJ, Schwarzer C, Dai F et al (2012) Sprouty2 and -4 regulate axon outgrowth by hippocampal neurons. Hippocampus 22:434–441
Marvaldi L, Thongrong S, Kozlowska A, Irschick R, Pritz CO, Bäumer B, Ronchi G, Geuna S et al (2015) Enhanced axon outgrowth and improved long-distance axon regeneration in sprouty2 deficient mice. Dev Neurobiol 75(3):217–231
Saffell JL, Williams EJ, Mason IJ, Walsh FS, Doherty P (1997) Expression of a dominant negative FGF receptor inhibits axonal growth and FGF receptor phosphorylation stimulated by CAMs. Neuron 18(2):231–242
Lom B, Hopker V, McFarlane S, Bixby JL, Holt CE (1998) Fibroblast growth factor receptor signaling in Xenopus retinal axon extension. J Neurobiol 37(4):633–641
Niethammer P, Delling M, Sytnyk V, Dityatev A, Fukami K, Schachner M (2002) Cosignaling of NCAM via lipid rafts and the FGF receptor is required for neuritogenesis. J Cell Biol 157(3):521–532
Chevet E, Lemaitre G, Janjic N, Barritault D, Bikfalvi A, Katinka MD (1999) Fibroblast growth factor receptors participate in the control of mitogen-activated protein kinase activity during nerve growth factor-induced neuronal differentiation of PC12 cells. J Biol Chem 274(30):20901–20908
Pot C, Simonen M, Weinmann O, Schnell L, Christ F, Stoeckle S, Berger P, Rulicke T et al (2002) Nogo-A expressed in Schwann cells impairs axonal regeneration after peripheral nerve injury. J Cell Biol 159(1):29–35
Hunt D, Mason MR, Campbell G, Coffin R, Anderson PN (2002) Nogo receptor mRNA expression in intact and regenerating CNS neurons. Mol Cell Neurosci 20(4):537–552
Josephson A, Trifunovski A, Widmer HR, Widenfalk J, Olson L, Spenger C (2002) Nogo-receptor gene activity: cellular localization and developmental regulation of mRNA in mice and humans. J Comp Neurol 453(3):292–304
Auer M, Schweigreiter R, Hausott B, Thongrong S, Höltje M, Just I, Bandtlow C, Klimaschewski L (2012) Rho-independent stimulation of axon outgrowth and activation of the ERK and Akt signaling pathways by C3 transferase in sensory neurons. Front Cell Neurosci 6:43
Liu H, Wu QF, Li JY, Liu XJ, Li KC, Zhong YQ, Wu D, Wang Q et al (2015) Fibroblast growth factor 7 is a nociceptive modulator secreted via large dense-core vesicles. J Mol Cell Biol 7(5):466–475
Furusho M, Dupree JL, Bryant M, Bansal R (2009) Disruption of fibroblast growth factor receptor signaling in nonmyelinating Schwann cells causes sensory axonal neuropathy and impairment of thermal pain sensitivity. J Neurosci 29(6):1608–1614
Baum P, Vogt MA, Gass P, Unsicker K, Halbach O VB u (2016) FGF-2 deficiency causes dysregulation of Arhgef6 and downstream targets in the cerebral cortex accompanied by altered neurite outgrowth and dendritic spine morphology. Int J Dev Neurosci 50:55–64
Yamanaka H, Obata K, Kobayashi K, Dai Y, Fukuoka T, Noguchi K (2007) Activation of fibroblast growth factor receptor by axotomy, through downstream p38 in dorsal root ganglion, contributes to neuropathic pain. Neuroscience 150(1):202–211
Bansal R, Pfeiffer SE (1997) Regulation of oligodendrocyte differentiation by fibroblast growth factors. Adv Exp Med Biol 429:69–77
Murtie JC, Zhou Y-X, Le TQ, Vana AC, Armstrong RC (2005) PDGF and FGF2 pathways regulate distinct oligodendrocyte lineage responses in experimental demyelination with spontaneous remyelination. Neurobiol Dis 19(1):171–182
Furusho M, Dupree JL, Nave KA, Bansal R (2012) Fibroblast growth factor receptor signaling in oligodendrocytes regulates myelin sheath thickness. J Neurosci 32(19):6631–6641
Ishii A, Furusho M, Dupree JL, Bansal R (2014) Role of ERK1/2 MAPK signaling in the maintenance of myelin and axonal integrity in the adult CNS. J Neurosci 34(48):16031–16045
Oh LYS, Denninger A, Colvin JS, Vyas A, Tole S, Ornitz DM, Bansal R (2003) Fibroblast growth factor receptor 3 signaling regulates the onset of oligodendrocyte terminal differentiation. J Neurosci 23(3):883–894
Lindner M, Thümmler K, Arthur A, Brunner S, Elliott C, McElroy D, Mohan H, Williams A et al (2015) Fibroblast growth factor signalling in multiple sclerosis: inhibition of myelination and induction of pro-inflammatory environment by FGF9. Brain 38:1875–1893
Mohan H, Friese A, Albrecht S, Krumbholz M, Elliott CL, Arthur A, Menon R, Farina C et al (2014) Transcript profiling of different types of multiple sclerosis lesions yields FGF1 as a promoter of remyelination. Acta Neuropath Comm 2(1):178
Sarchielli P, Di Filippo M, Ercolani MV, Chiasserini D, Mattioni A, Bonucci M, Tenaglia S, Eusebi P et al (2008) Fibroblast growth factor-2 levels are elevated in the cerebrospinal fluid of multiple sclerosis patients. Neurosci Lett 435(3):223–228
Butt AM, Dinsdale J (2005) Fibroblast growth factor 2 induces loss of adult oligodendrocytes and myelin in vivo. Exp Neurol 192(1):125–133
Bribián A, Barallobre MJ, Soussi-Yanicostas N, de Castro F (2006) Anosmin-1 modulates the FGF-2-dependent migration of oligodendrocyte precursors in the developing optic nerve. Mol Cell Neurosci 33(1):2–14
Clemente D, Ortega MC, Arenzana FJ, de Castro F (2011) FGF-2 and Anosmin-1 are selectively expressed in different types of multiple sclerosis lesions. J Neurosci 31(42):14899–14909
Bribián A, Medina-Rodríguez EM, Josa-Prado F, García-Álvarez I, Machín-Díaz I, Esteban PF, Murcia-Belmonte V, Vega-Zelaya L et al (2020) Functional heterogeneity of mouse and human brain OPCs: relevance for preclinical studies in multiple sclerosis. J Clin Med 9(6):1681
Nakamura S, Todo T, Motoi Y, Haga S, Aizawa T, Ueki A, Ikeda K (1999) Glial expression of fibroblast growth factor-9 in rat central nervous system. Glia 28(1):53–65
Cohen RI, Chandross KJ (2000) Fibroblast growth factor-9 modulates the expression of myelin related proteins and multiple fibroblast growth factor receptors in developing oligodendrocytes. J Neurosci Res 61(3):273–287
Kuroda M, Muramatsu R, Maedera N, Koyama Y, Hamaguchi M, Fujimura H, Yoshida M, Konishi M et al (2017) Peripherally derived FGF21 promotes remyelination in the central nervous system. J Clin Invest 127(9):3496–3509
Harirchian MH, Tekieh AH, Modabbernia A, Aghamollaii V, Tafakhori A, Ghaffarpour M, Sahraian MA, Naji M et al (2012) Serum and CSF PDGF-AA and FGF-2 in relapsing-remitting multiple sclerosis: a case–control study. Eur J Neurol 19(2):241–247
Furusho M, Roulois AJ, Franklin RJM, Bansal R (2015) Fibroblast growth factor signaling in oligodendrocyte-lineage cells facilitates recovery of chronically demyelinated lesions but is redundant in acute lesions. Glia 63(10):1714–1728
Armstrong RC, Le TQ, Frost EE, Borke RC, Vana AC (2002) Absence of fibroblast growth factor 2 promotes oligodendroglial repopulation of demyelinated white matter. J Neurosci 22(19):8574–8585
Armstrong RC, Le TQ, Flint NC, Vana AC, Zhou Y-X (2006) Endogenous cell repair of chronic demyelination. J Neuropath Exp Neurol 65(3):245–256
Zhou Y-X, Pannu R, Le TQ, Armstrong RC (2012) Fibroblast growth factor 1 (FGFR1) modulation regulates repair capacity of oligodendrocyte progenitor cells following chronic demyelination. Neurobiol Dis 45(1):196–205
Mierzwa AJ, Zhou YX, Hibbits N, Vana AC, Armstrong RC (2013) FGF2 and FGFR1 signaling regulate functional recovery following cuprizone demyelination. Neurosci Lett 548:280–285
Rajendran R, Giraldo-Velasquez M, Stadelmann C, Berghoff M (2018) Oligodendroglial fibroblast growth factor receptor 1 gene targeting protects mice from experimental autoimmune encephalomyelitis through ERK/AKT phosphorylation. Brain Pathol 28(2):212–224
Ruffini F, Furlan R, Poliani PL, Brambilla E, Marconi PC, Bergami A, Desina G, Glorioso JC et al (2001) Fibroblast growth factor-II gene therapy reverts the clinical course and the pathological signs of chronic experimental autoimmune encephalomyelitis in C57BL/6 mice. Gene Ther 8(16):1207–1213
Kiyota T, Ingraham KL, Jacobsen MT, Xiong H, Ikezu T (2011) FGF2 gene transfer restores hippocampal functions in mouse models of Alzheimer’s disease and has therapeutic implications for neurocognitive disorders. Proc Natl Acad Sci USA 108(49):E1339–E1348
Rajendran R, Giraldo Velásquez M, Stadelmann C, Berghoff M (2017) Oligodendroglial fibroblast growth factor receptor 1 gene targeting protects mice from experimental autoimmune encephalomyelitis through ERK/AKT phosphorylation. Brain Pathol 28(2):212–224
Figueiredo C, Pais TF, Gomes JR, Chatterjee S (2008) Neuron-microglia crosstalk up-regulates neuronal FGF-2 expression which mediates neuroprotection against excitotoxicity via JNK1/2. J Neurochem 107(1):73–85
Rottlaender A, Villwock H, Addicks K, Kuerten S (2011) Neuroprotective role of fibroblast growth factor-2 in experimental autoimmune encephalomyelitis. Immunology 133(3):370–378
Zhou W, Du X, Song F, Zheng H, Chen K, Zhang W, Yang J (2016) Prognostic roles for fibroblast growth factor receptor family members in malignant peripheral nerve sheath tumor. Oncotarget 17(16):22234–22244
Rand V, Huang J, Stockwell T, Ferriera S, Buzko O, Levy S, Busam D, Li K et al (2005) Sequence survey of receptor tyrosine kinases reveals mutations in glioblastomas. Proc Natl Acad Sci U S A 102(40):14344–14349
Ohashi R, Matsuda Y, Ishiwata T, Naito Z (2014) Downregulation of fibroblast growth factor receptor 2 and its isoforms correlates with a high proliferation rate and poor prognosis in high-grade glioma. Oncol Rep 32(3):1163–1169
Cloughesy TF, Cavenee WK, Mischel PS (2013) Glioblastoma: from molecular pathology to targeted treatment. Ann Rev Pathol 9:1–25
Holland EC, Celestino J, Dai C, Schaefer L, Sawaya RE, Fuller GN (2000) Combined activation of Ras and Akt in neural progenitors induces glioblastoma formation in mice. Nat Genet 25(1):55–57
Park JW, Wollmann G, Urbiola C, Fogli B, Florio T, Geley S, Klimaschewski L (2018) Sprouty2 enhances the tumorigenic potential of glioblastoma cells. Neuro Oncol. 20(8):1044–1054
Maxwell M, Naber SP, Wolfe HJ, Hedley-Whyte ET, Galanopoulos T, Neville-Golden J, Antoniades HN (1991) Expression of angiogenic growth factor genes in primary human astrocytomas may contribute to their growth and progression. Cancer Res 51(4):1345–1351
Yamaguchi F, Saya H, Bruner JM, Morrison RS (1994) Differential expression of two fibroblast growth factor-receptor genes is associated with malignant progression in human astrocytomas. Proc Natl Acad Sci USA 91(2):484–488
Loilome W, Joshi AD, Rhys CM, Piccirillo S, Vescovi AL, Gallia GL, Riggins GJ (2009) Glioblastoma cell growth is suppressed by disruption of fibroblast growth factor pathway signaling. J Neurooncol 94(3):359–366
Morrison RS, Yamaguchi F, Saya H, Bruner JM, Yahanda AM, Donehower LA, Berger M (1994) Basic fibroblast growth factor and fibroblast growth factor receptor I are implicated in the growth of human astrocytomas. J Neurooncol 18(3):207–216
Cuevas P, Carceller F, Angulo J, Gonzblez-Corrochano R, Cuevas-Bourdier A, Gimqnez-Gallego G (2011) Antiglioma effects of a new, low molecular mass, inhibitor of fibroblast growth factor. Neurosci Lett 491(1):1–7
Wang Q, Du J, Xu B, Xu L, Wang X, Liu J, Wang J (2016) Silence of bFGF enhances chemosensitivity of glioma cells to temozolomide through the MAPK signal pathway. Acta Biochim Biophys Sin 48(6):501–508
Auguste P, Gürsel DB, Lemiere S, Reimers D, Cuevas P, Carceller F, Di Santo JP, Bikfalvi A (2001) Inhibition of fibroblast growth factor/fibroblast growth factor receptor activity in glioma cells impedes tumor growth by both angiogenesis-dependent and -independent mechanisms. Cancer Res 61(4):1717–1726
Kargiotis O, Rao JS, Kyritsis AP (2006) Mechanisms of angiogenesis in gliomas. J Neurooncol 78(3):281–293
Toyoda K, Tanaka K, Nakagawa S, Thuy D, Ujifuku K, Kamada K, Hayashi K, Matsuo T et al (2013) Initial contact of glioblastoma cells with existing normal brain endothelial cells strengthen the barrier function via fibroblast growth factor 2 secretion: a new in vitro blood-brain barrier model. Cell Mol Neurobiol 33(4):489–501
Gouaze-Andersson V, Delmas C, Taurand M, Martinez-Gala J, Evrard S, Mazoyer S, Toulas C, Cohen-Jonathan Moyal E (2016) FGFR1 induces glioblastoma radioresistance through the PLCγ/Hif1α pathway. Cancer Res 76(10):3036–3044
Graham BM, Richardson R (2011) Memory of fearful events: the role of fibroblast growth factor-2 in fear acquisition and extinction. Neuroscience 189:156–169
Turner CA, Watson S, Akil H (2012) The fibroblast growth factor family: neuromodulation of affective behavior. Neuron 76(1):160–174
Terwisscha van Scheltinga AF, Bakker SC, Kahn RS, Kas MJH (2013) Fibroblast growth factors in neurodevelopment and psychopathology. Neuroscientist 19(5):479–494
Graham BM, Richardson R (2015) FIbroblast growth factor 2 as a new approach to fighting fear. JAMA Psy 72(10):959–960
FFv H, Leiter I, Rumpel R, Langenhagen A, Wedekind D, Häger C, Bleich A, Palme R et al (2019) FGF-2 isoforms influence the development of dopaminergic neurons in the murine substantia nigra, but not anxiety-like behavior, stress susceptibility, or locomotor behavior. Behav Brain Res 374:112113
Flajolet M, Wang Z, Futter M, Shen W, Nuangchamnong N, Bendor J, Wallach I, Nairn AC et al (2008) FGF acts as a co-transmitter through adenosine A2A receptor to regulate synaptic plasticity. Nat Neurosci 11:1402
Chaudhury S, Aurbach EL, Sharma V, Blandino P, Turner CA, Watson SJ, Akil H (2014) FGF2 is a target and a trigger of epigenetic mechanisms associated with differences in emotionality: partnership with H3K9me3. Proc Natl Acad Sci 111(32):11834–11839
Kao CY, Hsu YC, Liu JW, Lee DC, Chung YF, Chiu IM (2013) The mood stabilizer valproate activates human FGF1 gene promoter through inhibiting HDAC and GSK-3 activities. J Neurochem 126(1):4–18
Shi Y, Li Z, Xu Q, Wang T, Li T, Shen J, Zhang F, Chen J et al (2011) Common variants on 8p12 and 1q24.2 confer risk of schizophrenia. Nat Genet 43:1224
Hashimoto K, Shimizu E, Komatsu N, Nakazato M, Okamura N, Watanabe H, Kumakiri C, Shinoda N et al (2003) Increased levels of serum basic fibroblast growth factor in schizophrenia. Psych Res 120(3):211–218
Pieper AA, Wu X, Han TW, Estill SJ, Dang Q, Wu LC, Reece-Fincanon S, Dudley CA et al (2005) The neuronal PAS domain protein 3 transcription factor controls FGF-mediated adult hippocampal neurogenesis in mice. Proc Natl Acad Sci USA 102(39):14052–14057
Evans SJ, Choudary PV, Neal CR, Li JZ, Vawter MP, Tomita H, Lopez JF, Thompson RC et al (2004) Dysregulation of the fibroblast growth factor system in major depression. Proc Natl Acad Sci USA 101(43):15506–15511
Birey F, Kloc M, Chavali M, Hussein I, Wilson M, Christoffel Daniel J, Chen T, Frohman Michael A et al (2015) Genetic and stress-induced loss of NG2 glia triggers emergence of depressive-like behaviors through reduced secretion of FGF2. Neuron 88(5):941–956
Elsayed M, Banasr M, Duric V, Fournier NM, Licznerski P, Duman RS (2012) Antidepressant effects of fibroblast growth factor-2 in behavioral and cellular models of depression. Biol Psych 72(4):258–265
Mallei A, Shi B, Mocchetti I (2002) Antidepressant treatments induce the expression of basic fibroblast growth factor in cortical and hippocampal neurons. Mol Pharmacol 61(5):1017–1024
Kajitani N, Hisaoka-Nakashima K, Morioka N, Okada-Tsuchioka M, Kaneko M, Kasai M, Shibasaki C, Nakata Y et al (2012) Antidepressant acts on astrocytes leading to an increase in the expression of neurotrophic/growth factors: differential regulation of FGF-2 by noradrenaline. PLOS ONE 7(12):e51197
Bachis A, Mallei A, Cruz MI, Wellstein A, Mocchetti I (2008) Chronic antidepressant treatments increase basic fibroblast growth factor and fibroblast growth factor-binding protein in neurons. Neuropharmacology 55(7):1114–1120
Tang MM, Lin WJ, Pan YQ, Li YC (2018) Fibroblast Growth Factor 2 modulates hippocampal microglia activation in a neuroinflammation induced model of depression. Front Cell Neurosci 12:255
Williams AJ, Yee P, Smith MC, Murphy GG, Umemori H (2016) Deletion of fibroblast growth factor 22 (FGF22) causes a depression-like phenotype in adult mice. Behav Brain Res 307:11–17
Hafenbreidel M, Twining RC, Todd CR, Mueller D (2015) Blocking infralimbic basic fibroblast growth factor (bFGF or FGF2) facilitates extinction of drug seeking after cocaine self-administration. Neuropsychopharmacology 40(13):2907–2915
Fornstedt B, Pileblad E, Carlsson A (1990) In vivo autoxidation of dopamine in guinea pig striatum increases with age. J Neurochem 55(2):655–659
Flores C, Samaha AN, Stewart J (2000) Requirement of endogenous basic fibroblast growth factor for sensitization to amphetamine. J Neurosci 20(2):RC55–RC55
Mueller D, Chapman CA, Stewart J (2006) Amphetamine induces dendritic growth in ventral tegmental area dopaminergic neurons in vivo via basic fibroblast growth factor. Neuroscience 137(3):727–735
Li R, Zou S, Wu Y, Li Y, Khor S, Mao Y, He H, Xu K et al (2017) Heparin-based coacervate of bFGF facilitates peripheral nerve regeneration by inhibiting endoplasmic reticulum stress following sciatic nerve injury. Oncotarget 8(29):48086–48097
Ryu W, Huang Z, Prinz FB, Goodman SB, Fasching R (2007) Biodegradable micro-osmotic pump for long-term and controlled release of basic fibroblast growth factor. J Control Release 124(1-2):98–105
Hansen RK, Christensen C, Korshunova I, Kriebel M, Burkarth N, Kiselyov VV, Olsen M, Ostergaard S et al (2007) Identification of NCAM-binding peptides promoting neurite outgrowth via a heterotrimeric G-protein-coupled pathway. J Neurochem 103(4):1396–1407
Li S, Christensen C, Kiselyov VV, Kohler LB, Bock E, Berezin V (2008) Fibroblast growth factor-derived peptides: functional agonists of the fibroblast growth factor receptor. J Neurochem 104(3):667–682
Thomas TP, Shukla R, Kotlyar A, Kukowska-Latallo J, Baker JR Jr (2010) Dendrimer-based tumor cell targeting of fibroblast growth factor-1. Bioorg Med Chem Lett 20(2):700–703
Chen B, He J, Yang H, Zhang Q, Zhang L, Zhang X, Xie E, Liu C et al (2015) Repair of spinal cord injury by implantation of bFGF-incorporated HEMA-MOETACL hydrogel in rats. Sci Rep 5:9017
Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, Prior TW, Lowes L, Alfano L et al (2017) Single-dose gene-replacement therapy for spinal muscular atrophy. New Engl J Med 377(18):1713–1722
Wong LF, Goodhead L, Prat C, Mitrophanous KA, Kingsman SM, Mazarakis ND (2006) Lentivirus-mediated gene transfer to the central nervous system: therapeutic and research applications. Hum Gene Ther 17(1):1–9
Bharali DJ, Klejbor I, Stachowiak EK, Dutta P, Roy I, Kaur N, Bergey EJ, Prasad PN et al (2005) Organically modified silica nanoparticles: a nonviral vector for in vivo gene delivery and expression in the brain. Proc Natl Acad Sci U S A 102(32):11539–11544
Code Availability
Not applicable
Funding
Open access funding provided by University of Innsbruck and Medical University of Innsbruck.
Author information
Authors and Affiliations
Contributions
All authors whose names appear on the submission:
(1)Made substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data; or the creation of new software used in the work
(2)Drafted the work or revised it critically for important intellectual content
(3)Approved the version to be published
(4)Agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved
Corresponding author
Ethics declarations
Consent to Participate
Not applicable.
Consent for Publication
All authors whose names appear on the submission agreed the version to be published.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Klimaschewski, L., Claus, P. Fibroblast Growth Factor Signalling in the Diseased Nervous System. Mol Neurobiol 58, 3884–3902 (2021). https://doi.org/10.1007/s12035-021-02367-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-021-02367-0