
Abstract
Glioblastomas (GBMs), are the most common intrinsic brain tumors in adults and are almost universally fatal. Despite the progresses made in surgery, chemotherapy, and radiation over the past decades, the prognosis of patients with GBM remained poor and the average survival time of patients suffering from GBM was still short. Discovering robust gene signatures toward better understanding of the complex molecular mechanisms leading to GBM is an important prerequisite to the identification of novel and more effective therapeutic strategies. Herein, a comprehensive study of genome-scale mRNA expression data by combining GBM and normal tissue samples from 48 studies was performed. The 147 robust gene signatures were identified to be significantly differential expression between GBM and normal samples, among which 100 (68%) genes were reported to be closely associated with GBM in previous publications. Moreover, function annotation analysis based on these 147 robust DEGs showed certain deregulated gene expression programs (e.g., cell cycle, immune response and p53 signaling pathway) were associated with GBM development, and PPI network analysis revealed three novel hub genes (RFC4, ZWINT and TYMS) play important role in GBM development. Furthermore, survival analysis based on the TCGA GBM data demonstrated 38 robust DEGs significantly affect the prognosis of GBM in OS (p < 0.05). These findings provided new insights into molecular mechanisms underlying GBM and suggested the 38 robust DEGs could be potential targets for the diagnosis and treatment.
Similar content being viewed by others

Introduction
Glioblastomas (GBMs) are the most common and highly aggressive malignant brain tumors1,2. Worldwide, in developed countries, an estimated 3~5 GBM cases per 100,000 inhabitants are diagnosed each year1,3. 10,000 new cases of malignant GBM are diagnosed each year in the United States3. Despite enormous advances in knowledge and therapies over the decades, survival of patients diagnosed with GBM has not significantly improved, only around 5.1% of glioblastoma patients have a 5-year survival rate4,5. Therefore, understanding the molecular mechanism of GBMs is an important prerequisite for discovering a novel and effective therapeutic strategy5,6,7,8.
High-throughput genomic technologies have been widely applied to facilitate to understand the mechanisms involved in the genesis of disease processes9. Among which, DNA microarray is recognized as very important and powerful tool for identifying the diversity of functional genes and identifying in-depth characterization of changes in gene expression because it can provide invaluable information on gene transcription by simultaneously measuring expression of thousands of genes within a particular biological sample10. For example, Li et al.11 identified that EZH2 could regulate neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications using DNA microarrays. And Dmitriy et al. discovered listeria species based on the iap gene sequence12.
Numerous studies have examined gene expression profiles of individuals with GBM compared with healthy controls and demonstrated that highly proliferation13,14, migration13, and invasion15 nature of GBM cell are key factors hindering effective treatment of gliomas. However, owing highly complexity and intrinsically heterogeneity of GBMs at a molecular level, the specific molecular mechanisms underlying GBM are still poorly understood16,17. Recent studies have shown that a robust signature comprising of genes can provide essential basis to study molecular mechanisms that underpin the process of disease18. It is reported that a robust signature critically depended on the sample sizes studied19, and even need thousands of samples. However, the number of normal control samples in public gene expression databases are disproportionally small compared to tumor samples in a variety of datasets20. In other words, the number of normal samples is inadequate for directly identifying the robust differential expression genes associated with GBM.
Herein, the most comprehensive set of genome-scale mRNA expression data was constructed by combining GBM and normal samples from multiple studies. In total, thousands of samples were analyzed to compile the accurate and robust relevant genes towards insight into the molecular mechanisms. In particular, a list of genes with well robustness significantly different between GBM and normal tissue samples was firstly identified. Secondly, functional analysis base on these robust gene sets was performed and certain deregulated gene expression programs (e.g. cell cycle, immune response, p53 signaling pathway) are identified in glioblastoma process. Moreover, enrichment analysis of transcription factors and targeted miRNAs revealed three novel hub genes including RFC4, ZWINT, and TYMS and three transcriptional factors TATA, E2F4DP1 and HFH4, and two microRNA hsa-mir-519E and hsa-mir-527 driving GBM tumorgenicity. Furthermore, survival analysis was applied for evaluating the prognostic performance of these robust differential expression genes using the clinical information of TCGA GBM data. In sum, the identified robust genes may facilitate the understanding of glioblastoma’s etiology and the discovery of novel hub genes, transcriptional factors and two microRNA driving GBM tumorgenicity would have therapeutic implications.
Materials and Methods
Data collection and pre-processing
Genome-wide expression data sets were collected based on Human Genome U133 Plus 2.0 Array from Affymetrix GeneChip. In particularly, all raw CEL files of analyzed samples were directly downloaded from two well-known Gene Expression Omnibus (GEO) and ArrayExpress (AE) databases. Annotations information of each sample studied was carefully inspected, including GSM files from GEO database and sdrf files from AE database. All CEL files then were processed using single-channel array normalization (SCAN) method by SCAN.UPC package21 with default option in R software. In addition, version 17.0 of BrainArray was used for addressing expression of the same gene with several probes. For gene expression of duplicated samples and only one sample was retained. The final data matrix consisted of expression values for 22215 probes sets and 1588 samples. All detailed descriptions could be found in Lee’s pioneer study20.
Statistical modelling for robust gene signature (RGS) between GBMs and normal samples
Gene expression matrix after combining all studies was furtherly analyzed using bioinformatic methods. Firstly, computing test statistics for expression value of each gene using mt.teststat function by multtest package of the R statistical computing environment. Secondly, p-value of each gene set were computed using one-sided tests. Then, the resulting p-value for the up and downregulated genes were further adjusted for multiple testing using Benjamini and Hochberg method (BH). Moreover, to identify robust DEGs between GBM and normal controls, the samples size 722 (361 samples each groups)22 were randomly selected from a data set of 1,588 samples, and a gene set was prepared by selecting top 500 genes with the lowest p-value from t test analysis. The random sampling was performed 200 times. Secondly, an overlap between two gene-sets was computed for each pair of 200 gene sets. Overlap is the fraction of shared features that appear on both two lists of markers which determined the robustness of the identified markers by measure the similarity of two lists of identified markers23. All procedures aimed to identify robust gene signature (RGS) between GBM and normal control samples. Genes repeatedly selected during random sampling are defined as robust22,24.
Hierarchical clustering based on the robust gene signature (RGS)
To determine the specificity of RGS between GBM and normal samples, unsupervised hierarchical clustering analysis25 (HCA) was utilized for clustering distinct sample groups. The GBMs and normal samples were clustered by HCA based on the manhattan distance, and the ctc packages in R was furtherly used for converting hclust objects to newick format file. Then, the resulting output was used by the version 3 of Interactive Tree Of Life (iTOL) software to generate the associated heatmap and clustering dendrogram26.
Functional category enrichment analysis
In order to explore biological functions of these differential expression genes, gene set enrichment analysis27 (GSEA) was performed based on the 1% most up- and downregulated genes between GBMs and normal samples. GSEA is a computational method that measures whether a known gene set shows significant differences between different biological conditions. Particularly, gene ontology term enrichment analysis was first conducted based the 160 DEGs, which including enrichment for GO ‘Biological Process’, ‘Molecular Function’ and ‘Cellular Component’ terms. Secondly, KEGG pathways enrichment analysis based these genes were also implemented. To investigate the top enriched biological functions and pathways of up and down-regulated genes. One thousand random permutations were performed for each analysis and the threshold of false discovery rate (FDR) was set at 0.05 to allow for investigative discovery.
Transcription factor and target miRNAs enrichment analysis
GSEA based on DEGs was also carried out to elucidate the significant enriched transcription factor (TF) and miRNAs. One thousand random permutations were performed for each analysis. In addition, TFmiR28 was applied for performing integrated analysis of transcription factors (TFs), microRNAs (miRNAs) and genes.
Construction of gene/protein interaction network and analysis
The Search Tool for the Retrieval of Interacting Genes (STRING) database29 has been widely used for exploring protein-protein interactions (PPI). Therefore, the PPI network of DEGs between GBMs and normal samples was constructed and visualized using the STRING online tool, which only included interactions with combined score ≥ 0.429,30. Secondly, the property of PPI network was analyzed using the NetworkAnalyzer module in Cytoscape v3.6.1 software31, which could be useful in visualizing biological networks and integrating PPI data. The nodes of PPI network indicated genes and degree suggested the number of interactions of the gene with other genes. In PPI network established, these genes with large degrees (connectivity degree > 5) and high betweenness centrality32 were selected and regarded as the hub genes.
Results and Discussion
A large-scale GBMs and normal tissues samples is completely collected in this work
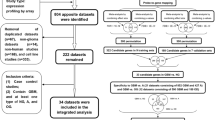
Thousands of samples were needed for generating robust differential expression genes associated with disease, which could contribute to understand molecular pathologies and mechanisms of disease33. Normal tissues samples can be widely used for cancer-associated studies by providing in invaluable clue for abnormal gene expression patterns in cancer compared to normal20. However, the number of normal sample were often small, while the number of cancer samples are relatively large. The unbalance of sample size of distinct groups may limit the study of the disease. Thus, we systematically searched two databases GEO and AE containing publicly-available microarray data sets to obtain the most complete datasets of GBMs and normal tissues samples. The overall numbers of GBMs and normal tissues samples integrating two well-known GEO and ArrayExpress databases was illustrated in Fig. 1. In sum, we collected a total of 48 expression profiling studies, including 723 samples of GBMs and 865 samples of normal controls. Among which, the majority of studies either contain GBMs or normal tissue samples separately, e.g. GSE7307 only contained 174 GBMs samples, and GSE68848 only consisted of 228 normal samples.

Statistics of datasets studied in this work. Expression profiles of all analyzed samples were collected by Gene Expression Omnibus (GEO) and ArrayExpress (AE) databases. E-MTAB- indicates the AE source; GSE indicates the GEO source. Datasets were ascending ordered by their total number of samples.
Sample size consideration
Statistical power analysis was performed for demonstrating the statistical power of this study. As known, sample size that is too small could reduce the power of a study and increases the probability of error, which can render the study meaningless34. Thus, in transcriptomic study, statistical power analysis was typically used in estimating sufficient sample sizes to achieve adequate power35,36. For the studied dataset, it has over 90% power to detect differential expression genes at an overall significance level of 0.01 with Bonferroni’s adjustment. As reported in Mapstone’s pioneer study, a power of 0.9 is well suited for detecting differential genes in transcriptomic study35. Therefore, the statistical power analysis suggested that the studied dataset (sample size) is well suited for identifying the discriminating genes between GBM cases and normal controls.
Identify robust and reliable change in gene expression based on thousands of samples
Identification of robust and reliable differential expression genes could provide immense help for understanding molecular mechanisms underlying complex disease. To facilitate the identification of differential expression genes between GBMs and normal samples, p-values were estimated to identify gene sets that were differential expressed between GBMs and control samples. After multiple test correction, the 160 DEGs were selected using the 1% most up and downregulated genes at a false discovery rate of 0.001. The top 10 most significantly up- or down-regulated DEGs were provided in Table 1. Based on the analysis of robust genes above description, a median overlap value was obtained (greater than 0.9), which suggested the identified DEGs are likely to be well robust. In tol, 678 robust genes were repeatedly selected during random sampling. Among of these 678 genes, 147 genes were common identified in the 160 gene-set (1% most up- and downregulated genes between GBMs and normal samples). Namely, 147 robust gene signatures were identified. The relationship between these 147 robust DEGs with GBM were investigated using manually searching PubMed database. We found that 68% (100 of the 147 unique genes have known connections to GBM (Supplementary Table S1). Among 100 robust DEGs associated with GBM, 80 genes were identified to were differential expression in GBM samples, which included 60 were overexpressed or upregulated in GBM samples,12 were downregulated in GBM samples and 8 were differential expression in GBM without the upregulated or downregulated information. The relationship between the top 10 most significant genes and GBMs was listed in Table 2. These genes have been reported to be associated with the survival, growth, invasive and proliferation characteristics of GBMs cells, for example, suppressing of TMEM45A expression in glioma cells remarkably suppressed cell migration and cell invasion, and GJB6, also known as Cx30 has the potential to influence growth, proliferation and migration of glioma cells. Moreover, downregulated two neural subtype expressed markers were identified such as GABRA1 (Gamma-Amino Butyric Acid A Receptor, Alpha1) and SLC12A5 (solute carrier family 12 member 5)37. In addition, some genes have not been reported in GBMs-associated studies such as the down-regulated gene TMEM130 with the lowest P-value (P = 2.51E-286). However, recent studies have shown that overexpression of transmembrane protein could increase migration capacity toward glioblastoma cells such as TMEM1838. Thus, gene TMEM130 could be associated with GBM and need further validation in the future.
Hierarchical clustering analysis of differentially expressed GBMs signature genes
Unsupervised hierarchical clustering analysis is one of the most powerful methods for the exploratory analysis of gene expression data and was widely used to reflecting distinct gene expression patterns or modules of highly co-expressed genes. Therefore, in this work, hierarchical clustering with ward algorithm39 was applied to cluster the expression profile of differentially expressed genes in each sample group based on these 147 robust DEGs including upregulated and downregulated genes in GBMs. As shown in Fig. 2, two subtypes of all samples studied were identified by unsupervised hierarchical clustering. The heatmap demonstrated the most of GBMs and normal samples could be separated based these DEGs.

Heatmap of 723 glioblastoma and 865 normal samples based on identified 147 robust differential expression (up and downregulated) genes. The highest expression values of DEGs are displayed in green and the lower gradually fading toward black color. The lowest expression values of DEG are shown in red, higher ones gradually fading toward black color. Glioblastoma samples were highlighted with red; Normal control samples were highlighted with blue.
Functional analysis of differentially expressed GBMs signature genes
Functional analysis is secondary analysis of differential expression genes identified and can collectively provide biological function underlying these genes. Understanding dysregulated biological process and pathway in cancer cells are essential for the development of complex diseases40, and can provided immense assistance in the understanding the pathology41. Therefore, GSEA was performed to investigate biological function and pathways of genes associated with GBM. As shown in Fig. 3, the BP terms of GO (Fig. 3A) showed that the up-regulated genes were enriched over 50 terms and the top 10 terms were associated with cell cycle and immune response. And the down-regulated genes showed 13 terms enrichment for cell signaling, anion transport, neurotransmitter transport and so on. The CC terms of GO (Fig. 3B) showed that the up-regulated genes were significantly enriched in 32 terms and the top 10 terms were associated with extracellular space, extracellular matrix, complex of collagen trimer, cell surface, golgi apparatus, collagen trimer and banded collagen fibril. And the down-regulated genes showed 31 terms enrichment for neuron projection, cell projection, intermediate filament and synapse, and so on. The MF terms of GO (Fig. 3C) showed that downregulated genes could be associated with transporter activity, anion channel activity, receptor activity and structural molecule activity, whereas the most upregulated genes were associated with protein complex, receptor, growth factor, enzyme binding. In addition, the KEGG pathways analysis based on these DEGs suggested that downregulated genes were significantly enriched in ecm receptor interaction, complement and coagulation cascades, p53 signaling pathway, focal adhesion, immune network and so on. And the down-regulated DEGs showed 2 pathways enrichment for neuroactive ligand receptor interaction and amyotrophic lateral sclerosis als.

Functional enrichment analysis of gene ontology terms and kegg biological pathway enrichment analysis of DEGs. Gene Ontology covers three domains: cellular component, molecular function and biological process. A-C GO analysis according to biological process, cellular component and molecular function, respectively. (A) Enrichment for GO ‘Biological Process’ terms of genes detected. The y-axis displays the fraction relative to all GO Biological Process terms. (B) Enrichment for GO ‘Molecular Function’ main terms of genes detected. The y-axis displays the fraction relative to all GO Cellular Component terms. (C) Enrichment for GO ‘Molecular Function’ main terms of genes detected. The y-axis displays the fraction relative to all GO Molecular Function terms. The figure shows terms on the x-axis that are significantly enriched (FDR < 0.05). (D) Enrichment for kegg ‘Biological Pathway’ terms of genes detected.
Transcription factor and target miRNAs analysis
Transcription factor (TF) and microRNA (miRNA) are essential for regulating the expression of gene42. Differentially expressed TFs in GBM, and their downstream gene targets, may be potential therapeutic biomarkers of GBM. Therefore, we perform the transcription factors enrichment analysis based on these DEGs. As the demonstrated Supplementary Table S2, top 10 TFs based unregulated DEGs and top 10 TFs based on downregulated DEGs are enrichment and listed by GSEA software at FDR < 0.05. The most of TFs reported that the directly associated GBMs. For example, the identified transcription factors FOXD3 could inhibit proliferation, migration, and invasion of GBM cells42. Moreover, numerous studies showed that miRNAs played important roles in development of cancer and could be potential targets for therapy5. Therefore, to investigate the regulatory mechanisms, we also performed miRNAs enrichment analysis based on these DEGs. As the demonstrated Supplementary Table S3, 16 miRNAs sets are enriched and list by GSEA software at FDR < 0.05. Similarity, the most of miRNAs has reported that the directly associated GBMs. For example, miR-196b was upregulated in GBM compared with normal control samples and associated with cell proliferation42.
In addition, combination analysis of TFs and miRNAs play important roles in understanding the pathogenic mechanisms associated with GBM tumorigenesis28. Therefore, we constructed and analyzed co-regulatory network based on enriched TFs and miRNAs using well known web server TFmiR28. As the illustrated Fig. 4, a total of 55 regulatory interactions were identified which included 29 nodes (miRNAs and TFs/genes). Among which, 28 interactions were experimentally validated. The over representation analysis of the full interaction network showed 9 targeted miRNAs including hsa-mir-106a, hsa-mir-130a, hsa-mir-196b, hsa-mir-20a, hsa-mir-20b, hsa-mir-29a, hsa-mir-29c, hsa-mir-381 and hsa-mir-19a involvement in cancerogenesis of GBMs.

Glioblastoma-specific miRNA/transcription factor co-regulatory networks. The miRNAs are from the enrichment result based on DEGs (top 1% upregulated) at a false discovery rate of 0.05. Green hexagon indicates the transcript factor, the yellow circle represents miRNA, the orange quadrilateral suggests target gene.
PPI network construction
Deciphering the structure of complex network of protein-protein interactions (PPI) can facilitate to understand of molecular mechanism behind the disease43. The hub genes of whole PPI network identified play a vital role in this signal transduction network. Therefore, in this work, we preformed PPI network analysis by choosing a well-known high-throughput STRING dataset44, which can further promote to select reliable edges of network. Particularly, the PPI network was constructed and visualized based on 1% up and down regulated DEGs, which included 158 nodes and 378 edges. As shown in Supplementary Fig. S1, nodes with high betweenness centrality and large degree (connectivity degree > 5) are selected as hub genes and were displayed in Table 3. Table 3 showed the top 10 crucial hub genes involved in the development of GBMs, which included TOP2A, RFC4, ZWINT, NUF2, UBE2C, TYMS, MYC, PBK, MELK and MCM2. Overall expression values of these hub genes were visualized by boxplot for the E-MTAB-3892 dataset. The obvious gene expression difference between GBM and normal samples could be seen in Fig. 5. Among which, the most of hub genes have been experimentally validated in GBM-associated studies, which reflected the hub genes identified is well reproducibility with previous findings. To be more specific, three hub genes including PBK (role in cell cycle), MELK (stem cell marker), and TOP2A (proliferation marker) have been validated in previous GBM-associated studies45. PBK was candidate can be a promising molecular target for GBM treatment46. MELK was identified for encoding other ABC transporters as well as Akt3 kinase in developing resistance of GBM to TMZ47. TOP2A was the hub protein of whole network, which have been demonstrated to its expression was correlated with aggressive and highly proliferating cancers, which were accordance with Horvath et a’ work. In addition, recent studies have showed that overexpression of MCM2 gene could be highly associated with survival of GBM48. Upregulated UBE2C gene was associated with the aggressive progression of GBM49. And siRNA-mediated knockdown against NUF2 may be a potential therapeutic method for treatment of GBM50.

Box plot of intensities after Scan normalization based on top 10 hub genes. Box plot showing median, interquartile range, minimum and maximum intensities with GBMs (blue boxes) compared to those with normal tissue sample (yellow boxes). Corresponding intensities values are displayed as dots. The p-value indicated significant differences between the distinct groups, which is calculated using t-test based on stat_compare_mean function in R ggpubr library.
However, the association between three hub genes including RFC4, ZWINT and TYMS expression and GBMs has not been reported. A recent study by Jiang et al.51 showed that miR-127-3p and its targeted gene SKI could be promising targets for GBM therapy. The present study revealed that hub gene ZWINT also is a target of miR-127-3p, which has functional annotation related to cell cycle, cell division and nuclear division. Therefore, the gene may be a key regulator in GBM development.
Survival analysis
Investigating the clinical significance (e.g. prognosis) of gene expression in GBM is crucial important for diagnosis and molecular target therapy of GBM52. As known, survival analysis was widely applied method to evaluate the prognostic performance of new biomarkers using the clinical data of oncological patients53. The Cancer Genome Atlas (TCGA) project is one of the largest available resources that accumulates genomic, transcriptomic and methylomic data for several types of cancer54. The TCGA provide a useful source of information for identification of prognostic markers55. Therefore, to investigate the oncogenic role of the robust differential expression genes in GBM progression, survival analysis by Kaplan-Meier estimates stratified by their expression was made based on the data of 521 GBM cases provided by TCGA. The expression values of these genes were classified as either high (expression value ≥ median) or low (expression value < median). As the demonstrated Supplementary Fig. S2, we found 38 robust DEGs were significantly related to the prognosis of GBM (OS, P < 0.05) based on Kaplan-Meier estimates (log-rank test). Figure 6 only listed top six significant genes. Thus, these genes are possible candidate genes for diagnosis and molecular target therapy of GBM. Moreover, these 38 robust DEGs significantly associated with overall survival (p < 0.05) in TCGA were retained for further analysis. Cox multivariate model was carried out with function “coxph” in the R package “survival” to develop the risk score model. As demonstrated Table 4, 20 robust DEGs were identified to be with positive coefficients, which could indicate their high expression positively correlated the risk score value, thus, these genes might be tumor genes. While 18 robust DEGs were identified to be with negative coefficients, which could indicate their high expression negatively correlated the risk score value, thus, these genes might be tumor suppressor genes. The performance of the risk score was evaluated by dividing the GBM samples in the TCGA into two subgroups, high-risk and low-risk, using the median risk score as a cutoff (2.992). As illustrated Supplementary Fig. S3, the survival time of the low risk score group is significantly longer than the high-risk score group.

Univariate survival analysis in GBM stratified by robust differential expression gene expression based on the TCGA data as determined by Kaplan-Meier estimates. 521 GBM cases with full data of both clinical and gene expression were collected from the TCGA database. The expression values of these genes were classified as either high (expression value ≥ median) or low (expression value < median). Kaplan-Meier estimates (log-rank test) were made and found 38 genes expression were significantly affect the prognosis of GBM in OS (p < 0.05) (only listed top six genes). More relevant genes were shown in Supplementary Fig. S2.
Conclusions
The most comprehensive set of genome-scale mRNA expression data was constructed by combining GBM and normal control samples from 48 studies, resulting thousands of samples for generating robust genes signature. Based on large-scale gene expression data of GBMs, we have identified 147 robust differential expression genes, which showed the underlying gene expression level differences between NC and GBMs samples. Moreover, the most of identified robust DEGs (67%) were reported that closed to associated with GBM, which suggested high reproducibility with published papers. Furthermore, the GO term and KEGG pathway enrichment results based these robust DEGs may contribute to better understand the molecular mechanisms of GBM. More importantly, based on these robustness DEGs, three new hub genes including RFC4, ZWINT, and TYMS and three top transcriptional factors TATA, E2F4DP1 and HFH4, and two miRNA hsa-mir-519E and hsa-mir-527 were identified in the present study. Furthermore, survival analysis based on the TCGA GBM data revealed 38 genes expression significantly affect the prognosis of GBM in OS (p < 0.05). In sum, these hub genes, transcriptional factors and microRNAs may be potential molecular targets for therapies of GBMs.
References
Martinez-Garcia, M. et al. SEOM clinical guidelines for diagnosis and treatment of glioblastoma (2017). Clin Transl Oncol. 20, 22–28 (2018).
Ferrandez, E., Gutierrez, O., Segundo, D. S. & Fernandez-Luna, J. L. NFkappaB activation in differentiating glioblastoma stem-like cells is promoted by hyaluronic acid signaling through TLR4. Sci Rep. 8, 6341 (2018).
Cloughesy, T. F., Cavenee, W. K. & Mischel, P. S. Glioblastoma: from molecular pathology to targeted treatment. Annu Rev Pathol. 9, 1–25 (2014).
Liu, T. et al. Identification of genes and pathways potentially related to PHF20 by gene expression profile analysis of glioblastoma U87 cell line. Cancer Cell Int. 17, 87 (2017).
Song, H. et al. miR-92b regulates glioma cells proliferation, migration, invasion, and apoptosis via PTEN/Akt signaling pathway. J Physiol Biochem. 72, 201–211 (2016).
Ernst, A. et al. Genomic and expression profiling of glioblastoma stem cell-like spheroid cultures identifies novel tumor-relevant genes associated with survival. Clin Cancer Res 15, 6541–6550 (2009).
Dieterich, L. C. et al. Transcriptional profiling of human glioblastoma vessels indicates a key role of VEGF-A and TGFbeta2 in vascular abnormalization. J Pathol. 228, 378–390 (2012).
Vastrad, B., Vastrad, C., Godavarthi, A. & Chandrashekar, R. Molecular mechanisms underlying gliomas and glioblastoma pathogenesis revealed by bioinformatics analysis of microarray data. Med Oncol. 34, 182 (2017).
Roh, S. W., Abell, G. C., Kim, K. H., Nam, Y. D. & Bae, J. W. Comparing microarrays and next-generation sequencing technologies for microbial ecology research. Trends Biotechnol. 28, 291–299 (2010).
Park, H. E., Park, H. T., Jung, Y. H. & Yoo, H. S. Gene expression profiles of immune-regulatory genes in whole blood of cattle with a subclinical infection of Mycobacterium avium subsp. paratuberculosis. PLoS One. 13, e0196502 (2018).
Li, Z. et al. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene. 37, 2714–2727 (2018).
Volokhov, D., Rasooly, A., Chumakov, K. & Chizhikov, V. Identification of Listeria species by microarray-based assay. J Clin Microbiol. 40, 4720–4728 (2002).
Scrideli, C. A. et al. Gene expression profile analysis of primary glioblastomas and non-neoplastic brain tissue: identification of potential target genes by oligonucleotide microarray and real-time quantitative PCR. J Neurooncol. 88, 281–291 (2008).
Gu, J. J., Zhang, J. H., Chen, H. J. & Wang, S. S. TPX2 promotes glioma cell proliferation and invasion via activation of the AKT signaling pathway. Oncol Lett. 12, 5015–5022 (2016).
Sugiyama, M. et al. Paired related homeobox 1 is associated with the invasive properties of glioblastoma cells. Oncol Rep. 33, 1123–1130 (2015).
Dong, H. et al. Investigation gene and microRNA expression in glioblastoma. BMC Genomics. 11(Suppl 3), S16 (2010).
Wang, L. et al. Screening of differentially expressed genes associated with human glioblastoma and functional analysis using a DNA microarray. Mol Med Rep. 12, 1991–1996 (2015).
Clarke, K. E. et al. A robust and reproducible human pluripotent stem cell derived model of neurite outgrowth in a three-dimensional culture system and its application to study neurite inhibition. Neurochem Int. 106, 74–84 (2017).
Kim, S. Y. Effects of sample size on robustness and prediction accuracy of a prognostic gene signature. BMC Bioinformatics. 10, 147 (2009).
Lee, J. & Choi, C. Oncopression: gene expression compendium for cancer with matched normal tissues. Bioinformatics. 33, 2068–2070 (2017).
Piccolo, S. R. et al. A single-sample microarray normalization method to facilitate personalized-medicine workflows. Genomics. 100, 337–344 (2012).
Ein-Dor, L., Zuk, O. & Domany, E. Thousands of samples are needed to generate a robust gene list for predicting outcome in cancer. Proc Natl Acad Sci USA 103, 5923–5928 (2006).
Wang, C. et al. The concordance between RNA-seq and microarray data depends on chemical treatment and transcript abundance. Nat Biotechnol. 32, 926–932 (2014).
Nyamundanda, G., Gormley, I. C., Fan, Y., Gallagher, W. M. & Brennan, L. MetSizeR: selecting the optimal sample size for metabolomic studies using an analysis based approach. BMC Bioinformatics. 14, 338 (2013).
Lauber, C., Klink, B. & Seifert, M. Comparative analysis of histologically classified oligodendrogliomas reveals characteristic molecular differences between subgroups. BMC Cancer. 18, 399 (2018).
Letunic, I. & Bork, P. Interactive tree of life (iTOL)v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–245 (2016).
Subramanian, A. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550 (2005).
Hamed, M., Spaniol, C., Nazarieh, M. & Helms, V. TFmiR: a web server for constructing and analyzing disease-specific transcription factor and miRNA co-regulatory networks. Nucleic Acids Res. 43, W283–288 (2015).
Szklarczyk, D. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45, D362–D368 (2017).
Gonzalez, O. & Zimmer, R. Contextual analysis of RNAi-based functional screens using interaction networks. Bioinformatics. 27, 2707–2713 (2011).
Shannon, P. et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504 (2003).
Liu, W. et al. Gene expression profile analysis of the bone microenvironment in patients with spinal metastases. Oncol Lett. 15, 61–68 (2018).
Womack, C. & Mager, S. R. Human biological sample biobanking to support tissue biomarkers in pharmaceutical research and development. Methods. 70, 3–11 (2014).
Ayeni, O., Dickson, L., Ignacy, T. A. & Thoma, A. A systematic review of power and sample size reporting in randomized controlled trials within plastic surgery. Plast Reconstr Surg. 130, 78e–86e (2012).
Mapstone, M. et al. Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med. 20, 415–418 (2014).
Billoir, E., Navratil, V. & Blaise, B. J. Sample size calculation in metabolic phenotyping studies. Brief Bioinform. 16, 813–819 (2015).
Ranjit, M., Motomura, K., Ohka, F., Wakabayashi, T. & Natsume, A. Applicable advances in the molecular pathology of glioblastoma. Brain Tumor Pathol. 32, 153–162 (2015).
Jurvansuu, J. et al. Transmembrane protein 18 enhances the tropism of neural stem cells for glioma cells. Cancer Res. 68, 4614–4622 (2008).
Ultsch, A. & Lotsch, J. Machine-learned cluster identification in high-dimensional data. J Biomed Inform. 66, 95–104 (2017).
Itadani, H., Mizuarai, S. & Kotani, H. Can systems biology understand pathway activation? Gene expression signatures as surrogate markers for understanding the complexity of pathway activation. Curr Genomics. 9, 349–360 (2008).
Wang, J. et al. Pathway and network approaches for identification of cancer signature markers from omics data. J Cancer. 6, 54–65 (2015).
Chen, Z. H. et al. Down-regulation of long non-coding RNA FOXD3 antisense RNA 1 (FOXD3-AS1) inhibits cell proliferation, migration, and invasion in malignant glioma cells. Am J Transl Res. 8, 4106–4119 (2016).
Srinivasa Rao, V., Srinivas, K., Kumar, G. N. & Sujin, G. N. Protein interaction network for Alzheimer’s disease using computational approach. Bioinformation. 9, 968–972 (2013).
Szklarczyk, D. et al. STRINGv10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 43, D447–452 (2015).
Arimappamagan, A., Kruthika, Dawn, R., Paturu, K. & Santosh, V. 332 Molecular Characteristics of Tumor Infiltrating Front in Glioblastoma: Insights Into Molecular Heterogeneity and Implications on Targeted Therapy. Neurosurgery. 63(Suppl 1), 197 (2016).
Joel, M. et al. Targeting PBK/TOPK decreases growth and survival of glioma initiating cells in vitro and attenuates tumor growth in vivo. Mol Cancer. 14, 121 (2015).
Stavrovskaya, A. A., Shushanov, S. S. & Rybalkina, E. Y. Problems of Glioblastoma Multiforme Drug Resistance. Biochemistry (Mosc). 81, 91–100 (2016).
Hua, C., Zhao, G., Li, Y. & Bie, L. Minichromosome Maintenance (MCM) Family as potential diagnostic and prognostic tumor markers for human gliomas. BMC Cancer. 14, 526 (2014).
Bredel, M. et al. Functional network analysis reveals extended gliomagenesis pathway maps and three novel MYC-interacting genes in human gliomas. Cancer Res. 65, 8679–8689 (2005).
Huang, S. K. et al. SiRNA-mediated knockdown against NUF2 suppresses tumor growth and induces cell apoptosis in human glioma cells. Cell Mol Biol (Noisy-le-grand). 60, 30–36 (2014).
Jiang, H. et al. Next generation sequencing analysis of miRNAs: MiR-127-3p inhibits glioblastoma proliferation and activates TGF-beta signaling by targeting SKI. OMICS. 18, 196–206 (2014).
Shi, Q. et al. Progression-Free Survival as a Surrogate End Point for Overall Survival in First-Line Diffuse Large B-Cell Lymphoma: An Individual Patient-Level Analysis of Multiple Randomized Trials (SEAL). J Clin Oncol. 5, JCO2018779124, https://doi.org/10.1200/JCO.2018.77.9124 (2018).
Shiozawa, Y. et al. Gene expression and risk of leukemic transformation in myelodysplasia. Blood. 130, 2642–2653 (2017).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 455, 1061–1068 (2008).
Kim, Y. W. et al. Identification of prognostic gene signatures of glioblastoma: a study based on TCGA data analysis. Neuro Oncol. 15, 829–839 (2013).
Gao, Y. F. et al. COL3A1 and SNAP91: novel glioblastoma markers with diagnostic and prognostic value. Oncotarget. 7, 70494–70503 (2016).
Lan, J. et al. TOP2A overexpression as a poor prognostic factor in patients with nasopharyngeal carcinoma. Tumour Biol. 35, 179–187 (2014).
Rasmussen, R. D. et al. BRCA1-regulated RRM2 expression protects glioblastoma cells from endogenous replication stress and promotes tumorigenicity. Nat Commun. 7, 13398 (2016).
Tso, C. L. et al. Distinct transcription profiles of primary and secondary glioblastoma subgroups. Cancer Res. 66, 159–167 (2006).
Amlin-Van Schaick, J. C. et al. Arlm1 is a male-specific modifier of astrocytoma resistance on mouse Chr 12. Neuro Oncol. 14, 160–174 (2012).
Jiang, L. et al. Overexpression of SMC4 activates TGFbeta/Smad signaling and promotes aggressive phenotype in glioma cells. Oncogenesis. 6, e301 (2017).
Sun, W. et al. Knockdown of TMEM45A inhibits the proliferation, migration and invasion of glioma cells. Int J Clin Exp Pathol. 8, 12657–12667 (2015).
Tung, J. N. et al. Inhibition of pentraxin 3 in glioma cells impairs proliferation and invasion in vitro and in vivo. J Neurooncol. 129, 201–209 (2016).
Arun, S., Vanisree, A. J. & Ravisankar, S. Connexin 30 downregulates Insulin-like growth factor receptor-1, abolishes Erk and potentiates effects of an IGF-R inhibitor in a glioma cell line. Brain Res. 1643, 80–90 (2016).
Masilamani, A. P. et al. KLF6 depletion promotes NF-kappaB signaling in glioblastoma. Oncogene. 36, 3562–3575 (2017).
Wei, B. et al. Identification of differentially expressed genes regulated by transcription factors in glioblastomas by bioinformatics analysis. Mol Med Rep. 11, 2548–2554 (2015).
D’Urso, P. I. et al. miR-155 is up-regulated in primary and secondary glioblastoma and promotes tumour growth by inhibiting GABA receptors. Int J Oncol. 41, 228–234 (2012).
Ferrarese, R. et al. Lineage-specific splicing of a brain-enriched alternative exon promotes glioblastoma progression. J Clin Invest. 124, 2861–2876 (2014).
Peng, G. et al. miR-25 promotes glioblastoma cell proliferation and invasion by directly targeting NEFL. Mol Cell Biochem. 409, 103–111 (2015).
Acknowledgements
This work was funded by the research support of the Lanzhou Science and Technology Bureau Program Funds (2017-RC-16, 2018-1-110) and Gansu Science and Technology Fund Grant (ID: 17ZD2FA009).
Author information
Authors and Affiliations
Contributions
Y.Z. and D.H. conceived the idea and supervised the work. J.T., P.Y. and J.H. analyzed data and results. J.T. and Y.Z. wrote the manuscript. All authors reviewed and approved the manuscript.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tang, J., He, D., Yang, P. et al. Genome-wide expression profiling of glioblastoma using a large combined cohort. Sci Rep 8, 15104 (2018). https://doi.org/10.1038/s41598-018-33323-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33323-z
- Springer Nature Limited
Keywords
This article is cited by
-
Evaluation of the role of KPNA2 mutations in breast cancer prognosis using bioinformatics datasets
BMC Cancer (2022)
-
SOX1 Is a Backup Gene for Brain Neurons and Glioma Stem Cell Protection and Proliferation
Molecular Neurobiology (2021)