
Abstract
Glioblastoma multiforme (GBM) is the most aggressive and common primary central nervous system tumour. Despite extensive therapy, GBM patients usually have poor prognosis with a median survival of 12–15 months. Novel molecular biomarkers that can improve survival prediction and help with treatment strategies are still urgently required. Here we aimed to robustly identify a gene signature panel for improved survival prediction in primary GBM patients. We identified 2166 differentially expressed genes (DEGs) using meta-analysis of microarray datasets comprising of 955 samples (biggest primary GBM cohort for such studies as per our knowledge) and 3368 DEGs from RNA-seq dataset with 165 samples. Based on the 1443 common DEGs, using univariate Cox and least absolute shrinkage and selection operator (LASSO) with multivariate Cox regression, we identified a survival associated 4-gene signature panel including IGFBP2, PTPRN, STEAP2 and SLC39A10 and thereafter established a risk score model that performed well in survival prediction. High-risk group patients had significantly poorer survival as compared with those in the low-risk group (AUC = 0.766 for 1-year prediction). Multivariate analysis demonstrated that predictive value of the 4-gene signature panel was independent of other clinical and pathological features and hence is a potential prognostic biomarker. More importantly, we validated this signature in three independent GBM cohorts to test its generality. In conclusion, our integrated analysis using meta-analysis approach maximizes the use of the available gene expression data and robustly identified a 4-gene panel for predicting survival in primary GBM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Globally, there were about 330000 incident cases of central nervous system (CNS) cancers with a significant increase in age-standardized incidence rate (17.3%) between 1990 and 2016. However, there was no significant change in age-standardized death rate (2.2%) globally between 1990 and 2016 when about 227,000 deaths were reported due to CNS cancers [1]. In particular, CNS cancer incidence was about 5053 in the UK in 2016 with a 21.6% change in age-standardized incidence rates between 1990 and 2016 [1]. Among these cancers, brain tumour incidence rates in the UK are expected to rise by 6% between 2014 and 2035 [2]. Glioblastoma multiforme (GBM), classified as a grade IV glioma (a brain tumour sub-type) as per the World Health Organization (WHO) classification is the most common and aggressive primary CNS tumour [3, 4]. About 2500 new GBM cases are diagnosed each year in England alone [5]. Currently, standard treatments for GBM include surgical resection followed by radiotherapy and adjuvant chemotherapy [6]. Despite recent advances in treatment strategies, the median survival of GBM patients is still about 12–15 months shorter than most of the other major cancers, e.g. breast cancer [7]. The poor outcome for GBM patients is the worst 5-year overall survival (OS) rate among all human cancers [8, 9].
Over the last decade, an increased focus has been on elucidating the molecular pathogenesis of GBM by identifying its specific molecular signatures and pathways [10, 11]. Some of these molecular genetic alterations, for example, isocitrate dehydrogenase 1 and 2 (IDH1/2) mutation and O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, have been recognized as more appropriate diagnostic and prognostic markers, respectively, in GBM than histological appearance alone [11, 12]. However, given the dismal prognosis of GBM, novel molecular signatures that can improve survival prediction and treatment response to better prognostic and therapeutic success are still urgently required.
Recently, large amounts of high-throughput genomic data generated using microarrays and next-generation sequencing (NGS) techniques have been archived on public databases such as Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/), ArrayExpress (https://www.ebi.ac.uk/arrayexpress/), The Cancer Genome Atlas (TCGA, https://portal.gdc.cancer.gov) and Chinese Glioma Genome Atlas (CGGA, http://www.cgga.org.cn). These provide us the opportunity and resources to explore, integrate and reanalyse the already existing data for new biomarker discovery and validation. In addition, previous studies have reported a correlation between differentially expressed genes (DEGs), microRNAs, long non-coding RNAs and differentially methylated genes and GBM prognosis and have indicated prognostic value using bioinformatic analysis [13,14,15,16,17,18,19,20,21,22,23], but no consistent model exists. For instance, Zuo et al. (2019) [15] and Cao et al. (2019) [16] identified a panel of 6 and 4 genes, respectively, for prognosis prediction with no genes in common. Multiple studies have also focused on establishing solitary gene-GBM relationship without considering the potential advantage of gene combination which may have limited prognostic and predictive power [13, 14].
To improve prognostic and predictive power, a number of recent studies considered multiple mRNA expression datasets and have identified panels of genes to predict prognosis in GBM patients [15,16,17,18,19,20,21]. Despite this, these studies have limited focus on a few datasets. Some studies lacked validation of their panels or models in independent cohorts, whereas there is no proper assessment of sensitivity and specificity of the prognostic models in others. Inclusion of more available datasets as well as application of meta-analysis methods [24, 25] to increase the statistical power of studies as a result of larger sample size can lead to a more robust selection of genes [26]. A robust selection of genes has the potential to improve prognosis prediction and treatment response in GBM. Moreover, these studies are also constrained by small number of normal samples. Furthermore, the majority of GBMs (~90%) develop de novo, i.e. they are primary GBM, and have worse prognosis than secondary GBMs which progress from lower-grade astrocytomas [27]. Hence, considering them separately is important.
In this study, we aimed to robustly identify a gene signature panel for improved survival prediction in primary GBM patients by conducting an integrated analysis on mRNA expression data available on public databases including TCGA, GEO and ArrayExpress. Here, DEGs were discovered from collected microarray datasets by using a novel meta-analysis approach we proposed previously [24], while DEGs from TCGA mRNA sequencing (RNA-seq) dataset were identified by RNA-seq analysis. Based on the common DEGs between microarray and RNA-seq datasets, prognosis-related genes were screened by univariate Cox regression. Among these, by using least absolute shrinkage and selection operator (LASSO) approach with multivariate Cox [28], we identified a survival associated 4-gene signature panel and established a risk score model for survival prediction in primary GBM. Moreover, we assessed the sensitivity and specificity of the model using time-dependent receiver operating characteristic (ROC) curves and validated this signature in three independent primary GBM cohorts.
Results
Differential Expression of Genes in GBM
Meta-analysis identified 2166 DEGs (hereby called as meta-DEGs) in GBM compared with normal brain tissues of which 707 were upregulated and 1459 downregulated. Similarly, 3368 genes were found to be DEGs (hereby called as RNA-seq DEGs) in the TCGA RNA-seq dataset of which 1086 and 2282 were up- and down-regulated, respectively (Fig. 1a). Between meta-DEGs and RNA-seq DEGs, 1443 DEGs (66.62% of meta-DEGs and 42.84% of RNA-seq DEGs) were common (Fig. 1b). Fisher’s exact test (P-value <2.2 × 10−16) showed that the overlap was statistically significant. All except three overlapped DEGs were regulated in the same direction (up or down) in both approaches suggesting that results were consistent among different techniques (Fig. 1c).

Differentially expressed genes between GBM and normal brain tissues. (a) Tabular diagram showing the number of up- and down-regulated DEGs of GBM in meta-analysis for microarray data and TCGA RNA-seq analysis. (b) Venn diagram representing the total number of overlapped DEGs between the meta-analysis for microarray data and TCGA RNA-seq analysis. (c) Number of up- and down-regulated DEGs in the overlapped DEGs between the two DEGs list (meta-analysis and TCGA RNA-seq analysis)
Prognostic Gene Signature Identification for GBM
By applying univariate Cox regression, we evaluated each common DEG for prognostic significance. Out of 1443 common DEGs (see supplementary file 1, Table S1), 123 were found to be associated with overall survival (OS, Cox P-value < 0.05). Thereafter, by using LASSO on these 123 genes, we identified STEAP2 metalloreductase (STEAP2), insulin-like growth factor binding protein 2 gene (IGFBP2), midkine (MDK), protein tyrosine phosphatase receptor type N2 (PTPRN2), solute carrier family 43 member 3 (SLC43A3), protein tyrosine phosphatase receptor type N (PTPRN), SIN-HDAC complex associated factor (SINHCAF), methylenetetrahydrofolate dehydrogenase (NADP+dependent) 2, methenyltetrahydrofolate cyclohydrolase (MTHFD2), granulin precursor (GRN), and solute carrier family 39 member 10 (SLC39A10) as the top 10 survival-related genes. The results were similar when LASSO was applied to all those 1443 common DEGs. However, only IGFBP2, PTPRN and STEAP2 remained consistently significant (Cox P-value < 0.05) for OS after multivariate Cox regression analysis was conducted on the top (5–10) survival-associated genes.
In order to robustly select a panel of genes among these top 10 genes, we fixed the top three IGFBP2, PTPRN and STEAP2 and looked for others that significantly improved survival prediction. After multivariate Cox regression was conducted on these combinations, we assessed their performance in survival prediction using the risk score model for each combination and time-dependent ROC curves. Area Under Curve (AUC) at 6 months and 1-year time points was calculated. Finally, a 4-gene signature (IGFBP2, PTPRN, STEAP2 and SLC39A10) that optimally predicted the OS of GBM patients (Table 1) was identified where SLC39A10 had negative effect while other three all had positive effects.
Risk Score Model Based on the 4-Gene Signature Predicts Survival in TCGA GBM Cohort
To assess GBM prognosis based on the 4-gene signature, a risk score model was established to compute risk scores (r) for each patient using the following formula (Figure 2a):
where egene is the expression value of a gene in TCGA GBM cohort. Then, based on the median value of risk scores defined by the above formula, the patients in TCGA GBM cohort were divided into a low-risk (79 patients) and high-risk (79 patients) group. In the high-risk group, IGFBP2, PTPRN and STEAP2 exhibited a higher expression than in the low-risk group, whereas a lower expression was observed in the high-risk group for SLC39A10 (Fig. 2b). Kaplan-Meier analysis with log-rank test revealed a shorter survival for patients in the high-risk group than that of the low-risk group (P-value < 0.0001, Fig. 2c) suggesting that there might be an adverse association between OS and the risk scores.

Association between the 4-gene signature and overall survival of GBM patients. (a) The distribution of risk scores ordered from low to high. (b) Heatmap showing the expression of the four prognostic genes. The expression change from left to right corresponds to the risk score from left to right. (c) Survival curves using Kaplan-Meier analysis of overall survival when patients are divided into two risk groups based on median of the risk scores
Time-dependent ROC curves showed that the risk scores were capable of predicting survival with high specificity and sensitivity as seen in Fig. 3a and 3b. AUC for the 6 months and 1-year survival prediction were 0.693 and 0.766. The patient’s division into high- and low-risk groups was further improved by using the optimal cut-off selected by maximizing the Youden’s index [29] in the ROC curve which in turn optimizes sensitivity and specificity. For 6 months and 1-year survival prediction, the cut-off was 2.27 and 2.36, respectively. Survival curves constructed using Kaplan-Meier method with log-rank test further suggested a marked difference in OS between the two risk groups (P-value < 0.0001) (Fig. 3c and 3d).

Survival prediction by the 4-gene prognostic signature. (a)–(b) ROC curves for 6 months and 1 year survival prediction by the four gene signature. Points marked in black represent the optimal cut-off selected for dividing patients into high and low risk groups based on optimization of sensitivity and specificity by maximizing Youden’s index. (c)–(d) Kaplan-Meier curves of overall survival of the high- and low-risk groups based on optimal cut-off for 6 months (2.27) and 1 year (2.36), respectively
Prognostic Gene Signature and Pathoclinical Factors in TCGA GBM Cohort
Clinical and pathological factors (including age, gender, IDH mutation status and MGMT methylation status) for which the information was present were studied to assess if the prognostic value of the 4-gene signature was independent of these factors. By using univariate Cox regression analysis, IDH mutation status (HR = 0.302, 95%CI: 0.123–0.744, P-value = 0.009), MGMT methylation status (HR = 0.553, 95%CI: 0.360–0.848, P-value = 0.007), and the prognostic signature-based risk score (HR = 2.709, 95%CI: 2.004–3.662, P-value < 0.001) were found to be significantly associated with OS whereas age and gender (P-value > 0.05) were not (see Table 2). Furthermore, multivariate Cox regression analysis by considering IDH mutation status, MGMT methylation status and risk score as covariates showed that only risk score (HR = 2.410, 95% CI: 1.569-3.700, P-value < 0.001) was significantly associated with patient prognosis (see Table 2b). All these indicated that the 4-gene signature-based risk score was an independent adverse prognostic factor. Overall survival time was considered for both univariate and multivariate Cox regression analysis for all the covariates.
Multivariate analysis with IDH mutation status, MGMT methylation status and risk status at different time points revealed that IDH mutation status and risk status were associated with OS (see Table 2b). Here, risk status refers to the risk group a patient is classified in based on the cut-off. As IDH mutation status was identified as OS-associated, the patients were stratified based on this. In the TCGA GBM cohort, since there are very small number of IDH mutant patients (9 in total), we only considered IDH-wt patients for further stratification analysis to reduce bias. The IDH-wt patients were sub-divided into high- and low-risk groups using the optimal cut-off points. Risk score and IDH-wt combined survival analysis showed that the IDH-wt patients in high-risk group had considerably poor prognosis than the low-risk IDH-wt patients (Fig. 4a and 4b).

Kaplan-Meier analysis of overall survival of IDH-wt GBM patients in TCGA GBM cohort. (a) High- and low-risk groups based on optimal cut-off for 6 months (2.27) and (b) 1 year (2.36), respectively
Risk Score Model Validation in Independent Primary GBM Cohorts
CGGA RNA-seq data including 84 primary GBM samples and two microarray datasets, GSE16011 comprising of 155 primary GBM cases and GSE43378 (32 primary GBM cases) were used for validation of the risk score model. The risk scores were calculated using the same parameters (βk's) as that of the TCGA GBM cohort. Patients were divided into different risk groups based on the re-estimated cut-off points using time-dependent ROC curve analysis at 6 months and 1 year time points (CGGA: 2.42 and 2.50, GSE16011: 2.54 and 3.01 & GSE43378: 1.33 and 1.51). Performance of the risk scores were then evaluated by Kaplan-Meier analysis. As shown in Fig. 5a–5f, the risk model works well in predicting OS in both GSE16011, GSE43378 and CGGA GBM cohorts and suggested a significantly shorter survival for high-risk group patients compared with that in the low-risk group (P-value = 0.00023, 0.0017 and 0.016, respectively, at 1 year).

Kaplan-Meier analyses of the four-gene panel in validation datasets. (a)–(b) For the GSE16011 dataset with optimal cut-off points estimated at 6 months (2.54) and 1 year (3.01) time points, respectively. (c)–(d) For the CGGA RNA-seq dataset with optimal cut-off points estimated at 6 months (2.42) and 1 year (2.50). (e)–(f) For the GSE43378 dataset with optimal cut-off points estimated at 6 months (1.33) and 1 year (1.51)
Discussion
Several recent reports indicate a potential application of a gene panel-derived risk model in predicting GBM prognosis. Zuo et al. (2019) [15] highlighted the implications of a gene panel as a prognostic predictor in GBM by establishing a six-gene signature risk score model using RNA-seq data from TCGA and CGGA databases but lacked an independent validation of the prognostic signature model. Cao et al. (2019) [16] demonstrated that a 4-gene signature-derived risk score model can predict prognosis and treatment response in GBM patients by conducting a combination analysis on GBM mRNA expression data from two GEO datasets and TCGA, but the sensitivity and specificity of the gene panel in survival prediction were not reported. Yin et al. (2019) [17] identified a 5-gene signature for prognosis prediction in GBM using TCGA RNA-seq cohort and a dataset from GEO database (GSE7696). However, the sensitivity and specificity were assessed using time-independent ROC curves in which the event (disease) status and marker value were considered fixed over time for an individual. In practice, both the disease status and marker value change over time. Moreover, the validation carried out in this study could not be claimed as an independent validation because the TCGA cohort used for validation contained all the samples from TCGA RNA-seq training cohort.
In our study, by integrating multiple gene expression datasets generated by different techniques, i.e. microarray and NGS, and conducting meta-analysis and RNA-seq analysis, we identified four important DEGs, namely, IGFBP2, PTPRN, STEAP2 and SLC39A10 in primary GBM which were also significantly associated to OS. IGFBP2 was up-regulated and inversely correlated with OS indicating it may act as an oncogene. SLC39A10 which was down-regulated and positively associated with OS may act as a tumour suppressor gene in GBM. However, PTPRN and STEAP2 were down-regulated and inversely correlated with OS. The risk-score model based on this 4-gene signature performs well in survival prediction for the TCGA GBM cohort and in three independent validation cohorts. Our 4-gene signature-derived risk score model performed better (AUC = 0.766 for 1-year prediction) at classifying the patients into high- and low-risk groups than the 6-gene signature derived and 5-gene signature-derived risk model described in Zuo et al. (2019) [15] (AUC = 0.699 and 0.718 for CGGA and TCGA for 1-year prediction) and Yin et al. (2019) [17] (AUC = 0.708), respectively. It also performed better than the integrated classifier reported in Cheng et al. (2019) [18] (AUC = 0.734 for 1 year).
Of the four genes identified in our prognostic panel, IGFBP2, located on the human chromosome 2 (2q35), is a member of the insulin-like growth factor binding protein family and has established roles in GBM. It has been increasingly recognized as a glioma oncogene and a therapeutic target [30, 31]. Overexpression of IGFBP2 has been found to promote GBM cell migration and invasion and contributes to glioma progression, recurrence, and poor survival in GBM [31]. Holmes et al. (2012) [31] demonstrated that IGFBP2 expression is closely linked to genes in the integrin and integrin-linked kinase and that these genes are associated with prognosis. Moreover, Liu et al. (2019) [30] established that IGFBP2 promotes vasculogenic mimicry (VM) formation in glioma cells via regulating CD144 and MMP2 expression. VM has been considered as one of the reasons that GBM becomes resistant to anti-VEGF therapy [30]. Our study confirmed significant up-regulation of IGFBP2 and predicted a poor outcome for patients as shown in previous studies [17, 31,32,33], thus providing more evidence for further research into its functional roles during GBM progression. PTPRN is also located on the human chromosome 2 (2q35) but is interestingly down-regulated in GBM tissue and was associated with poor prognosis as the expression increased. Recent reports have also found that a higher expression of PTPRN in GBM tissues is associated with a shorter survival of GBM patients albeit it being down-regulated which was in line with our finding [14, 17, 34]. In oncogenesis, Xu et al. (2016) [35] showed that a high expression of PTPRN is associated to tumour growth and proliferation in small cell lung cancer (SCLC). The study further demonstrated that PTPRN is a target of the miR-342 and that miR-342 mimics suppressed the expression of PTPRN which lead to substantial decrease in SCLC growth. However, the expression level of both miR-342-3p (previously known as miR-342) and PTPRN has been reported to be decreased in GBM samples [36, 37], thus warranting future elucidation of other molecular mechanisms involved in PTPRN expression and its role in GBM growth and progression. STEAP2 is located on chromosome 7q21.13 close to STEAP1 and STEAP4 genes and plays a role in iron and copper reduction [38]. Its role has been confirmed in prostate and breast cancer in previous studies [39, 40] but has not been studied in GBM. STEAP2 expression has been found to be significantly increased in prostate cancer, and its knockdown reduced the invasive potential of prostate cancer cells [41]. On the other hand, it is down-regulated in breast cancer tissues, and its low expression was associated with malignant phenotype and poor prognosis [40]. In our study, STEAP2 was significantly down-regulated in GBM tissues and inversely associated with survival. Moreover, STEAP2 down-regulation could promote cell proliferation and invasion by activating the PI3K/AKT/mTOR signalling pathway which is also an activated pathway involved in GBM tumorigenesis [40, 42]. This indicates a new research objective for future studies exploring the role of STEAP2 in GBM growth, progression and prognosis. SLC39A10 is located on chromosome 2q32.3, and the encoded protein belongs to a subfamily of proteins that show structural characteristics of zinc transporters [43]. High expression levels of SLC39A10 have been reported to be correlated with invasive behaviour by stimulating cell migration in breast cancer cells [44]. Similar observations have been made in the case of colorectal cancer [45] and renal cell carcinoma [46]. Cao et al. (2019) [16] demonstrated that SLC39A10 was down-regulated in GBM tissues and positively associated with survival which is consistent with our finding in this study, but its role in GBM progression is poorly understood and requires further exploration in future studies.
A limitation of this study is that in some datasets used for meta-analysis, epilepsy and white matter samples have been deemed as control samples in order to have a considerable number of control samples as compared with the number of tumour samples. The relatively small number of control samples might lead to missing out some potential DEGs. Nevertheless, our meta-analysis will outperform individual microarray studies. Another limitation is that the independent validation is constrained by availability of very few primary GBM datasets that have large sample size (> 50). Nonetheless, our results were validated in the two of the largest independent dataset (to our knowledge) available. For validation datasets, patients were divided into different risk groups based on the re-estimated cut-off. Ideally, the same cut-off for both the discovery and validation datasets should be used, but given the difference in expression values for genes across different datasets generated using different platforms, re-estimation of the cut-off is needed. Furthermore, the role and function of the identified genes in GBM prognosis should be further elucidated in wet-lab experiments.
To summarize, the biological functions and molecular mechanisms in oncogenesis involving the four genes identified have provided hints towards understanding their roles in GBM progression and prognostic and treatment significance of the derived risk score. Moreover, future experimental work is needed to better understand their roles and functions in GBM. The 4-gene panel has promising practical value in the treatment of primary GBM apart from being robust for predicting the survival in primary GBM. In future integrated analysis, we propose to understand the practical value in survival prediction by combining this gene signature with clinical risk factors and other prognostic indices by applying machine learning techniques.
In conclusion, our integrated analysis using meta-analysis approach and two different gene expression techniques maximizes the use of the available gene expression data and robustly identified a 4-gene panel for predicting survival in primary GBM. Multivariate analysis demonstrated that the predictive value of the gene panel-derived risk score was independent of other clinical and pathological features. Hence, the 4-gene panel is a potential prognostic biomarker of primary GBM. Moreover, our findings provide new insights into GBM pathogenesis and prognosis and necessitate future studies.
Methods
Gene Expression Data Collection
As for the discovery datasets, public databases GEO (https://www.ncbi.nlm.nih.gov/geo/), TCGA (https://portal.gdc.cancer.gov/) and arrayExpress (https://www.ebi.ac.uk/arrayexpress/) were searched for all primary GBM-related mRNA expression studies of human brain tissue. Studies were selected for analysis if they: (a) used clinically diagnosed adult primary GBM patients and (b) had at least three control and three tumour samples in their study cohort. Only one biological sample was used for a certain patient in case there were replicates or multiple samples from the same patient. Using our search and selection criteria, we found eight microarray gene expression data sets (GSE4290, GSE12657, GSE13276, GSE19728, GSE90886, GSE108474, GSE116520 and TCGA microarray) with a total of 955 (865 case and 90 control) samples as well as the TCGA RNA-seq dataset with 160 GBM and 5 control samples (Table 3). Seven of the eight microarray datasets were Affymetrix chip generated, whereas one was Illumina chip produced. Corresponding clinical information was also downloaded for these selected studies.
Data Pre-processing
For the identified microarray datasets, raw CEL and non-normalized expression files were obtained for the Affymetrix platforms and Illumina HumanHT-12 V4.0 expression beadchip platform generated data, respectively. Each dataset was prepared individually for the meta-analysis starting with removal of outlier samples using box and density plots. Data acquisition and pre-processing were done according to the framework prescribed in Ramasamy et al. (2008) [47]. The datasets were then normalized using the Robust Multi-array Average (RMA) approach [48]. Annotation of the probesets to Entrez Gene IDs and gene symbols was carried out using the manufacturer supplied annotation files. The probesets that did not map to any Entrez Gene ID were removed. Probesets that mapped to multiple genes were removed as well. For genes that matched to more than one probesets, the one with the largest absolute estimated effect size was kept [24]. To remove low expression data noise, a two-step filtering was applied to each dataset. First, a pre-filtering was done using the present/absent call (affy MAS5.0 algorithm) such that the probesets that are present in at least 10% of the samples are kept. For the perfect-match arrays only, probesets with average expression level less than three were discarded. Second, we removed the bottom 5% of average expression values across samples for each dataset. Additionally, for GSE108474 and TCGA microarray dataset, batch-effect correction was applied using the ComBat function in R (sva, version: 3.32.1) as they comprise of a collection of data generated at various centres.
For GBM RNA-seq data, raw counts were downloaded from TCGA database and were annotated by mapping Ensembl IDs to Entrez Gene IDs and gene symbols (org.Hs.eg.db package in R, version 3.8.2). We filtered out the one with no Entrez ID and in the case of multiple matchings, we selected the one with highest aggregated count. Counts per million (CPM) filtering were used to reduce the number of low expressed transcripts [49]. We removed a transcript if five or more samples had less than 0.85 CPM for that transcript. This is analogous to removing any transcripts with less than 40 mapped reads across all samples. The remaining transcripts were then normalized using the trimmed mean of M values (TMM) normalization method and common and tag-wise dispersion were estimated. The edgeR package in R [50] was used for RNA-seq analysis.
Differential Expression Analysis
We explored DEGs in both microarray and RNA-seq datasets separately.
Meta-analysis for Microarray Studies
Differential expression analysis for all the microarray data sets was performed using the novel meta-analysis method described in Li et al. (2015) [24] and implemented as metaUnion package in R (accessed from https://github.com/chingtoe365/metaUnion). Using this method, a combined effect size across studies was computed to identify DEGs assuming normality of the data. We used this approach over the other existing ones [51] because this method accounts for the combined gene sets from all studies included in the meta-analysis. The DEGs between control and tumour samples were selected based on \( \sum \limits_{i=1}^n\mid {\log}_2\mathrm{F}{\mathrm{C}}_{\mathrm{i}}\mid /\mathrm{n}>1 \) and Bonferroni P-value < 0.05 criteria where n denotes the number of datasets in which a particular gene was present. However, we only consider a gene as DEG for our final analysis if it was present in at least two datasets included in the meta-analysis to improve the robustness.
DEGs from RNA-seq Data
Once the transcripts were normalized and both common and tag-wise dispersion estimated, a negative binomial generalized log-linear model was fitted to the read counts using the glmFit function in R under the edgeR package. DEGs were then selected based on |log2FC| > 1 and Bonferroni P-value < 0.05 criteria. To obtain the final expression level for each gene, we computed the transcripts per million (TPM) values as log2(TPM + 1). A constant factor one was added to account for genes with zero read count in some cases [17].
Common DEGs and Survival Analysis
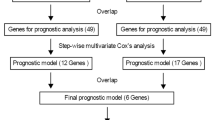
To search for robust DEGs related to GBM, we selected the DEGs that are common in the two DEG lists obtained from microarray meta-analysis and RNA-seq analysis. A two-tailed Fisher’s exact test was used to determine the significance of overlap between these two DEG lists. We also checked for consistency of our results by comparing what percentage of DEGs was regulated in the same direction in the two lists. Next, to evaluate the association of common DEGs with OS, we first conducted univariate Cox proportional hazard regression analysis for each of them in the TCGA GBM (RNA-seq) cohort. The proportional hazard assumption was also checked and found to be appropriate in our case. Second, for the significant genes (Cox P-value < 0.05) from the univariate analysis, we used LASSO with multivariate Cox proportional hazards model to robustly and optimally select a panel of genes which were key DEGs associated with OS [28] (Fig. 6b). Pathoclinical features were also assessed for association with the overall survival using the univariate and multivariate Cox regression model. The workflow and schematics of our study are shown in Fig. 6.

Study workflow. (a) Overall steps in the integrated analysis to identify a 4-gene prognostic signature. (b) Feature selection step in the workflow. The predictive performance step was carried out for each possible combination of genes from the top 10 survival-related genes
Risk Score Model
Based on the selected survival associated gene signature, we established a risk score (r) model [15,16,17] for prognosis, i.e.
where m is the total number of genes in the selected gene signature and βk and ek are the multivariate Cox regression coefficient and expression value of the kth gene in the signature respectively. r was computed for each patient using the above formula, and patients were divided into low-risk and high-risk groups. The split into these two risk groups was first based on the median of risk scores and then optimized by choosing the optimal cut-off determined by time-dependent ROC curve analysis using Youden’s index [29]. Kaplan-Meier method with log-rank test was used to analyse survival differences and plot the survival curves for these two risk groups.
Risk Score Model Validation
For validation purposes, three independent primary GBM gene expression datasets (microarray and RNA-seq) with survival information were downloaded from GEO database (GSE16011, GSE43378) and the CGGA (http://www.cgga.org.cn/), respectively. These datasets were processed by the same workflow as the one used for discovery datasets described above. We used the same βk’s as the one for TCGA GBM cohort for constructing the risk score model for these datasets. Time-dependent ROC curves and Kaplan-Meier method were used to validate the prognostic value of the 4-gene signature for primary GBM patients.
Abbreviations
- GBM:
-
Glioblastoma Multiforme
- DEGs:
-
Differentially expressed genes
- LASSO:
-
Least absolute shrinkage and selection operator
- CNS:
-
Central nervous system
- UK:
-
United Kingdom
- WHO:
-
World Health Organization
- IDH1/2:
-
Isocitrate dehydrogenase 1 and 2
- MGMT:
-
O6-methylguanine-DNA methyltransferase
- NGS:
-
Next-generation sequencing
- GEO:
-
Gene Expression Omnibus
- TCGA:
-
The Cancer Genome Atlas
- CGGA:
-
Chinese Glioma Genome Atlas
- ROC:
-
Receiver operating characteristic
- IGFBP2:
-
Insulin-like growth factor binding protein 2
- MDK:
-
Midkine
- PTPRN2:
-
Protein tyrosine phosphatase receptor type N2
- PTPRN:
-
Protein tyrosine phosphatase receptor type N
- SLC43A3:
-
Solute carrier family 43 member 3
- STEAP2:
-
STEAP2 metalloreductase
- SINHCAF:
-
SIN-HDAC complex-associated factor
- MTHFD2:
-
Methylenetetrahydrofolate dehydrogenase (NADP+dependent) 2, methenyltetrahydrofolate cyclohydrolase
- GRN:
-
Granulin precursor
- SLC39A10:
-
Solute carrier family 39 member 10
- OS:
-
Overall survival
- AUC:
-
Area Under Curve
- CD144:
-
Cadherin 5
- MMP2:
-
Matrix Metalloproteinase 2
- VM:
-
Vasculogenic mimicry
- RMA:
-
Robust multi-array approach
- TMM:
-
Trimmed mean of M values
References
Patel AP, Fisher JL, Nichols E, Abd-Allah F, Abdela J, Abdelalim A, Abraha HN, Agius D et al (2019) Global, regional, and national burden of brain and other CNS cancer, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 18(4):376–393
Smittenaar CR, Petersen KA, Stewart K, Moitt N (2016) Cancer incidence and mortality projections in the UK until 2035. Br J Cancer 115(9):1147–1155
Louis DN, Perry A, Reifenberger G, Von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD et al (2016 Jun 1) The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol 131(6):803–820
Tanaka S, Louis DN, Curry WT, Batchelor TT, Dietrich J (2013 Jan) Diagnostic and therapeutic avenues for glioblastoma: no longer a dead end? Nat Rev Clin Oncol 10(1):14–26
Philips A, Henshaw DL, Lamburn G, O’Carroll MJ (2018) Brain tumours: rise in glioblastoma multiforme incidence in England 1995–2015 suggests an adverse environmental or lifestyle factor. J Environ Public Health 2018
Omuro A, DeAngelis LM (2013 Nov 6) Glioblastoma and other malignant gliomas: a clinical review. Jama. 310(17):1842–1850
Gittleman H, Lim D, Kattan MW, Chakravarti A, Gilbert MR, Lassman AB, Lo SS, Machtay M et al (2017) An independently validated nomogram for individualized estimation of survival among patients with newly diagnosed glioblastoma: NRG Oncology RTOG 0525 and 0825. Neuro-oncology. 19(5):669–677
Tykocki T, Eltayeb M (2018 Aug 1) Ten-year survival in glioblastoma. A systematic review. J Clin Neurosci 54:7–13
Lapointe S, Perry A, Butowski NA (2018) Primary brain tumours in adults. Lancet 392(10145):432–446
Mao H, LeBrun DG, Yang J, Zhu VF, Li M (2012) Deregulated signaling pathways in glioblastoma multiforme: molecular mechanisms and therapeutic targets. Cancer Investig 30(1):48–56
Brandner S, Jaunmuktane Z (2018) Neurological update: gliomas and other primary brain tumours in adults. J Neurol 265(3):717–727
Brandner S, von Deimling A (2015) Diagnostic, prognostic and predictive relevance of molecular markers in gliomas. Neuropathol Appl Neurobiol 41(6):694–720
Di Jia SL, Li D, Xue H, Yang D, Mining LY (2018) TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging (Albany NY) 10(4):592
Shergalis A, Bankhead A, Luesakul U, Muangsin N, Neamati N (2018) Current challenges and opportunities in treating glioblastoma. Pharmacol Rev 70(3):412–445
Zuo S, Zhang X, Wang L (2019) A RNA sequencing-based six-gene signature for survival prediction in patients with glioblastoma. Sci Rep 9(1):1–10
Cao M, Cai J, Yuan Y, Shi Y, Wu H, Liu Q, Yao Y, Chen L et al (2019) A four-gene signature-derived risk score for glioblastoma: prospects for prognostic and response predictive analyses. Cancer Biol Med 16(3):595–605
Yin W, Tang G, Zhou Q, Cao Y, Li H, Fu X, Wu Z, Jiang X (2019) Expression profile analysis identifies a novel five-gene signature to improve prognosis prediction of glioblastoma. Front Genet 10:419
Cheng Q, Huang C, Cao H, Gong X, Li J, Chen Y, Tian Z, Fang Z et al (2019) A Novel Prognostic Signature of Transcription Factors for the Prediction in Patients With GBM. Front Genet 10:906
Hsu JB, Chang TH, Lee GA, Lee TY, Chen CY (2019) Identification of potential biomarkers related to glioma survival by gene expression profile analysis. BMC Med Genet 11(7):34
Wang Z, Gao L, Guo X, Feng C, Lian W, Deng K, Xing B (2019) Development and validation of a nomogram with an autophagy-related gene signature for predicting survival in patients with glioblastoma. Aging (Albany NY) 11(24):12246
Zhao J, Wang L, Kong D, Hu G, Wei B (2019) Construction of Novel DNA Methylation-Based Prognostic Model to Predict Survival in Glioblastoma. J Comput Biol
Liu Q, Qi C, Li G, Su W (2019) Prediction of the Outcome for Patients with Glioblastoma with lncRNA Expression Profiles. Biomed Res Int 2019:1–10
Xian J, Zhang Q, Guo X, Liang X, Liu X, Feng Y (2019) A prognostic signature based on three non-coding RNA s for prediction of the overall survival of glioma patients. FEBS Open Bio 9(4):682–692
Li X, Long J, He T, Belshaw R, Scott J (2015) Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer’s disease. Sci Rep 5:12393
Tseng GC, Ghosh D, Feingold E (2012) Comprehensive literature review and statistical considerations for microarray meta-analysis. Nucleic Acids Res 40(9):3785–3799
Wang S, Liu F, Wang Y, Fan W, Zhao H, Liu L, Cen C, Jiang X et al (2019) Integrated analysis of 34 microarray datasets reveals CBX3 as a diagnostic and prognostic biomarker in glioblastoma. J Transl Med 17(1):179
Ohgaki H, Kleihues P (2013) The definition of primary and secondary glioblastoma. Clin Cancer Res 19(4):764–772
Tibshirani R (1997) The lasso method for variable selection in the Cox model. Stat Med 16(4):385–395
Perkins NJ, Schisterman EF (2006) The inconsistency of “optimal” cutpoints obtained using two criteria based on the receiver operating characteristic curve. Am J Epidemiol 163(7):670–675
Liu Y, Song C, Shen F, Zhang J, Song SW (2019) IGFBP2 promotes immunosuppression associated with its mesenchymal induction and FcγRIIB phosphorylation in glioblastoma. PLoS One 14(9)
Holmes KM, Annala M, Chua CY, Dunlap SM, Liu Y, Hugen N, Moore LM, Cogdell D et al (2012) Insulin-like growth factor-binding protein 2-driven glioma progression is prevented by blocking a clinically significant integrin, integrin-linked kinase, and NF-κB network. Proc Natl Acad Sci 109(9):3475–3480
Cai J, Chen Q, Cui Y, Dong J, Chen M, Wu P, Jiang C (2018) Immune heterogeneity and clinicopathologic characterization of IGFBP2 in 2447 glioma samples. Oncoimmunology. 7(5):e1426516
Yuan Q, Cai HQ, Zhong Y, Zhang MJ, Cheng ZJ, Hao JJ, Wang MR, Wan JH (2019) Overexpression of IGFBP2 mRNA predicts poor survival in patients with glioblastoma. Biosci Rep 28:39(6)
Xu P, Yang J, Liu J, Yang X, Liao J, Yuan F, Xu Y, Liu B et al (2018) Identification of glioblastoma gene prognosis modules based on weighted gene co-expression network analysis. BMC Med Genet 11(1):1–2
Xu H, Cai T, Carmona GN, Abuhatzira L, Notkins AL (2016) Small cell lung cancer growth is inhibited by miR-342 through its effect of the target gene IA-2. J Transl Med 14(1):278
Shao L, He Q, Liu Y, Liu X, Zheng J, Ma J, Liu L, Li H et al (2019) UPF1 regulates the malignant biological behaviors of glioblastoma cells via enhancing the stability of Linc-00313. Cell Death Dis 10(9):1–8
Wang Q, Li P, Li A, Jiang W, Wang H, Wang J, Xie K (2012) Plasma specific miRNAs as predictive biomarkers for diagnosis and prognosis of glioma. J Exp Clin Cancer Res 31(1):97
Ohgami RS, Campagna DR, McDonald A, Fleming MD (2006) The Steap proteins are metalloreductases. Blood. 108(4):1388–1394
Wang L, Jin Y, Arnoldussen YJ, Jonson I, Qu S, Mælandsmo GM, Kristian A, Risberg B et al (2010 Jul 15) STAMP1 is both a proliferative and an antiapoptotic factor in prostate cancer. Cancer Res 70(14):5818–5828
Yang Q, Ji G, Li J (2020) STEAP2 is down-regulated in breast cancer tissue and suppresses PI3K/AKT signaling and breast cancer cell invasion in vitro and in vivo. Cancer Biol Ther 21(3):278–291
Burnell SE, Spencer-Harty S, Howarth S, Bodger O, Kynaston H, Morgan C, Doak SH (2018) STEAP2 knockdown reduces the invasive potential of prostate cancer cells. Sci Rep 8(1):1–2
Pearson JR, Regad T (2017) Targeting cellular pathways in glioblastoma multiforme. Signal Transduction and Targeted Therapy 2(1):1–11
Taylor KM, Nicholson RI (2003) The LZT proteins; the LIV-1 subfamily of zinc transporters. Biochimica et Biophysica Acta (BBA)-Biomembranes 1611(1-2):16–30
Kagara N, Tanaka N, Noguchi S, Hirano T (2007) Zinc and its transporter ZIP10 are involved in invasive behavior of breast cancer cells. Cancer Sci 98(5):692–697
Barresi V, Valenti G, Spampinato G, Musso N, Castorina S, Rizzarelli E, Condorelli DF (2018) Transcriptome analysis reveals an altered expression profile of zinc transporters in colorectal cancer. J Cell Biochem 119(12):9707–9719
Pal D, Sharma U, Singh SK, Prasad R (2014) Association between ZIP10 gene expression and tumor aggressiveness in renal cell carcinoma. Gene. 552(1):195–198
Ramasamy A, Mondry A, Holmes CC, Altman DG (2008) Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med 5(9)
Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP (2003) Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 31(4):e15–e15
Raithel S, Johnson L, Galliart M, Brown S, Shelton J, Herndon N, Bello NM (2016) Inferential considerations for low-count RNA-seq transcripts: a case study on the dominant prairie grass Andropogon gerardii. BMC Genomics 17(1):140
Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 26(1):139–140
Hong F, Breitling RA (2008) Comparison of meta-analysis methods for detecting differentially expressed genes in microarray experiments. Bioinformatics. 24(3):374–382
Funding
This research was supported by AiPBAND (www.aipband-itn.eu), European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement 764281. BP is a Marie-Curie early stage research fellow of AiPBAND.
Author information
Authors and Affiliations
Contributions
Conception and design, BP and XL; Development of methodology, BP and XL; Data acquisition, BP; Analysis and interpretation of data, BP, XL and YT; Original draft preparation, BP; Review and editing, BP, YT and XL; Visualization, BP; Funding acquisition, XL. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval
Not applicable
Consent to participate
Not applicable
Consent for publication
Not applicable
Availability of data and materials
All the data used in this study is publically available on public databases GEO, TCGA and CGGA. Accession numbers of all the datasets used have been provided in Table 3 of the methods section.
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Additional file 1:
Table S1: Differentially expressed genes identified to be significantly associated to survival in TCGA RNA-seq GBM cohort using univariate Cox regression analysis. (PDF 109 kb)
Additional file 2:
Differentially expressed genes identified in our meta-analysis of microarray primary GBM datasets collected from GEO and TCGA databases. (CSV 88 kb)
Additional file 3:
Differentially expressed genes identified by RNA-seq analysis in the TCGA RNA-seq GBM dataset. (CSV 251 kb)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Prasad, B., Tian, Y. & Li, X. Large-Scale Analysis Reveals Gene Signature for Survival Prediction in Primary Glioblastoma. Mol Neurobiol 57, 5235–5246 (2020). https://doi.org/10.1007/s12035-020-02088-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-02088-w