
Abstract
Despite several compounds entering clinical trials for the negative and cognitive symptoms of schizophrenia, few have progressed beyond phase III. This is partly attributed to a need for improved preclinical models, to understand disease and enable predictive evaluation of novel therapeutics. To this end, one recent approach incorporates “dual-hit” neurodevelopmental insults like neonatal phencyclidine plus isolation rearing (PCP-Iso). Glutamatergic dysfunction contributes to schizophrenia pathophysiology and may represent a treatment target, so we used enzyme-based microsensors to evaluate basal- and drug-evoked glutamate release in hippocampal slices from rats that received neonatal PCP and/or isolation rearing. 5-HT6 antagonist-evoked glutamate release (thought to be mediated indirectly via GABAergic disinhibition) was reduced in PCP-Iso, as were cognitive effects of a 5-HT6 antagonist in a hippocampal glutamate-dependent novel object discrimination task. Yet mGlu7 antagonist-evoked glutamatergic and cognitive responses were spared. Immunohistochemical analyses suggest these findings (which mirror the apparent lack of clinical response to 5-HT6 antagonists in schizophrenia) are not due to reduced hippocampal 5-HT input in PCP-Iso, but may be explained by reduced calbindin expression. This calcium-binding protein is present in a subset of GABAergic interneurons receiving preferential 5-HT innervation and expressing 5-HT6 receptors. Its loss (in schizophrenia and PCP-Iso) would be expected to reduce interneuron firing and potentially prevent further 5-HT6 antagonist-mediated disinhibition, without impacting on responses of VIP-expressing interneurons to mGlu7 antagonism. This research highlights the importance of improved understanding for selection of appropriate preclinical models, especially where disease neurobiology impacts on cells mediating the effects of potential therapeutics.
Similar content being viewed by others

Introduction
Neurodevelopmental disorders like schizophrenia, attention deficit hyperactivity disorder, and autistic spectrum disorder have a complex etiology, involving combinations of early-life risk factors that trigger persistent long-term changes and disease emergence later in life [1]. Relatively poor management of negative and cognitive symptoms of schizophrenia by existing antipsychotics often prevents reintegration into society [2], and as a result, this disorder remains one of the top 10 causes of disability worldwide with an annual cost of over $158 billion in the USA alone [3]. 5-HT6 receptor antagonists and numerous other receptor- and transporter-selective compounds showed promising activity against seemingly relevant deficits in preclinical models, but disappointingly very few progressed beyond phase III clinical trials. This high attrition is partly attributed to a need for improved preclinical models, to further elucidate disease neurobiology and enable more predictive evaluation of novel therapeutics [4].
One approach to producing more comprehensive rodent models for neurodevelopmental disorders like schizophrenia involves “dual-hit” combinations of established perinatal and peripubertal interventions that each mirror different aspects of delayed symptom onset and multiple neurotransmitter involvement [5]. For example, neonatal NMDA receptor antagonist administration (between postnatal days 7–11 when sensitivity to their pro-apoptotic effects peaks [6]) followed by postweaning isolation rearing of gregarious rat pups induces more robust deficits than either manipulation alone [7,8,9,10]. Thus, combined neonatal phencyclidine (PCP) plus isolation rearing (PCP-Iso) produces more extensive cognitive impairment across a broader array of domains, including spatial reference and fear-motivated associative memory [7, 10], plus altered pro-social interaction and concomitant ultrasonic vocalizations [11, 12] that appear more akin to negative symptomatology than the increased aggression seen with single-hit isolation rearing [13]. These changes are accompanied by downregulation of hippocampal genes involved in glutamate metabolism, dopaminergic neurotransmission, and GABA receptor signaling, as well as those encoding parvalbumin and glutamic acid decarboxylase 67 (GAD67) [14]. Preliminary evidence suggests visual recognition memory deficits in the dual-hit model have some predictive validity, being reversed by the dopamine D3-preferring D2/D3 receptor partial agonist cariprazine [12] which is now approved by the FDA for management of schizophrenia [15], the atypical antipsychotic aripiprazole [12] that has modest cognitive benefit in some patients [16], and lamotrigine [14] which although not widely used may assist clozapine-resistant cases [17]. However, further insight into the molecular and neurochemical basis for differences between single and dual-hit models is essential to understand their potential utility in drug discovery for different patient subgroups or schizophrenia as a whole.
There is clear evidence for glutamatergic dysfunction in schizophrenia [18,19,20,21,22] and other disorders that feature cognitive impairment, and marked pharmaceutical interest in developing glutamate-based treatments [23]. Yet so far the possibility of more extensive glutamatergic deficits in combined versus separate neonatal PCP and isolation rearing models has not been studied at either a functional or protein expression level, and our initial experiment therefore focused on both these issues. We used enzyme-based microsensors to evaluate basal- and drug-evoked glutamate release in hippocampal slices from neonatal PCP-treated and/or isolation-reared rats, because this technique provides a direct method to selectively monitor extracellular glutamate on a second-by-second basis [24]. In addition, the ability to use separate slices from each individual to investigate a range of putative procognitive drugs that increase extracellular glutamate via different mechanisms (without the confounding influence of anesthesia required for magnetic resonance spectroscopy (MRS)) represents a marked contribution to the reduction component of the 3Rs initiative. Similar approaches have been applied to study epilepsy [25], spinal injury [26], and infection [27]. We focused on the hippocampus because glutamate hypofunction has been linked to declarative memory deficits in schizophrenia [28]. Although alterations within the dentate gyrus and CA3 are reported [29], we chose to record from CA1 due to evidence for volumetric and morphological abnormalities in early schizophrenia [30, 31], plus synaptic pathology in chronic cases [32]. We report that the expected glutamate release evoked by a 5-HT6 receptor antagonist was reduced in our dual-hit model, whereas glutamatergic responses to depolarization, reuptake inhibition, group III metabotropic receptor (mGlu) blockade, and mGlu7 allosteric antagonism all remained unaffected. Parallel western blot studies to investigate the underlying reasons for this focused on protein expression of vesicular glutamate transporters (VGLUT) 1–3 (required for presynaptic glutamate release), excitatory amino acid transporters (EAAT) 1–3 (responsible for glutamate reuptake), GAD67 (the GABA synthesis enzyme), vesicular GABA transporter (VGAT, required for presynaptic GABA release), plus 5-HT6 and mGlu7 receptors (that each regulate glutamate release via different mechanisms).
To determine the functional correlates of attenuated drug-evoked glutamate release in the slice preparation, our final experiment compared the cognitive-enhancing effects of 5-HT6 and mGlu7 antagonists in rats that received both PCP and isolation to those in single-hit isolation-only animals. We focused exclusively on novel object discrimination (NOD) because of its dependence on hippocampal glutamate [33,34,35,36], our previous findings with cariprazine, aripiprazole, and lamotrigine suggesting potential predictive validity of this test when combined with this model [12, 14], and the ability to perform repeated testing using a cross-over design to reduce animal numbers and further comply with the 3Rs initiative. Having noted a selective absence of 5-HT6 antagonist-mediated cognitive effects in the dual-hit model, we performed immunohistochemical analyses of 5-HT input to the hippocampus, plus the calbindin-positive cells that are its preferential target [37] and the main subset of 5-HT6 receptor-expressing GABAergic interneurons [38], in an attempt to provide mechanistic insight.
Materials and Methods
Animals
This research used a total of 113 male Lister-hooded rats (Charles River UK) maintained under controlled conditions (21 ± 2 °C, 55 ± 10% humidity, 12-h light-dark cycle; on at 07:00 h). For initial microsensor/western blot characterization of glutamatergic deficits, 69 pups from 14 litters were obtained with dams on postnatal day (PND) 3, randomized (by drawing lots) to receive saline vehicle (Veh; 1 ml/kg s.c.) or PCP HCl (10 mg/kg base) on PND 7, 9, and 11 [9], then housed in mixed treatment groups (3–4; Gr) or isolation from weaning on PND 21 (with allocation balanced across litters). Resultant Veh-Gr, PCP-Gr, Veh-Iso, and PCP-Iso were randomized (as above) to microsensor (n = 7–9) or western blot (n = 8–10) subgroups, with three age-matched drug-naïve group-housed rats (423–469 g; Charles River UK) for in-house microsensor validation. Subsequent NOD assessment of pharmacological sensitivity followed by immunohistochemistry used 41 pups from 6 litters (n = 13–14; Fig. 1).

Summary of the experimental design. Two separate cohorts of male Lister hooded rats that received saline (1 ml/kg s.c.; Veh) or PCP (10 mg/kg) on PND 7, 9, and 11 were housed in social groups (Gr) or isolation (Iso) from weaning on PND 21. The first cohort a underwent locomotor activity, NOD, and PPI (n = 15–18 per treatment-housing combination) before balanced allocation to microsensor (n = 7–9) or western blot (n = 8–10) subgroups. The second cohort b underwent NOD on three occasions at 1–2-week intervals to receive acute vehicle (0.5% methylcellulose 1% Tween 80; 1 ml/kg i.p. 30 min before the familiarization trial), SB-399885 (10 mg/kg), or MMPIP (10 mg/kg) on separate test days in a pseudorandom order (n = 13–14 per neurodevelopmental condition), before tissue collection for immunohistochemistry
Dams with litters were housed in individually ventilated cages with standard environmental enrichment. After weaning cages (Gr 32 × 51 cm, Iso 25 × 42 cm) contained only sawdust with grid lids to ensure maintenance of visual, olfactory, and auditory contact [39]. Handling was restricted to a single weekly cage change and body weight measurement until behavioral testing. Neonatal injections and behavioral testing occurred in the light phase (10:00–11:00 h and 09:00–16:00 h, respectively). All procedures were conducted in accordance with the Animals (Scientific Procedures) Act, 1986 and ARRIVE guidelines [40, 41], with University of Nottingham Local Ethical Committee approval. Group sizes were based on previous studies employing these techniques [12, 24, 42]. Data were obtained by trained observers unaware of neurodevelopmental history or any acute treatment.
Microsensor and Western Blot Characterization of Glutamatergic Deficits
Prior Confirmation of Neonatal PCP and Isolation Rearing-Induced Behavioral Phenotype
To confirm expected development of the previously reported behavioral phenotype before tissue collection, rats underwent a short battery of tasks [14] selected for translational relevance to the positive and cognitive symptoms of schizophrenia [43,44,45] and which map to the arousal, cognitive, and sensorimotor sections of the Research Domain Criteria (RDoC) [46]. Tests were ordered from least to most aversive and are well established within the laboratory (e.g., [10, 12, 14, 47,48,49]) and described in detail elsewhere [42]. The length of time required to conduct microsensor recordings meant that more extensive behavioral evaluation including confirmation of previously reported deficits in fear-motivated associative memory [10, 12] was not possible, due to project license limits on the duration of isolation and weight restrictions for the required euthanasia method.
Locomotor activity [42, 47] was assessed (PND 56–58, with a balanced mix of housing and treatment groups each day) for 1 h in individual photobeam activity chambers (39 × 23.5 × 24.5 cm: San Diego instruments, CA, USA), where a single ambulation count was recorded for every two consecutive adjacent lower beam breaks, a single rearing count for every upper beam break, and a single fine movement count for each repeated lower beam break.
NOD [42, 48] was assessed the following day in the same arena. Rats received 3 min habituation, then two consecutive 3 min object exploration trials separated by 2 h. In the familiarization trial, rats encountered two identical bottles. For the choice trial, one was randomly replaced with a novel object (striped bottle). Object exploration (sniffing, licking, chewing, or having moving vibrissae while directing the nose towards and ≤ 1 cm) was timed and used to calculate the choice trial discrimination ratio (exploration of novel/total choice trial object exploration). The 2 h inter-trial interval was chosen to ensure intact memory in group-housed controls [48] and permit detection of neonatal PCP and/or isolation rearing-evoked deficits [10, 12, 14, 42]. However, we acknowledge this comparatively short interval makes the task less suitable for dissociating the effects of cognitive-enhancing test compounds on memory acquisition versus consolidation or retention, as can be performed with longer intervals [48].
Pre-pulse inhibition of the acoustic startle response (PPI [42, 49]) was assessed (PND 63) in SR-Lab startle response chambers (San Diego instruments, CA, USA) where rats received 5 min acclimatization to background white noise (62 dB), ten 120 dB startle trials, a pseudorandom mix of 50 trials with or without a preceding sub-threshold 72, 76, 80, or 84 dB prepulse, and then five final startle trials (all separated by 10–20 s). Individual whole-body startle responses were recorded for 100 ms after startle pulse onset and used to calculate area under the curve (AUC). For each trial type, the mean percentage PPI was calculated from mean AUC (after conditional elimination of values greater than two standard deviations from the mean, attributed to movement during startle delivery), using the equation % PPI = ((pulse alone AUC − prepulse AUC)/pulse alone AUC) × 100.
Glutamate Microsensor Recordings
Methods were modified from Oldenziel et al. [24]. Rats were euthanized by cervical dislocation and brains immersed in ice-cold artificial cerebrospinal fluid (aCSF; 126 mM NaCl, 3.0 mM KCl, 2.0 mM CaCl2, 2.0 mM MgCl2, 1.2 mM NaH2PO4, 26 mM NaHCO3, and 10 mM glucose) were gassed with 95% O2/5% CO2. Transverse hippocampal slices (400 μm) prepared using a vibrating microtome (Leica Biosystems, Nussloch, Germany) recovered for at least 1 h at room temperature in a brain slice prechamber (Harvard Apparatus, Cambridge, UK). Glutamate and null biosensors (50 μm diameter × 500 μm length: Sarissa Biomedical Ltd., Coventry, UK [50]) were rehydrated (100 mM NaCl, 1 mM MgCl2, 2 mM glycerol, 10 mM NaPi, pH 7.4 for 10 min then glycerol for 5 min), polarized (MicroC: WPI, Sarasota, FL, USA) to 500 mV versus an Ag/AgCl reference electrode in a brain slice interface chamber (Harvard Apparatus), cycled from − 500 to 500 mV at 100 mV/s for 10 cycles, and then checked for initial sensitivity to 10 μM L-glutamate (response > 0.1 nA). Test solutions and potential interfering agents (10 μM 5-HT, dopamine, glutamine, aspartate, and 100 μM ascorbic acid) were also examined in the absence of tissue. Signals were recorded using Clampex 9.2 (Molecular Devices, Wokingham, UK). Individual slices were transferred to the interface chamber where they were superfused with aCSF containing 2 mM glycerol (37 °C) and continuously aerated by humidified 95% O2/5% CO2. Glutamate and null sensors were inserted in close proximity (~ 200 μm; V-shaped form) into CA1 (Fig. 2c) and equilibrated for 30 min.

Characterization of the glutamate microsensor signal prior to studies in neurodevelopmentally manipulated rats. Representative a, d–h or mean ± SEM b difference in current output between glutamate and null sensors on addition of a exogenous glutamate or potential interfering agents, which demonstrates a selective response to glutamate, and b increasing concentrations of glutamate in the absence of tissue, which demonstrates a linear relationship, as well as following insertion of sensors into the CA1 region c of separate slices from drug-naïve group-housed rats (n = 3) where glutamate signals were increased by exposure to d elevated KCl, e the glutamate reuptake inhibitor DL-TBOA, f sodium channel activator veratridine, g antiport exchange substrate L-cysteine, or h α-latrotoxin which induces exocytosis from presynaptic vesicles, suggesting neuronal origin of the glutamate signal. Bars represent 10-min perfusion via the aCSF reservoir, and arrows local application to the interface chamber
Validation studies in tissue from drug-naïve group-housed animals examined stimulation of glutamate by KCl depolarization (30, 60, 120 mM, with corresponding reductions in NaCl to maintain osmotic balance), the glutamate reuptake inhibitor DL-TBOA (200 μM), sodium channel activator veratridine (10, 50, 100 μM), antiport exchange substrate L-cysteine (50, 100, 500 μM), and α-latrotoxin (300 nM) which induces exocytosis from presynaptic vesicles, plus inhibition of basal-, KCl-, and TBOA-evoked release by the voltage-dependent sodium channel blocker tetrodotoxin (TTX; 20 μM). Responses to KCl depolarization, TBOA inhibition of glutamate reuptake, the 5-HT6 antagonist SB-399885 (3 μM ± 120 mM KCl), group III mGlu antagonist CPPG (100 μM ± 120 mM KCl), and mGlu7 allosteric antagonist MMPIP (100 μM ± 120 mM KCl) were then compared in slices from the four different neurodevelopmental conditions (PND 73–112). Antagonist concentrations were similar to previous in vitro [51, 52] and hippocampal slice [53, 54] studies, and intentionally higher than achieved in vivo to allow for slice penetration [55]. Compounds (one per slice) were perfused via the aCSF reservoir (10 min or 15 min for 5-HT6/mGlu antagonists with high KCl) or applied direct to slices (α-latrotoxin). Point calibration to 10 μM L-glutamate was repeated postslice. The difference in current output (nA) between glutamate and null sensors was calculated off-line and used with calibration data to determine glutamate concentration. An average basal extracellular glutamate for each animal was derived from the five separate slices used to examine AUC responses to different glutamatergic manipulations, such that the final n for each neurodevelopmental condition represents the number of different animals each investigated with different pair of glutamate and null sensors.
Western Blots
Rats were euthanized on PND 64 by concussion and immediate decapitation, and western blotting was performed as previously described [42]. Portions of nitrocellulose membranes containing proteins > 40 kDa were incubated overnight (4 °C) with mouse monoclonal or rabbit polyclonal primary antibodies against VGLUT1, VGLUT2 (1:500; Merck Millipore, Watford, UK), VGLUT3 (1:500; Abcam, Cambridge, UK), EAAT1, EAAT2, EAAT3 (1:500; Alpha Diagnostic International, San Antonio, TX, USA), GAD67 (1:1000; Merck Millipore), VGAT (1:400; Abcam), 5-HT6 (1:500; Abcam), or mGlu7 (1:500; Merck Millipore). Corresponding portions containing proteins < 40 kDa were incubated with mouse monoclonal or rabbit polyclonal primary antibodies against the housekeeping protein GAPDH (1:20,000; Sigma-Aldrich, Poole, UK). After infrared secondary antibody (800 CW anti-mouse or anti-rabbit, 1:10,000; LI-COR, Cambridge, UK) incubation (1 h in the dark), bands were detected and quantified using a LI-COR Odyssey system and data expressed as a percentage of GAPDH.
NOD Assessment of Pharmacological Sensitivity, Followed by Immunohistochemistry
NOD
Consistent with previous observations [10], behavioral phenotyping of our microsensor/western blot cohort found no effect of single-hit neonatal PCP on NOD (Fig. S1d-e), so to comply with the 3Rs principle, we did not include a PCP-Gr subgroup (with predicted intact NOD) in the cohort comparing pharmacological reversal of NOD deficits in Veh-Iso versus PCP-Iso. Veh-Gr, Veh-Iso, and PCP-Iso underwent NOD (as described above) on three occasions at 1–2 week intervals (PND 57–80) to receive vehicle (0.5% methylcellulose 1% Tween 80; 1 ml/kg i.p. 30 min before familiarization), SB-399885 (10 mg/kg), or MMPIP (10 mg/kg) on separate days in a pseudorandom order and serve as their own controls. SB-399885 and MMPIP doses were selected from studies demonstrating behavioral activity, including reversal of NOD deficits in other models for schizophrenia [42, 56], without motor impairment [57,58,59].
Immunohistochemistry
Rats were euthanized after the final NOD (PND 78–80) by concussion and immediate decapitation. Brain hemispheres were immersed fixed in 4% paraformaldehyde then 30% sucrose (each overnight, 4 °C) and frozen in isopentane on dry ice. Serial coronal sections (60 μm) were obtained throughout the dorsal hippocampus using a freezing microtome (Anglia Scientific, Cambridge, UK) and stored in 30% glycerol, 30% ethylene glycol (− 20 °C) until free-floating immunohistochemistry. One PCP-Iso was excluded from the rest of the study due to technical difficulties during slicing. Six evenly spaced sections were selected from each rat, to span approximately Bregma − 2.44 to − 4.42 according to a digital atlas [60]. Sections were washed (4 × 5 min) in phosphate-buffered saline (PBS), incubated (1 h) in 2% goat or donkey serum in buffer 1 (0.5% BSA, 0.3% Triton X-100), then (overnight, 4 °C) rabbit or goat polyclonal primary antibodies against calbindin or 5-HT (Abcam: 1:500 in buffer 1), or buffer alone for negative control. Sections were washed (3 × 5 min) in buffer 2 (0.15% BSA, 0.1% Triton X-100), incubated (1 h in the dark) in Alexa Fluor 568 goat anti-rabbit or 594 donkey anti-goat secondary antibodies (Thermo-Fisher, Loughborough, UK: 1:500 in buffer 2), washed (2 × 5 min each) in buffer 2 and PBS, and then mounted on gelatin-subbed slides and air-dried. They were then rinsed with PBS, counterstained with DAPI nuclear stain (Sigma-Aldrich: 1:2000 in H2O; 30 s), rinsed with H2O, and cover slipped with DABCO (Sigma-Aldrich: 0.2% in 90% glycerol) then stored at 4 °C. With the expectation of DAPI, all solutions were prepared in PBS.
Sections were viewed on a Nikon EFD-3 fluorescence microscope and images obtained using an Insight QE camera and SPOT Imaging software (Diagnostic Instruments Inc., MI, USA). The number of calbindin-positive cells within strata oriens, radiatum, and lacunosum-moleculare of the subiculum/fasciola cinereum, CA1, and CA2/3 was counted from × 4 images manually reconstructed to cover the entire dorsal hippocampus. The intensity of calbindin and 5-HT immunoreactivity in consistently placed × 20 images of CA1 (encompassing strata oriens, pyramidale, and radiatum; Fig. 5a), and of 5-HT immunoreactivity in further × 40 images entirely within strata oriens or radiatum (Fig. 5e), was quantified using Fiji [61]. Anatomical boundaries were determined using a digital atlas [60].
Drugs
PCP HCl, L-glutamate, 5-HT, dopamine, ascorbic acid, and L-cysteine were purchased from Sigma-Aldrich, α-latrotoxin from Enzo Life Sciences (Exeter, UK), and all other compounds from Tocris (Bristol, UK). For microsensor studies, stock solutions of TBOA, SB-399885, CPPG, MMPIP, TTX (2–10 mM in saline), and α-latrotoxin (300 nM in 50% glycerol) were stored in aliquots at − 20 °C and diluted in aCSF on the day of use. All other solutions were prepared daily.
Statistical Analysis
All analyses were performed using GraphPad Prism (v7) or IBM SPSS (v24). After confirming normality and homogeneity of variance, data from the microsensor/western blot cohort were analyzed by three-way repeated measures ANOVA (with time, object, prepulse, applied drug concentration, or glutamate transporter subtype as a within-subject factor) or two-way ANOVA (total locomotor activity, NOD discrimination ratio, microsensor responses to single-drug concentrations, and remaining protein expression) with neonatal treatment and housing as between-subject factors. Data from the pharmacological NOD and immunohistochemistry cohort were analyzed by three-way repeated measures ANOVA with acute treatment and object, or hippocampal subfield and cell layer as within-subject factors and neurodevelopmental condition as a between-subject factor. NOD discrimination ratios were analyzed by two-way repeated measures ANOVA with acute treatment as a within-subject factor, and remaining immunohistochemical data by one-way ANOVA, with neurodevelopmental condition as a between-subject factor in each case. Pearson’s r correlation analyses were also performed between immunohistochemical and discrimination ratio data. Post hoc within- and between-subject comparisons used Sidak and Tukey tests, respectively. P < 0.05 was regarded as statistically significant and data are presented as mean ± SEM.
Results
Microsensor and Western Blot Characterization of Glutamatergic Deficits
Prior Confirmation of Neonatal PCP and Isolation Rearing-Induced Behavioral Phenotype
The time course of ambulation in a novel arena showed a time × housing interaction (F(11,605) = 3.055; P < 0.001), and although there were no time × treatment or time × treatment × housing interactions, PCP-Iso had higher ambulation than Veh-Gr controls and single-hit PCP-Gr or Veh-Iso at multiple time points (Fig. S1a). Total ambulation showed treatment (F(1,55) = 3.872; P < 0.05) and housing (F(1,55) = 3.817; P < 0.05) effects and was higher in PCP-Iso (but not Veh-Iso or PCP-Gr) than in Veh-Gr (P < 0.05; Fig. S1b). Similar patterns were observed for rearing and fine movement (data not shown).
NOD testing confirmed an effect of object (F(1,65) = 37.016; P < 0.001) and an object × housing interaction (F(1,65) = 18.979, P < 0.001) during the choice trial. Veh-Gr and PCP-Gr were both able to discriminate the novel from familiar object (P < 0.0001 and P < 0.001, respectively), but Veh-Iso and PCP-Iso were not (P > 0.05; Fig. S1d). This isolation-induced impairment was further supported by a housing effect on the discrimination ratio (F(1,65) = 22.37, P < 0.0001), which was lower in both Veh-Iso (P < 0.001) and PCP-Iso (P < 0.01) than in Veh-Gr (Fig. S1e). Importantly, these changes occurred without any spatial preference between identical objects during the familiarization trial (Fig. S1c) or any differences in total object exploration (data not shown).
PPI testing showed the expected effect of prepulse volume (F(2,130) = 122.534; P < 0.001), although the prepulse × housing interaction just failed to reach statistical significance (F(2,130) = 2.921, P = 0.057; Fig. S1f). There were no differences in startle reactivity (data not shown).
Taken together, these findings suggest the current PCP-Iso cohort had slightly more marked locomotor hyperactivity than previous studies, and the expected robust NOD impairment, but a smaller PPI deficit than when these tests were performed after an extra week of isolation [10, 14], which was not compatible with the current microsensor study design. We therefore progressed these animals to planned microsensor and western blot studies, given the novelty of these proposed ex vivo measures following isolation rearing.
Effect of Neonatal PCP and Isolation Rearing on Glutamate Release from Hippocampal Slices
Validation studies confirmed that in the absence of tissue, sensors responded to exogenous glutamate (Fig. 2a) in a linear manner (Fig. 2b), but not to test compounds (data not shown) or potential interfering agents (Fig. 2a). Basal extracellular glutamate in slices from drug-naïve group-housed rats (2.43 ± 0.52 μM) was reduced by TTX (1.40 ± 0.27 μM; − 42%; P < 0.05) and elevated by KCl (Fig. 2d), TBOA (Fig. 2e), veratridine (Fig. 2f), L-cysteine (Fig. 2g), and α-latrotoxin (Fig. 2h). KCl- and TBOA-evoked responses were both sensitive to TTX (data not shown).
Slices from neurodevelopmentally manipulated rats revealed a main effect of housing on basal hippocampal glutamate (F(1,28) = 4.567; P < 0.05) which was 32% lower in Iso than Gr (Fig. 3a) but did not reach post hoc significance for Veh-Iso or PCP-Iso compared to Veh-Gr or PCP-Gr. KCl-evoked (F(2,56) = 21.834; P < 0.001) and TBOA-evoked (F(1,28) = 15.665; P < 0.001) increases were both unaffected by neurodevelopmental history (data not shown). Neither SB-399885, CPPG, nor MMPIP influenced extracellular glutamate levels when administered alone (data not shown), but of particular note, the release evoked by the 5-HT6 antagonist in the presence of KCl (F(1,28) = 71.528; P < 0.001) interacted with neonatal PCP treatment (F(1,28) = 6.213; P < 0.05) and was attenuated in PCP-Iso compared to both Veh-Gr (P < 0.05) and Veh-Iso (P < 0.01; Fig. 3b). No such reduction was seen for release evoked by the group III mGlu antagonist (F(1,28) = 56.539; P < 0.001; Fig. 3c) or mGlu7 allosteric antagonist (F(1,28) = 66.418; P < 0.001; Fig. 3d) with KCl, which remained unaffected by treatment or housing.

Effect of neonatal PCP and isolation rearing on glutamate release from hippocampal slices. Mean ± SEM (left-hand y-axis) a basal extracellular glutamate concentration and evoked AUC responses to b 3 μM SB-399885, c 100 μM CPPG, and d 100 μM MMPIP in the presence of 120 mM KCl for 15 min, together with (b–d right-hand y-axis) representative difference in current output between glutamate and null sensors. Bars represent 15-min perfusion via the aCSF reservoir. Male Lister hooded rats that received saline (1 ml/kg s.c.; Veh) or PCP (10 mg/kg) on PND 7, 9, and 11 were housed in groups (Gr) or isolation (Iso) from weaning on PND 21, with hippocampal slices obtained on PND 73–112 (n = 7–9 per treatment-housing combination). There were main effects of a isolation on basal extracellular glutamate levels (P < 0.05), and glutamate release was evoked by the combination of elevated KCl with b SB-399885 (P < 0.001), c CPPG (P < 0.001), or d MMPIP (P < 0.001). Of note, the release evoked by b SB-399885 plus high KCl interacted with neonatal PCP treatment (P < 0.05) and was reduced in PCP-Iso compared to Veh-Gr or Veh-Iso, whereas release evoked by c CPPG plus high KCl or d MMPIP plus high KCl remained unaffected by neonatal PCP treatment or isolation rearing. ~P < 0.05 versus Gr (two-way ANOVA); *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 versus basal in the same slice (three-way repeated measures ANOVA with Sidak post hoc); †P < 0.05 versus Veh-Gr; ##P < 0.01 versus Veh-Iso (two-way ANOVA with Tukey post hoc)
Effect of Neonatal PCP and Isolation Rearing on Hippocampal Expression of Glutamatergic and GABAergic Markers, plus 5-HT6 and mGlu7 Receptors
Protein expression data are shown in Fig. S2 and Fig. S3. There was a subtype × treatment × housing interaction for VGLUT expression (F(2,56) = 4.306; P < 0.05). Despite no treatment × housing interaction for VGLUT1 (F(1,32) = 3.095; P = 0.0881; Fig. S2a), there were interactions for the ratio of VGLUT1:VGLUT2 (F(1,32) = 5.473; P < 0.05; Fig. S3a) and VGLUT1 as a proportion of total VGLUT (F(1,32) = 4.952; P < 0.05; Fig. S3b), which were lower in PCP-Iso than in PCP-Gr (P < 0.05; − 35% and − 22% respectively). There was no subtype × treatment × housing interaction for EAATs, but a treatment × housing interaction for EAAT2 (F(1,32) = 4.303; P < 0.05) which was lower in PCP-Iso than in Veh-Iso (P < 0.05, − 20%; Fig. S2e). There was also a treatment × housing interaction for 5-HT6 expression (F(1,32) = 5.127; P < 0.05; Fig. S2i) but between-group differences did not reach post hoc significance.
NOD Assessment of Pharmacological Sensitivity, Followed by Immunohistochemistry
Effect of Neonatal PCP and Isolation Rearing on the Cognitive Effects of 5-HT6 and mGlu7 Receptor Antagonists in the NOD Task
There was an acute treatment effect on the duration of familiarization trial object exploration (F(2,76) = 12.684; P < 0.001) which was decreased by SB-399885 and MMPIP, but importantly no spatial preference between identical objects nor any neurodevelopmental condition × acute treatment interaction (Fig. 4a). In contrast, there were choice trial effects of object (F(1,38) = 69.167; P < 0.001) plus an object × neurodevelopmental condition interaction (F(2,38) = 6.382, P < 0.01). Veh-Gr discriminated the novel from familiar object following acute vehicle (P < 0.01), inferring intact memory, and this was not modified by SB-399885 (P < 0.05) or MMPIP (P < 0.01), consistent with a ceiling effect. Veh-Iso and PCP-Iso were both unable to discriminate following acute vehicle (P > 0.05), suggesting an inability to remember experiences from the familiarization trial 2 h previously. Memory was restored by SB-399885 in single-hit Veh-Iso (P < 0.001) but not dual-hit PCP-Iso (P > 0.05), whereas MMPIP remained effective in both models (P < 0.001 and P < 0.01, respectively; Fig. 4b). Discrimination ratios support this pattern, being lower in both Veh-Iso and PCP-Iso than in Veh-Gr following acute vehicle (P < 0.05), and of particular note also lower in PCP-Iso than the two other neurodevelopmental conditions following acute SB-399885 (P < 0.01; Fig. 4c).

Impact of combined neonatal PCP and isolation rearing on the ability of a 5-HT6 (but not mGlu7) antagonist to reverse isolation-induced cognitive deficits in the NOD task. Mean ± SEM time spent exploring a two identical objects during the familiarization trial and b the novel and familiar object during the choice trial 2 h later, as well as c choice trial discrimination ratio (time exploring novel/total choice trial object exploration). Male Lister hooded rats that received saline (1 ml/kg s.c.; Veh) or PCP (10 mg/kg) on PND 7, 9, and 11 were housed in groups (Gr; Veh only) or isolation (Iso; Veh and PCP) from weaning on PND 21, then underwent NOD on three occasions at 1–2-week intervals (PND 57–80) to receive acute vehicle (Veh; 0.5% methylcellulose 1% Tween 80; 1 ml/kg i.p. 30 min before the familiarization trial), SB-399885 (SB; 10 mg/kg), or MMPIP (MP; 10 mg/kg) on separate test days in a pseudorandom order and serve as their own controls (n = 13–14 per neurodevelopmental condition). In the familiarization trial a, there was a main effect of acute treatment on the duration of object exploration (P < 0.001), which was decreased by SB-399885 and MMPIP, but importantly, no spatial preference between identical objects or any neurodevelopmental condition × treatment interaction. In the choice trial b, there was an effect of object (P < 0.001) and an object × neurodevelopmental condition interaction (P < 0.01). Veh-Gr discriminated the novel object following acute vehicle and this intact memory was not further enhanced by acute treatment, consistent with a ceiling effect. Impaired memory was restored by SB-399885 in single-hit Veh-Iso but not dual-hit PCP-Iso, whereas MMPIP remained effective in both models. Accordingly, discrimination ratios (c) were lower in Veh-Iso and PCP-Iso than in Veh-Gr following acute vehicle, and of note also lower in PCP-Iso than the other two neurodevelopmental conditions following acute SB-399885. *P < 0.05; **P < 0.01; ***P < 0.001 versus the familiar object following the same acute treatment in the same rats (three-way repeated measures ANOVA with Sidak post hoc); †P < 0.05 versus acute vehicle in Veh-Gr, ‡‡P < 0.01 versus acute SB-399885 in Veh-Gr; ##P < 0.01 versus acute SB-399885 in Veh-Iso (two-way ANOVA with Tukey post hoc)
Effect of Neonatal PCP and Isolation Rearing on Hippocampal Calbindin and 5-HT Immunoreactivity
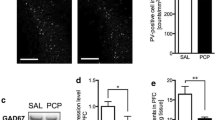
There was typical intense calbindin labeling in the dentate gyrus and stratum pyramidale, which prevented cell counting within these regions. Remaining cells matched the distribution of GABA interneurons [62] (Fig. 5a, b) and their counts were influenced by neurodevelopmental history (F(2,37) = 6.795; P < 0.01) and reduced in PCP-Iso (80 ± 3) compared to Veh-Gr (101 ± 5; P < 0.01) or Veh-Iso (95 ± 3; P < 0.05). This was predominantly due to changes in CA1 (PCP-Iso versus Veh-Gr P < 0.0001 and Veh-Iso P < 0.05), where labeling intensity was also affected by neurodevelopmental history (F(2,37) = 3.294; P < 0.05) and lower in PCP-Iso (91 ± 4) than in Veh-Gr (105 ± 4; P < 0.05). Reduced counts in Veh-Iso were restricted to strata oriens of CA1 but extended in PCP-Iso to strata radiatum, plus strata oriens of remaining subfields (Fig. 5c). Significant positive correlations were observed between calbindin immunoreactivity and NOD discrimination ratios when rats received SB-399885 (Fig. 5d), but there was no link between calbindin immunoreactivity and cognitive performance when the same rats received either vehicle or MMPIP (data not shown).

Impact of combined neonatal PCP and isolation rearing on hippocampal calbindin and 5-HT immunoreactivity in the dorsal hippocampus. Representative a, b calbindin immunoreactivity throughout a all subfields and b part of CA1 (indicated in a by a solid border), together with e higher magnification images of typical 5-HT immunoreactivity in CA1 strata oriens (s.o.) and radiatum (s.r.) locations (indicated in a by dashed borders). Red represents a, b calbindin or e 5-HT, and blue in a and the top portion of each image in e represents DAPI nuclear counterstain; the bottom portion of each image in e is a duplicate presented without the nuclear counterstain. Scale bars are equivalent to 100 μm a, b or 10 μm e. Mean ± SEM c number of calbindin-positive cells in s.o., s.r., and stratum lacunosum-moleculare (s.l-m) of the dorsal hippocampal subiculum/fasciola cinereum (sub/fc), CA1, and CA2/3, and f intensity of 5-HT immunofluorescence in s.o. and s.r. of dorsal hippocampal CA1 and CA3 and molecular (mol) and polymorphic (pol) layers of the dentate gyrus (DG), plus correlation analyses of d calbindin and g 5-HT immunofluorescence intensity in CA1 versus the NOD choice trial discrimination ratio (time exploring novel/total choice trial object exploration) following acute SB-399885. Male Lister hooded rats that received saline (1 ml/kg s.c.; Veh) or PCP (10 mg/kg) on PND 7, 9, and 11 were housed in groups (Gr; Veh only) or isolation (Iso; Veh and PCP) from weaning on PND 21, then underwent NOD on three occasions (PND 57–80) before tissue collection (PND 78–80), to receive vehicle (0.5% methylcellulose 1% Tween 80; 1 ml/kg i.p. 30 min before the familiarization trial), SB-399885 (10 mg/kg), or MMPIP (10 mg/kg) on separate test days in a pseudorandom order and serve as their own controls (n = 13–14 per neurodevelopmental condition). Calbindin immunoreactivity a was strongest in the dentate gyrus and stratum pyramidale, and remaining cells matched the distribution of GABA interneurons. The b intensity of calbindin immunoreactivity in CA1 and c number of calbindin-positive cells throughout dorsal hippocampal subfields and cell layers were both influenced by neurodevelopmental condition (P < 0.05 and P < 0.01, respectively), and although there were some reductions in single-hit Veh-Iso, these were much more extensive in PCP-Iso and correlated with d NOD performance following acute administration of SB-399885. Patterns of e 5-HT immunoreactivity were consistent with labeling of fine varicose axons (marked by arrowheads) but labeling intensity f was not influenced by neurodevelopmental condition and g did not correlate with NOD flowing SB-399885 administration. †P < 0.05 Veh-Iso and †P < 0.05; ††P < 0.01; ††††P < 0.0001 PCP-Iso versus Veh-Gr; #P < 0.05 PCP-Iso versus Veh-Iso (three-way repeated measures ANOVA with Tukey post hoc)
Labeling of varicose 5-HT axons (Fig. 5e, which are known to preferentially contact calbindin-positive interneurons [37, 63]), showed the expected effect of cell layer [64] but was not influenced by neurodevelopmental history (Fig. 5f) and did not correlate with NOD following SB-399885 (Fig. 5g) or any other acute treatment (data not shown).
Discussion
Combining two established rodent neurodevelopmental models for schizophrenia, neonatal PCP and isolation rearing, produces a wider range of behavioral and neurochemical alterations akin to the core pathophysiology of schizophrenia than either alone [7, 10,11,12]. Our locomotor data again support this more pronounced change in PCP-Iso, and the reliable NOD impairment in 100% of 9 PCP-Iso cohorts now examined in our laboratory confirm this deficit in visual recognition memory is more reproducible than the reported 70% following single-hit isolation [39]. PPI deficits seen in two previous PCP-Iso cohorts [10, 14] were not observed here, in only the third cohort to undergo this test, perhaps due to the slightly earlier timing of this assessment to accommodate subsequent microsensor studies while remaining within project license limits on the duration of isolation and weight restrictions for the required euthanasia method. Nevertheless, preliminary suggestions of a PPI deficit in 67% of the three PCP-Iso cohorts examined to date may represent some improvement on the approximately 55% of 18 single-hit isolate cohorts examined within our laboratory, particularly if the PCP-Iso deficit can be maximized by closely controlling timing of the assessment. Although assessment of fear-motivated memory could not be included within the current test battery, the reliable impairment in 100% of five PCP-Iso cohorts now examined in our laboratory ([10, 12], and unpublished observations) suggests this deficit is also more robust than observed in 67% of 12 single-hit isolation cohorts. Accumulating behavioral data from the PCP-Iso dual-hit model therefore support an additive or cumulative stress hypothesis [65] as also observed for maternal separation [66] or neonatal AMPA/kainate receptor agonism [67] plus isolation rearing, and these newer models allow scope to investigate how complex interactions between early-life risk factors contribute to the neurobiology of neurodevelopmental disorders like schizophrenia. Models involving social manipulations like maternal separation and isolation rearing appear to have excellent construct validity for schizophrenia, since parental separation or loss, frequent relocation during adolescence, and social disadvantage or exclusion extending into later life are all risk factors [68]. While we acknowledge that developmental exposure to glutamate receptor ligands is less common [69,70,71] it appears the long-term consequences of such exposure in rodents as part of a dual-hit approach still afford good face validity. The availability of more robust preclinical models with which to test novel therapeutics has crucial relevance for drug discovery, but to our knowledge the pharmacological sensitivity of combined versus separate PCP and isolation models has not been directly compared before. Our principal finding is that dual-hit PCP-Iso are less sensitive to the 5-HT6 antagonist SB-399885, in terms of glutamate release from hippocampal slices, and this translates to reduced cognitive effect of SB-399885 in a NOD task. Reductions in hippocampal calbindin expression could potentially underlie both of these observations.
We recognize that the relevance of glutamate microsensor measurements is open to interpretation, because their invasive nature permits direct comparison with data from patients versus healthy controls. However, it is important to note that direct comparison between human and animal data are similarly lacking even in cases where the same MRS approach to study glutamatergic neurotransmission is used across species, due to requirements for general anesthesia in rodents. For compounds in the earlier phases of development glutamate microsensor measures in brain slices, or ultimately in freely moving animals, therefore provide a valuable direct indication of extracellular glutamate with high temporal resolution [24]. Basal extracellular glutamate levels in slices from control animals were similar to those obtained using alternative sensors [24], and components of the signal met several criteria for neuronal origin. These include sensitivity to KCl, α-latrotoxin, and TTX, with decreases to the latter in line with previous microsensor reports [72]. Neuronal glutamate sources in CA1 include CA3 Schaffer collateral synapses (onto basal and proximal apical pyramidal dendrites in strata oriens and radiatum) and entorhinal cortex temporoammonic projections (onto distal apical dendrites in stratum lacunosum-moleculare [73]). Precise replication of sensor placement between slices and animals was unattainable but typically within the region of apical dendrites and so receptive to both pathways. Reduced basal glutamate in isolates matches MRS findings [74] and mirrors the pattern of NOD impairments, consistent with dependence of this type of memory on glutamatergic neurotransmission within CA1 [33,34,35,36]. Although the consensus is that schizophrenia begins with hypofunction of NMDA receptors on GABAergic interneurons leading to disinhibition of pyramidal cells and excitotoxic damage to CA1 [75], MRS generally shows little hippocampal change in early schizophrenia [76]. Our NOD studies began at the accepted onset of adulthood, which is approximately 3 weeks after typical emergence of isolation-induced dopaminergic changes and hyperlocomotion [77]. Since our microsensor studies continued for almost 3 months after this first emergence it can be argued that current findings more closely mirror chronic disease where MRS can reveal decreased glutamate in patients [19], potentially due to reduced synaptic density [20] and VGLUT1 expression [21, 22]. Indeed, similar changes are reported in isolates within the time frame of our microsensor measurements [78, 79] and although we did not observe outright VGLUT deficits it is possible that subfield- and/or lamina-specific changes in expression were masked by analysis of whole homogenates. The altered ratio of VGLUT1:VGLUT2 may also contribute to the current isolation effect on basal glutamate, since these transporters respectively localize to synapses with low release probability that exhibit long-term potentiation or high release probability that exhibit long-term depression [80].
5-HT6 antagonists elevate hippocampal glutamate efflux in vivo [81] but the absence of similar effects when applied alone to synaptosomes [82] or slices is readily attributed to loss of 5-HT tone, while the lack of mGlu7 allosteric antagonist effect [83] is explained by relatively low affinity of glutamate for this receptor and consequent activation only during high-frequency firing [84]. Elevated extracellular K+ concentrations which occur during such discharge [85] are routinely used to induce neurotransmitter release ex vivo, and because KCl-evoked responses were unaffected by neurodevelopmental history, this stimulus appeared suitable for further examination of receptor-selective compounds. 5-HT6 blockade augments KCl-evoked glutamate release from slices in a TTX-sensitive manner [86] and we are the first to show this effect is attenuated in slices from PCP-Iso. We are also the first to reveal the potential functional correlates of this observation, since an SB-399885 dose capable of reversing isolation-induced NOD deficits was inactive in the combined neurodevelopmental model. While 5-HT6 antagonists’ effects on NOD ultimately depend on NMDA receptor-mediated glutamatergic mechanisms [48, 87, 88] and there is extensive colocalization of hippocampal 5-HT6 and VGLUT1 mRNA, any direct effect of excitatory Gαs protein-coupled 5-HT6 receptors on principal neurons is played down by suggestions of little tonic 5-HT input to this population of 5-HT6 receptors [89]. Instead, GABAergic disinhibition is the main mechanism proposed to underlie 5-HT6 antagonist-induced glutamate release. The 5-HT6-expressing GABA interneurons appear to be largely calbindin-positive—not parvalbumin-positive (60 versus 5% mRNA co-localization, respectively [38]), which is consistent with preferential serotonergic innervation of the calbindin-positive interneurons that in turn arborize with proximal apical dendrites of pyramidal cells and mediate feedforward inhibition [37, 63]. In contrast, the mGlu7 antagonist-stimulated glutamate release that was maintained in PCP-Iso is likely to be largely independent of calbindin-positive interneurons, since Gαi/o protein-coupled inhibitory mGlu7 autoreceptors and heteroreceptors are expressed on VIP-positive interneurons and appear to be preferentially found at synapses onto somatostatin-positive interneurons in stratum oriens that mediate feedback inhibition of distal pyramidal dendrites [90, 91]. It therefore appears plausible that dysfunction of calbindin-positive interneurons could account for attenuated glutamatergic and cognitive responses to SB-399885 and sparing of responses to MMPIP in PCP-Iso. The correlation between calbindin expression and NOD performance following SB-399885 does not confirm causality but at least appears consistent with our working hypothesis. We certainly saw no evidence for reduced 5-HT6 receptor expression in the dual-hit model, nor for reduced 5-HT innervation of the dorsal hippocampus that would influence 5-HT6 receptor tone. Admittedly, SERT expression or indicators of tonic 5-HT release in PCP-Iso remain unexplored, but any serotonergic dysfunction in schizophrenia [92], isolation-reared [93,94,95] or neonatal NMDA antagonist-treated rats [96, 97] appears far less extensive than that produced by intentional median raphe lesions that abolished 5-HT6 antagonists’ effect on NOD during dissociation of relevant neuroanatomical substrates [98].
There is evidence that some patients with schizophrenia do have reduced calbindin expression and a disordered pattern of calbindin interneurons in the hippocampus [99, 100, but see 101], while the VIP-positive interneurons that express mGlu7 remain unaffected in schizophrenia [102]. PCP-Iso therefore appear to have better relevance, in terms of face validity, for calbindin-deficient patient subgroups than single-hit isolation-reared rats or indeed other non-neurodevelopmental models. There has previously been some conflict whether reduced calcium-binding protein immunoreactivity in preclinical models and patients with schizophrenia reflects a selective posttranslational decrease in calcium-binding protein expression [103] or actual loss of that cell population [104]. Our PCP-Iso-induced calbindin decrease without any change in GAD67, VGAT, or 5-HT6 markers known to be present in the same cell types appears to support the former. Reduced calcium-binding protein expression would be expected to reduce interneuron firing [105], potentially preventing further 5-HT6 antagonist-mediated disinhibition. Future high-priority studies beyond scope of this manuscript should employ calbindin-deficient mice or RNA interference-mediated calbindin knockdown to test the hypothesis that a selective reduction in calbindin expression (of the order associated with schizophrenia) is actually sufficient to impact on 5-HT6 antagonist-mediated responses. It may become necessary to explore alternative explanations including alterations in frontal cortical dopamine, which is also important for NOD [106] and modulated by 5-HT6 antagonists [107] but as yet unexplored in PCP-Iso. Such alternatives may underlie the ability of cariprazine to reverse NOD deficits in PCP-Iso, since in vivo microdialysis shows this novel antipsychotic normalizes acute PCP-induced dopamine and glutamate efflux in the medial prefrontal cortex [108] and elevates dopamine efflux in the nucleus accumbens and hippocampus without having any effect on hippocampal glutamate [109]. On the basis of this latter observation, cariprazine would not be expected to influence the signal in our current microsensor studies and we therefore instead prioritized the inclusion of other putative procognitive drugs with established effects on glutamatergic neurotransmission within the hippocampus.
A diverse array of compounds, including 5-HT6 antagonists [42, 110,111,112], an mGlu2/3 agonist [49], NMDA receptor glycine modulatory site partial agonist [113], α4β2 and α7 nicotinic receptor agonists [114], donepezil [115], risperidone [47], and fluoxetine [116], all reverse “single-hit” isolation-induced NOD deficits. Some of these approaches are associated with clinical benefit in other psychiatric or neurodegenerative disorders but ultimately lack marked clinical benefit in schizophrenia [117,118,119]. For example, 5-HT6 antagonists appear to improve cognition in mild to moderate Alzheimer’s disease [120, 121] which interestingly, and consistent with our working hypothesis, does not appear to involve calbindin deficits in CA1 [122,123,124]. Sparse clinical data for cognitive effects of 5-HT6 antagonists in schizophrenia [125] together with anecdotal indications that most pharmaceutical companies have suspended trials in this area seem to imply that previous findings in normal rats and other models for this disorder may actually represent false positives. In support of this, it is worth noting the earlier failure of three independent groups to replicate procognitive effects of 5-HT6 antagonists in non-neurodevelopmental studies [126,127,128]. The current absence of any “gold standard” treatment for the cognitive impairment associated with schizophrenia makes it difficult to ascertain the true predictive validity of PCP-Iso. However, our previous findings with cariprazine, aripiprazole, and lamotrigine [12, 14], together with current observations with a 5-HT6 receptor antagonist, suggest wider adoption of PCP-Iso may aid more reliable preclinical evaluation of novel compounds to manage the symptoms of schizophrenia or even modulate disease onset. Any progress towards improved early distinction of promising from less suitable pharmacological approaches using the PCP-Iso model has clear relevance from streamlining drug discovery.
Paradoxically, 5-HT6 receptor agonists have similar cognitive effects to antagonists of the same receptor, so prolong NOD memory in normal animals [129] and reverse a variety of acute NMDA receptor antagonist-induced NOD deficits [130, 131, 132]. The underlying neurochemical mechanisms remain unclear, since 5-HT6 agonists actually facilitate GABAergic and inhibit glutamatergic neurotransmission in the hippocampus and cortex [53, 55, 82, 133]. Future neurochemical and cognitive studies in PCP-Iso should therefore examine the extent to which 5-HT6 receptor agonist-mediated responses might be perturbed by glutamatergic deficits and interneuron dysfunction in schizophrenia. The relationship between mGlu7 and memory is also complex, with the mGlu7-negative modulator MMPIP both impairing NOD in normal mice [58] and recently proposed as a putative antipsychotic due to its ability to reverse acute MK-801-induced hyperactivity and cognitive deficits in mouse NOD and rat spatial delayed alternation tests [56]. It has been suggested mGlu7 may represent a new treatment target for neurodevelopmental disorders [134], and current neurochemical and behavioral findings certainly appear to justify continued preclinical evaluation of mGlu7 allosteric antagonists against a broader variety of PCP-Iso-induced cognitive impairments. This is important since the NOD test of visual recognition memory performed here has translational relevance to only one of several cognitive domains impaired in schizophrenia [45]. Future studies should also assess the integrity of VIP-expressing interneurons in PCP-Iso to test our current working hypothesis that cells mediating the effects of mGlu7 receptor ligands would be spared in this animal model and therefore mirror findings in patients with schizophrenia [102]. In conclusion, this research highlights the importance of improved understanding for selection of appropriate preclinical models, and for better stratification of patient subpopulations to different drug treatments, especially in cases where disease neurobiology impacts upon the cells mediating pharmacological effects of potential therapeutics.
References
Weinberger DR (2017) Future of days past: neurodevelopment and schizophrenia. Schizophr Bull 43:1164–1168. https://doi.org/10.1093/schbul/sbx118
Strassnig M, Kotov R, Fochtmann L, Kalin M, Bromet EJ, Harvey PD (2018) Associations of independent living and labor force participation with impairment indicators in schizophrenia and bipolar disorder at 20-year follow-up. Schizophr Res 197:150–155. https://doi.org/10.1016/j.schres.2018.02.009
MacEwan JP, Seabury S, Aigbogun MS, Kamat S, van Eijndhoven E, Francois C, Henderson C, Citrome L (2016) Pharmaceutical innovation in the treatment of schizophrenia and mental disorders compared with other diseases. Innov Clin Neurosci 13:17–25
Hutson PH, Clark JA, Cross AJ (2017) CNS target identification and validation: avoiding the valley of death or naive optimism? Annu Rev Pharmacol Toxicol 57:171–187. https://doi.org/10.1146/annurev-pharmtox-010716-104624
Möller M, Swanepoel T, Harvey BH (2015) Neurodevelopmental animal models reveal the convergent role of neurotransmitter systems, inflammation, and oxidative stress as biomarkers of schizophrenia: implications for novel drug development. ACS Chem Neurosci 6:987–1016. https://doi.org/10.1021/cn5003368
Ikonomidou C, Bosch F, Miksa M, Bittigau P, Vöckler J, Dikranian K, Tenkova TI, Stefovska V et al (1999) Blockade of NMDA receptors and apoptotic neurodegeneration in the developing brain. Science 283:70–74. https://doi.org/10.1126/science.283.5398.70
Gaskin PL, Alexander SP, Fone KC (2011) Combining rearing in social isolation with perinatal PCP treatment as a preclinical model of schizophrenia. J Psychopharmacol 25S:A70
Lim AL, Taylor DA, Malone DT (2012) A two-hit model: behavioural investigation of the effect of combined neonatal MK-801 administration and isolation rearing in the rat. J Psychopharmacol 26:1252–1264. https://doi.org/10.1177/0269881111430751
Gilabert-Juan J, Belles M, Saez AR, Carceller H, Zamarbide-Fores S, Moltó MD, Nacher J (2013) A “double hit” murine model for schizophrenia shows alterations in the structure and neurochemistry of the medial prefrontal cortex and the hippocampus. Neurobiol Dis 59:126–140. https://doi.org/10.1016/j.nbd.2013.07.008
Gaskin PL, Alexander SP, Fone KC (2014) Neonatal phencyclidine administration and post-weaning social isolation as a dual-hit model of ‘schizophrenia-like’ behaviour in the rat. Psychopharmacology 231:2533–2545. https://doi.org/10.1007/s00213-013-3424-y
Kohli S, Alberati D, Ballard TM, Steward LJ, King MV, Fone KCF (2016) The GlyT1 inhibitor RO4993850 alters social behavior and ultrasonic vocalization calls in a neonatal-phencyclidine isolation-reared rat model for schizophrenia. Eur Neuropsychopharmacol 26:S494–S495
Watson DJG, King MV, Gyertyán I, Kiss B, Adham N, Fone KCF (2016) The dopamine D3-preferring D2/D3 dopamine receptor partial agonist, cariprazine, reverses behavioural changes in a rat neurodevelopmental model for schizophrenia. Eur Neuropsychopharmacol 26:208–224. https://doi.org/10.1016/j.euroneuro.2015.12.020
Jones CA, Watson DJ, Fone KC (2011) Animal models of schizophrenia. Br J Pharmacol 164:1162–1194. https://doi.org/10.1111/j.1476-5381.2011.01386.x
Gaskin PL, Toledo-Rodriguez M, Alexander SP, Fone KC (2016) Down-regulation of hippocampal genes regulating dopaminergic, GABAergic, and Glutamatergic function following combined neonatal phencyclidine and post-weaning social isolation of rats as a neurodevelopmental model for schizophrenia. Int J Neuropsychopharmacol 19:1–13. https://doi.org/10.1093/ijnp/pyw062
McCormack PL (2015) Cariprazine: first global approval. Drugs 75:2035–2043. https://doi.org/10.1007/s40265-015-0494-7
Riedel M, Spellmann I, Schennach-Wolff R, Musil R, Dehning S, Cerovecki A, Opgen-Rhein M, Matz J et al (2010) Effect of aripiprazole on cognition in the treatment of patients with schizophrenia. Pharmacopsychiatry 43:50–57. https://doi.org/10.1055/s-0029-1239539
Tiihonen J, Wahlbeck K, Kiviniemi V (2009) The efficacy of lamotrigine in clozapine-resistant schizophrenia: a systematic review and meta-analysis. Schizophr Res 109:10–14. https://doi.org/10.1016/j.schres.2009.01.002
Hu W, MacDonald ML, Elswick DE, Sweet RA (2015) The glutamate hypothesis of schizophrenia: evidence from human brain tissue studies. Ann N Y Acad Sci 1338:38–57. https://doi.org/10.1111/nyas.12547
Stan AD, Ghose S, Zhao C, Hulsey K, Mihalakos P, Yanagi M, Morris SU, Bartko JJ et al (2015) Magnetic resonance spectroscopy and tissue protein concentrations together suggest lower glutamate signaling in dentate gyrus in schizophrenia. Mol Psychiatry 20:433–439. https://doi.org/10.1038/mp.2014.54
Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA (2005) Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse 57:47–55. https://doi.org/10.1002/syn.20153
Eastwood SL, Harrison PJ (2005) Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr Res 73:159–172. https://doi.org/10.1016/j.schres.2004.05.010
Sawada K, Barr AM, Nakamura M, Arima K, Young CE, Dwork AJ, Falkai P, Phillips AG et al (2005) Hippocampal complexin proteins and cognitive dysfunction in schizophrenia. Arch Gen Psychiatry 62:263–272. https://doi.org/10.1001/archpsyc.62.3.263
Girgis RR, Zoghbi AW, Javitt DC, Lieberman JA (2019) The past and future of novel, non-dopamine-2 receptor therapeutics for schizophrenia: a critical and comprehensive review. J Psychiatr Res 108:57–83. https://doi.org/10.1016/j.jpsychires.2018.07.006
Oldenziel WH, van der Zeyden M, Dijkstra G, Ghijsen WE, Karst H, Cremers TI et al (2007) Monitoring extracellular glutamate in hippocampal slices with a microsensor. J Neurosci Methods 160:37–44. https://doi.org/10.1016/j.jneumeth.2006.08.003
Butler CR, Boychuk JA, Pomerleau F, Alcala R, Huettl P, Ai Y, Jakobsson J, Whiteheart SW et al (2020) Modulation of epileptogenesis: a paradigm for the integration of enzyme-based microelectrode arrays and optogenetics. Epilepsy Res 159:106244. https://doi.org/10.1016/j.eplepsyres.2019.106244
Mazzone GL, Nistri A (2019) Modulation of extrasynaptic GABAergic receptor activity influences glutamate release and neuronal survival following excitotoxic damage to mouse spinal cord neurons. Neurochem Int 128:175–185. https://doi.org/10.1016/j.neuint.2019.04.018
Wippel C, Maurer J, Förtsch C, Hupp S, Bohl A, Ma J, Mitchell TJ, Bunkowski S et al (2013) Bacterial cytolysin during meningitis disrupts the regulation of glutamate in the brain, leading to synaptic damage. PLoS Pathog 9:e1003380. https://doi.org/10.1371/journal.ppat.1003380
Tamminga CA, Stan AD, Wagner AD (2010) The hippocampal formation in schizophrenia. Am J Psychiatry 167:1178–1193. https://doi.org/10.1176/appi.ajp.2010.09081187
Tamminga CA, Southcott S, Sacco C, Wagner AD, Ghose S (2012) Glutamate dysfunction in hippocampus: relevance of dentate gyrus and CA3 signaling. Schizophr Bull 38:927–935. https://doi.org/10.1093/schbul/sbs062
Kalmady SV, Shivakumar V, Arasappa R, Subramaniam A, Gautham S, Venkatasubramanian G, Gangadhar BN (2017) Clinical correlates of hippocampus volume and shape in antipsychotic-naïve schizophrenia. Psychiatry Res 263:93–102. https://doi.org/10.1016/j.pscychresns.2017.03.014
Sauras R, Keymer A, Alonso-Solis A, Díaz A, Molins C, Nuñez F, Rabella M, Roldán A et al (2017) Volumetric and morphological characteristics of the hippocampus are associated with progression to schizophrenia in patients with first-episode psychosis. Eur Psychiatry 45:1–5. https://doi.org/10.1016/j.eurpsy.2017.06.006
Matosin N, Fernandez-Enright F, Lum JS, Engel M, Andrews JL, Gassen NC, Wagner KV, Schmidt MV et al (2016) Molecular evidence of synaptic pathology in the CA1 region in schizophrenia. NPJ Schizophr 2:16022. https://doi.org/10.1038/npjschz.2016.22
Rampon C, Tang YP, Goodhouse J, Shimizu E, Kyin M, Tsien JZ (2000) Enrichment induces structural changes and recovery from nonspatial memory deficits in CA1 NMDAR1-knockout mice. Nat Neurosci 3:238–244. https://doi.org/10.1038/72945
Baker KB, Kim JJ (2002) Effects of stress and hippocampal NMDA receptor antagonism on recognition memory in rats. Learn Mem 9:58–65. https://doi.org/10.1101/lm.46102
Cohen SJ, Munchow AH, Rios LM, Zhang G, Asgeirsdóttir HN, Stackman RW Jr (2013) The rodent hippocampus is essential for nonspatial object memory. Curr Biol 23:1685–1690. https://doi.org/10.1016/j.cub.2013.07.002
King MV, Kurian N, Qin S, Papadopoulou N, Westerink BH, Cremers TI et al (2014) Lentiviral delivery of a vesicular glutamate transporter 1 (VGLUT1)-targeting short hairpin RNA vector into the mouse hippocampus impairs cognition. Neuropsychopharmacology 39:464–476. https://doi.org/10.1038/npp.2013.220
Freund TF, Gulyás AI, Acsády L, Görcs T, Tóth K (1990) Serotonergic control of the hippocampus via local inhibitory interneurons. Proc Natl Acad Sci U S A 87:8501–8505. https://doi.org/10.1073/pnas.87.21.8501
Helboe L, Egebjerg J, de Jong IE (2015) Distribution of serotonin receptor 5-HT6 mRNA in rat neuronal subpopulations: a double in situ hybridization study. Neuroscience 310:442–454. https://doi.org/10.1016/j.neuroscience.2015.09.064
Fone KC, Porkess MV (2008) Behavioural and neurochemical effects of post-weaning social isolation in rodents-relevance to developmental neuropsychiatric disorders. Neurosci Biobehav Rev 32:1087–1102. https://doi.org/10.1016/j.neubiorev.2008.03.003
Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG, NC3Rs Reporting Guidelines Working Group (2010) Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol 160:1577–1579. https://doi.org/10.1111/j.1476-5381.2010.00872.x
Percie du Sert N, Hurst V, Ahluwalia A, Alam S, Avey MT, Baker M et al (2019) The ARRIVE guidelines 2019: updated guidelines for reporting animal research. https://doi.org/10.1101/703181v1.
Shortall SE, Negm OH, Fowler M, Fairclough LC, Tighe PJ, Wigmore PM, King MV (2018) Characterization of behavioral, signaling and cytokine alterations in a rat neurodevelopmental model for schizophrenia, and their reversal by the 5-HT6 receptor antagonist SB-399885. Mol Neurobiol 55:7413–7430. https://doi.org/10.1007/s12035-018-0940-0
Geyer MA (2006) Are cross-species measures of sensorimotor gating useful for the discovery of procognitive cotreatments for schizophrenia? Dialogues Clin Neurosci 8:9–16
Fabricius K, Steiniger-Brach B, Helboe L, Fink-Jensen A, Wörtwein G (2011) Socially isolated rats exhibit changes in dopamine homeostasis pertinent to schizophrenia. Int J Dev Neurosci 29:347–350. https://doi.org/10.1016/j.ijdevneu.2010.09.003
Rajagopal L, Massey BW, Huang M, Oyamada Y, Meltzer HY (2014) The novel object recognition test in rodents in relation to cognitive impairment in schizophrenia. Curr Pharm Des 20:5104–5114. https://doi.org/10.2174/1381612819666131216114240
National Advisory Mental Health Council Workgroup on Changes to the Research Domain Criteria Matrix (2018). RDoC changes to the matrix (CMAT) workgroup update: addition of the sensorimotor domain. Bethesda, MD. https://www.nimh.nih.gov/about/advisory-boards-and-groups/namhc/reports/rdoc-changes-to-the-matrix-cmat-workgroup-update-proposed-positive-valence-domain-revisions.shtml. Accessed 19 February 2020
McIntosh AL, Ballard TM, Steward LJ, Moran PM, Fone KC (2013) The atypical antipsychotic risperidone reverses the recognition memory deficits induced by post-weaning social isolation in rats. Psychopharmacology 228:31–42. https://doi.org/10.1007/s00213-013-3011-2
King MV, Sleight AJ, Woolley ML, Topham IA, Marsden CA, Fone KC (2004) 5-HT6 receptor antagonists reverse delay-dependent deficits in novel object discrimination by enhancing consolidation—an effect sensitive to NMDA receptor antagonism. Neuropharmacology 47:195–204. https://doi.org/10.1016/j.neuropharm.2004.03.012
Jones CA, Brown AM, Auer DP, Fone KC (2011) The mGluR2/3 agonist LY379268 reverses post-weaning social isolation-induced recognition memory deficits in the rat. Psychopharmacology 214:269–283. https://doi.org/10.1007/s00213-010-1931-7
Tian F, Gourine AV, Huckstepp RT, Dale N (2009) A microelectrode biosensor for real time monitoring of L-glutamate release. Anal Chim Acta 645:86–91. https://doi.org/10.1016/j.aca.2009.04.048
Hirst WD, Stean TO, Rogers DC, Sunter D, Pugh P, Moss SF et al (2006) SB-399885 is a potent, selective 5-HT6 receptor antagonist with cognitive enhancing properties in aged rat water maze and novel object recognition models. Eur J Pharmacol 553:109–119. https://doi.org/10.1016/j.ejphar.2006.09.049
Toms NJ, Jane DE, Kemp MC, Bedingfield JS, Roberts PJ (1996) The effects of (RS)-alpha-cyclopropyl-4-phosphonophenylglycine ((RS)-CPPG), a potent and selective metabotropic glutamate receptor antagonist. Br J Pharmacol 119:851–854. https://doi.org/10.1111/j.1476-5381.1996.tb15750.x
West PJ, Marcy VR, Marino MJ, Schaffhauser H (2009) Activation of the 5-HT6 receptor attenuates long-term potentiation and facilitates GABAergic neurotransmission in rat hippocampus. Neuroscience 164:692–701. https://doi.org/10.1016/j.neuroscience.2009.07.061
Morishita W, Alger BE (2000) Differential effects of the group II mGluR agonist, DCG-IV, on depolarization-induced suppression of inhibition in hippocampal CA1 and CA3 neurons. Hippocampus 10:261–268. https://doi.org/10.1002/1098-1063(2000)10:3<261::AID-HIPO6>3.0.CO;2-1
Schechter LE, Lin Q, Smith DL, Zhang G, Shan Q, Platt B, Brandt MR, Dawson LA et al (2008) Neuropharmacological profile of novel and selective 5-HT6 receptor agonists: WAY-181187 and WAY-208466. Neuropsychopharmacology 33:1323–1335. https://doi.org/10.1038/sj.npp.1301503
Cieślik P, Woźniak M, Kaczorowska K, Brański P, Burnat G, Chocyk A, Bobula B, Gruca P et al (2018) Negative allosteric modulators of mGlu7 receptor as putative antipsychotic drugs. Front Mol Neurosci 11:316. https://doi.org/10.3389/fnmol.2018.00316
Wesołowska A, Nikiforuk A (2007) Effects of the brain-penetrant and selective 5-HT6 receptor antagonist SB-399885 in animal models of anxiety and depression. Neuropharmacology 52:1274–1283. https://doi.org/10.1016/j.neuropharm.2007.01.007
Hikichi H, Murai T, Okuda S, Maehara S, Satow A, Ise S, Nishino M, Suzuki G et al (2010) Effects of a novel metabotropic glutamate receptor 7 negative allosteric modulator, 6-(4-methoxyphenyl)-5-methyl-3-pyridin-4-ylisoxazonolo[4,5-c]pyridin-4(5H)-one (MMPIP), on the central nervous system in rodents. Eur J Pharmacol 639:106–114. https://doi.org/10.1016/j.ejphar.2009.08.047
Klakotskaia D, Ramsey AK, Fowler SW, Serfozo P, Simonyi A, Schachtman TR (2013) Effects of group II and III metabotropic glutamate receptor ligands on conditioned taste aversion learning. Behav Brain Res 253:9–16. https://doi.org/10.1016/j.bbr.2013.06.032
Kjonigsen LJ, Leergaard TB, Witter MP, Bjaalie JG (2011) Digital atlas of anatomical subdivisions and boundaries of the rat hippocampal region. Front Neuroinform 5:2. https://doi.org/10.3389/fninf.2011.00002
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. https://doi.org/10.1038/nmeth.2019
Freund TF, Buzsáki G (1996) Interneurons of the hippocampus. Hippocampus 6:347–470. https://doi.org/10.1002/(SICI)1098-1063(1996)6:4<347::AID-HIPO1>3.0.CO;2-I
Aznar S, Qian ZX, Knudsen GM (2004) Non-serotonergic dorsal and median raphe projection onto parvalbumin- and calbindin-containing neurons in hippocampus and septum. Neuroscience 124:573–581. https://doi.org/10.1016/j.neuroscience.2003.12.020
Oleskevich S, Descarries L (1990) Quantified distribution of the serotonin innervation in adult rat hippocampus. Neuroscience 34:19–33. https://doi.org/10.1016/0306-4522(90)90301-j
McEwen BS (1998) Stress, adaptation, and disease. Allostasis and allostatic load. Ann N Y Acad Sci 840:33–44. https://doi.org/10.1111/j.1749-6632.1998.tb09546.x
Vargas J, Junco M, Gomez C, Lajud N (2016) Early life stress increases metabolic risk, HPA axis reactivity, and depressive-like behavior when combined with postweaning social isolation in rats. PLoS One 11:e0162665. https://doi.org/10.1371/journal.pone.0162665
Marriott AL, Tasker RA, Ryan CL, Doucette TA (2016) Alterations to prepulse inhibition magnitude and latency in adult rats following neonatal treatment with domoic acid and social isolation rearing. Behav Brain Res 298:310–317. https://doi.org/10.1016/j.bbr.2015.11.009
Stilo SA, Murray RM (2010) The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin Neurosci 12:305–315
Rahbar F, Fomufod A, White D, Westney LS (1993) Impact of intrauterine exposure to phencyclidine (PCP) and cocaine on neonates. J Natl Med Assoc 85:349–352
Mvula MM, Miller JM Jr, Ragan FA (1999) Relationship of phencyclidine and pregnancy outcome. J Reprod Med 44:1021–1024
Scroggin TL, McMillin GA (2018) Quantitation of cocaine and metabolites, phencyclidine, butalbital and phenobarbital in meconium by liquid chromatography-tandem mass spectrometry. J Anal Toxicol 42:177–182. https://doi.org/10.1093/jat/bkx097
Moussawi K, Riegel A, Nair S, Kalivas PW (2011) Extracellular glutamate: functional compartments operate in different concentration ranges. Front Syst Neurosci 5:94. https://doi.org/10.3389/fnsys.2011.00094
Spruston N (2008) Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci 9:206–221. https://doi.org/10.1038/nrn2286
Shao Y, Yan G, Xuan Y, Peng H, Huang QJ, Wu R, Xu H (2015) Chronic social isolation decreases glutamate and glutamine levels and induces oxidative stress in the rat hippocampus. Behav Brain Res 282:201–208. https://doi.org/10.1016/j.bbr.2015.01.005
Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, Inbar BP, Corcoran CM et al (2013) Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron 78:81–93. https://doi.org/10.1016/j.neuron.2013.02.011
Iwata Y, Nakajima S, Plitman E, Mihashi Y, Caravaggio F, Chung JK, Kim J, Gerretsen P et al (2018) Neurometabolite levels in antipsychotic-naïve/free patients with schizophrenia: a systematic review and meta-analysis of 1H-MRS studies. Prog Neuro-Psychopharmacol Biol Psychiatry 86:340–352. https://doi.org/10.1016/j.pnpbp.2018.03.016
Goh J-Y, O’Sullivan SE, Shortall SE, Zordan N, Piccinini AM, Potter HG, Fone KCF, King MV (2020) Gestational poly(I:C) attenuates, not exacerbates, the behavioral, cytokine and mTOR changes caused by isolation rearing in a rat ‘dual-hit’ model for neurodevelopmental disorders. Brain Behav Immun. https://doi.org/10.1016/j.bbi.2020.05.076
Silva-Gómez AB, Rojas D, Juárez I, Flores G (2003) Decreased dendritic spine density on prefrontal cortical and hippocampal pyramidal neurons in postweaning social isolation rats. Brain Res 983:128–136. https://doi.org/10.1016/s0006-8993(03)03042-7
Harte MK, Piyabhan P, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP (2007) Reduced vesicular glutamate transporter-1 in the prefrontal cortex and hippocampus of isolation reared rats. J Psychopharmacol 21S:A56
Varoqui H, Schäfer MK, Zhu H, Weihe E, Erickson JD (2002) Identification of the differentiation-associated Na+/PI transporter as a novel vesicular glutamate transporter expressed in a distinct set of glutamatergic synapses. J Neurosci 22:142–155. https://doi.org/10.1523/JNEUROSCI.22-01-00142.2002
Dawson LA, Nguyen HQ, Li P (2001) The 5-HT6 receptor antagonist SB-271046 selectively enhances excitatory neurotransmission in the rat frontal cortex and hippocampus. Neuropsychopharmacology 25:662–668. https://doi.org/10.1016/S0893-133X(01)00265-2
Wang HY, Lu CW, Lin TY, Kuo JR, Wang SJ (2016) WAY208466 inhibits glutamate release at hippocampal nerve terminals. Eur J Pharmacol 781:117–127. https://doi.org/10.1016/j.ejphar.2016.04.010
Summa M, Di Prisco S, Grilli M, Usai C, Marchi M, Pittaluga A (2013) Presynaptic mGlu7 receptors control GABA release in mouse hippocampus. Neuropharmacology 66:215–224. https://doi.org/10.1016/j.neuropharm.2012.04.020
Schoepp DD, Jane DE, Monn JA (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38:1431–1476. https://doi.org/10.1016/s0028-3908(99)00092-1
Heinrich A, Andó RD, Túri G, Rózsa B, Sperlágh B (2012) K+ depolarization evokes ATP, adenosine and glutamate release from glia in rat hippocampus: a microelectrode biosensor study. Br J Pharmacol 167:1003–1020. https://doi.org/10.1111/j.1476-5381.2012.01932.x
Marcos B, Gil-Bea FJ, Hirst WD, García-Alloza M, Ramírez MJ (2006) Lack of localization of 5-HT6 receptors on cholinergic neurons: implication of multiple neurotransmitter systems in 5-HT6 receptor-mediated acetylcholine release. Eur J Neurosci 24:1299–1306. https://doi.org/10.1111/j.1460-9568.2006.05003.x
Pitsikas N, Zisopoulou S, Pappas I, Sakellaridis N (2008) The selective 5-HT6 receptor antagonist Ro 04-6790 attenuates psychotomimetic effects of the NMDA receptor antagonist MK-801. Behav Brain Res 188:304–309. https://doi.org/10.1016/j.bbr.2007.11.010
Asselot R, Simon-O'Brien E, Lebourgeois S, Nee G, Delaunay V, Duchatelle P et al (2017) Time-dependent impact of glutamatergic modulators on the promnesiant effect of 5-HT6R blockade on mice recognition memory. Pharmacol Res 118:111–118. https://doi.org/10.1016/j.phrs.2016.06.009
Fone KC (2008) An update on the role of the 5-hydroxytryptamine6 receptor in cognitive function. Neuropharmacology 55:1015–1022. https://doi.org/10.1016/j.neuropharm.2008.06.061
Shigemoto R, Kinoshita A, Wada E, Nomura S, Ohishi H, Takada M, Flor PJ, Neki A et al (1997) Differential presynaptic localization of metabotropic glutamate receptor subtypes in the rat hippocampus. J Neurosci 17:7503–7522. https://doi.org/10.1523/JNEUROSCI.17-19-07503.1997
Somogyi P, Dalezios Y, Luján R, Roberts JD, Watanabe M, Shigemoto R (2003) High level of mGluR7 in the presynaptic active zones of select populations of GABAergic terminals innervating interneurons in the rat hippocampus. Eur J Neurosci 17:2503–2520. https://doi.org/10.1046/j.1460-9568.2003.02697.x
Abi-Dargham A, Laruelle M, Aghajanian GK, Charney D, Krystal J (1997) The role of serotonin in the pathophysiology and treatment of schizophrenia. J Neuropsychiatr Clin Neurosci 9:1–17. https://doi.org/10.1176/jnp.9.1.1
Whitaker-Azmitia P, Zhou F, Hobin J, Borella A (2000) Isolation-rearing of rats produces deficits as adults in the serotonergic innervation of hippocampus. Peptides 21:1755–1759. https://doi.org/10.1016/s0196-9781(00)00327-2
Muchimapura S, Fulford AJ, Mason R, Marsden CA (2002) Isolation rearing in the rat disrupts the hippocampal response to stress. Neuroscience 112:697–705. https://doi.org/10.1016/s0306-4522(02)00107-0
Brenes JC, Fornaguera J (2009) The effect of chronic fluoxetine on social isolation-induced changes on sucrose consumption, immobility behavior, and on serotonin and dopamine function in hippocampus and ventral striatum. Behav Brain Res 198:199–205. https://doi.org/10.1016/j.bbr.2008.10.036
Gorter JA, Botterblom MH, Feenstra MG, Boer GJ (1992) Chronic neonatal NMDA receptor blockade with MK-801 alters monoamine metabolism in the adult rat. Neurosci Lett 137:97–100. https://doi.org/10.1016/0304-3940(92)90307-s
Kurumaji A, Aihara O, Yamada S, Toru M (2000) Increased DOI-induced head shakings in adult rats neonatally treated with MK-801. Brain Res Dev Brain Res 124:125–127. https://doi.org/10.1016/s0165-3806(00)00107-3
King MV, Spicer CH, Sleight AJ, Marsden CA, Fone KC (2009) Impact of regional 5-HT depletion on the cognitive enhancing effects of a typical 5-ht6 receptor antagonist, Ro 04-6790, in the novel object discrimination task. Psychopharmacology 202:111–123. https://doi.org/10.1007/s00213-008-1334-1
Iritani S, Kuroki N, Ikeda K, Kazamatsuri H (1999) Calbindin immunoreactivity in the hippocampal formation and neocortex of schizophrenics. Prog Neuro-Psychopharmacol Biol Psychiatry 23:409–421. https://doi.org/10.1016/s0278-5846(99)00005-6
Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y, Young TA, Bullard J et al (2005) Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry 58:85–96. https://doi.org/10.1016/j.biopsych.2005.03.031
Zhang ZJ, Reynolds GP (2002) A selective decrease in the relative density of parvalbumin-immunoreactive neurons in the hippocampus in schizophrenia. Schizophr Res 55:1–10. https://doi.org/10.1016/s0920-9964(01)00188-8
Roberts GW, Ferrier IN, Lee Y, Crow TJ, Johnstone EC, Owens DG et al (1983) Peptides, the limbic lobe and schizophrenia. Brain Res 288:199–211. https://doi.org/10.1016/0006-8993(83)90095-1
Kaalund SS, Riise J, Broberg BV, Fabricius K, Karlsen AS, Secher T, Plath N, Pakkenberg B (2013) Differential expression of parvalbumin in neonatal phencyclidine-treated rats and socially isolated rats. J Neurochem 124:548–557. https://doi.org/10.1111/jnc.12061
Benes FM, Kwok EW, Vincent SL, Todtenkopf MS (1998) A reduction of nonpyramidal cells in sector CA2 of schizophrenics and manic depressives. Biol Psychiatry 44:88–97. https://doi.org/10.1016/s0006-3223(98)00138-3
Eyles DW, McGrath JJ, Reynolds GP (2002) Neuronal calcium-binding proteins and schizophrenia. Schizophr Res 57:27–34. https://doi.org/10.1016/s0920-9964(01)00299-7
Watson DJ, Marsden CA, Millan MJ, Fone KC (2012) Blockade of dopamine D3 but not D2 receptors reverses the novel object discrimination impairment produced by post-weaning social isolation: implications for schizophrenia and its treatment. Int J Neuropsychopharmacol 15:471–484. https://doi.org/10.1017/S1461145711000435
Lacroix LP, Dawson LA, Hagan JJ, Heidbreder CA (2004) 5-HT6 receptor antagonist SB-271046 enhances extracellular levels of monoamines in the rat medial prefrontal cortex. Synapse 51:158–164. https://doi.org/10.1002/syn.10288
Kehr J, Yoshitake T, Ichinose F, Yoshitake S, Kiss B, Gyertyán I, Adham N (2018) Effects of cariprazine on extracellular levels of glutamate, GABA, dopamine, noradrenaline and serotonin in the medial prefrontal cortex in the rat phencyclidine model of schizophrenia studied by microdialysis and simultaneous recordings of locomotor activity. Psychopharmacology 235:1593–1607. https://doi.org/10.1007/s00213-018-4874-z
Huang M, He W, Kiss B, Farkas B, Adham N, Meltzer HY (2019) The role of dopamine D3 receptor partial agonism in cariprazine-induced neurotransmitter efflux in rat hippocampus and nucleus accumbens. J Pharmacol Exp Ther 371:517–525. https://doi.org/10.1124/jpet.119.259879
Marsden CA, King MV, Fone KC (2011) Influence of social isolation in the rat on serotonergic function and memory—relevance to models of schizophrenia and the role of 5-HT6 receptors. Neuropharmacology 61:400–407. https://doi.org/10.1016/j.neuropharm.2011.03.003
Meffre J, Chaumont-Dubel S, Mannoury la Cour C, Loiseau F, Watson DJ, Dekeyne A et al (2012) 5-HT6 receptor recruitment of mTOR as a mechanism for perturbed cognition in schizophrenia. EMBO Mol Med 4:1043–1056. https://doi.org/10.1002/emmm.201201410
de Bruin NMWJ, van Loevezijn A, Wicke KM, de Haan M, Venhorst J, Lange JHM, de Groote L, van der Neut MAW et al (2016) The selective 5-HT6 receptor antagonist SLV has putative cognitive- and social interaction enhancing properties in rodent models of cognitive impairment. Neurobiol Learn Mem 133:100–117. https://doi.org/10.1016/j.nlm.2016.06.020
Fone KCF, Watson DJG, Billiras RI, Sicard DI, Dekeyne A, Rivet JM, Gobert A, Millan MJ (2020) Comparative pro-cognitive and neurochemical profiles of glycine modulatory site agonists and glycine reuptake inhibitors in the rat: potential relevance to cognitive dysfunction and its management. Mol Neurobiol 57:2144–2166. https://doi.org/10.1007/s12035-020-01875-9
Khan A, Kendall DA, Fone KCF (2011) Varenicline, an α4β2 nicotinic acetylcholine receptor partial agonist, reverses cognitive deficits associated with the social isolation rearing model of schizophrenia. Program no 900.22 Society for Neuroscience meeting planner. https://www.abstractsonline.com/plan/start.aspx?mkey={8334BE29-8911-4991-8C31-32B32DD5E6C8}. Accessed 19 February 2020
Khan A, Kendall DA, Fone KCF (2011) The effects of the acetylcholinesterase inhibitor, donepezil, on isolation rearing-induced behavioural deficits in rats. http://bna.kinetixevents.co.uk/bna/bna21/abstract/A591_2527.PDF. Accessed 18 December 2017
Bianchi M, Fone KC, Shah AJ, Atkins AR, Dawson LA, Heidbreder CA et al (2009) Chronic fluoxetine differentially modulates the hippocampal microtubular and serotonergic system in grouped and isolation reared rats. Eur Neuropsychopharmacol 19:778–790. https://doi.org/10.1016/j.euroneuro.2009.06.005
Wierońska JM, Zorn SH, Doller D, Pilc A (2016) Metabotropic glutamate receptors as targets for new antipsychotic drugs: historical perspective and critical comparative assessment. Pharmacol Ther 157:10–27. https://doi.org/10.1016/j.pharmthera.2015.10.007
Smith RC, Amiaz R, Si TM, Maayan L, Jin H, Boules S, Sershen H, Li C et al (2016) Varenicline effects on smoking, cognition, and psychiatric symptoms in schizophrenia: a double-blind randomized trial. PLoS One 11:e0143490. https://doi.org/10.1371/journal.pone.0143490
Santos B, González-Fraile E, Zabala A, Guillén V, Rueda JR, Ballesteros J (2018) Cognitive improvement of acetylcholinesterase inhibitors in schizophrenia. J Psychopharmacol 32:1155–1166. https://doi.org/10.1177/0269881118805496
Maher-Edwards G, Zvartau-Hind M, Hunter AJ, Gold M, Hopton G, Jacobs G, Davy M, Williams P (2010) Double-blind, controlled phase II study of a 5-HT6 receptor antagonist, SB-742457, in Alzheimer’s disease. Curr Alzheimer Res 7:374–385. https://doi.org/10.2174/156720510791383831
Wilkinson D, Windfeld K, Colding-Jørgensen E (2014) Safety and efficacy of idalopirdine, a 5-HT6 receptor antagonist, in patients with moderate Alzheimer’s disease (LADDER): a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 13:1092–1099. https://doi.org/10.1016/S1474-4422(14)70198-X
Ferrer I, Tuñon T, Soriano E, del Rio A, Iraizoz I, Fonseca M, Guionnet N (1993) Calbindin D-28k immunoreactivity in the temporal neocortex in patients with Alzheimer’s disease. Clin Neuropathol 12:53–58
Maguire-Zeiss KA, Li ZW, Shimoda LM, Hamill RW (1995) Calbindin D28k mRNA in hippocampus, superior temporal gyrus and cerebellum: comparison between control and Alzheimer disease subjects. Brain Res Mol Brain Res 30:362–366. https://doi.org/10.1016/0169-328x(95)00035-q
Iritani S, Niizato K, Emson PC (2001) Relationship of calbindin D28K-immunoreactive cells and neuropathological changes in the hippocampal formation of Alzheimer’s disease. Neuropathology 21:162–167. https://doi.org/10.1046/j.1440-1789.2001.00393.x
Morozova M, Burminskiy D, Rupchev G, Lepilkina T, Potanin S, Beniashvili A, Lavrovsky Y, Vostokova N et al (2017) 5-HT6 receptor antagonist as an adjunct treatment targeting residual symptoms in patients with schizophrenia: unexpected sex-related effects (double-blind placebo-controlled trial). J Clin Psychopharmacol 37:169–175. https://doi.org/10.1097/JCP.0000000000000673
Russell MG, Dias R (2002) Memories are made of this (perhaps): a review of serotonin 5-HT6 receptor ligands and their biological functions. Curr Top Med Chem 2:643–654. https://doi.org/10.2174/1568026023393877
Lindner MD, Hodges DB Jr, Hogan JB, Orie AF, Corsa JA, Barten DM, Polson C, Robertson BJ et al (2003) An assessment of the effects of serotonin 6 (5-HT6) receptor antagonists in rodent models of learning. J Pharmacol Exp Ther 307:682–691. https://doi.org/10.1124/jpet.103.056002
Thur KE, Nelson AJ, Cassaday HJ (2014) Ro 04-6790-induced cognitive enhancement: no effect in trace conditioning and novel object recognition procedures in adult male Wistar rats. Pharmacol Biochem Behav 127:42–48. https://doi.org/10.1016/j.pbb.2014.10.006
Kendall I, Slotten HA, Codony X, Burgueño J, Pauwels PJ, Vela JM, Fone KCF (2011) E-6801, a 5-HT6 receptor agonist, improves recognition memory by combined modulation of cholinergic and glutamatergic neurotransmission in the rat. Psychopharmacology 213:413–430. https://doi.org/10.1007/s00213-010-1854-3
Nikiforuk A, Fijał K, Potasiewicz A, Popik P, Kos T (2013) The 5-hydroxytryptamine (serotonin) receptor 6 agonist EMD 386088 ameliorates ketamine-induced deficits in attentional set shifting and novel object recognition, but not in the prepulse inhibition in rats. J Psychopharmacol 27:469–476. https://doi.org/10.1177/0269881113480991
Rychtyk J, Partyka A, Gdula-Argasińska J, Mysłowska K, Wilczyńska N, Jastrzębska-Więsek et al (2019) 5-HT6 receptor agonist and antagonist improve memory impairments and hippocampal BDNF signaling alterations induced by MK-801. Brain Res 1722:146375. https://doi.org/10.1016/j.brainres.2019.146375
Vanda D, Soural M, Canale V, Chaumont-Dubel S, Satała G, Kos T, Funk P, Fülöpová V et al (2018) Novel non-sulfonamide 5-HT6 receptor partial inverse agonist in a group of imidazo[4,5-b]pyridines with cognition enhancing properties. Eur J Med Chem 144:716–729. https://doi.org/10.1016/j.ejmech.2017.12.053
Tassone A, Madeo G, Schirinzi T, Vita D, Puglisi F, Ponterio G, Borsini F, Pisani A et al (2011) Activation of 5-HT6 receptors inhibits corticostriatal glutamatergic transmission. Neuropharmacology 61:632–637. https://doi.org/10.1016/j.neuropharm.2011.05.004
Fisher NM, Seto M, Lindsley CW, Niswender CM (2018) Metabotropic glutamate receptor 7: a new therapeutic target in neurodevelopmental disorders. Front Mol Neurosci 11:387. https://doi.org/10.3389/fnmol.2018.00387
Funding
This research was funded by a University of Nottingham Early Career Research & Knowledge Transfer Award to MVK.
Author information
Authors and Affiliations
Contributions
MVK designed and conceived the study, collected data, performed statistical analysis, and wrote the first draft of the manuscript. SES assisted with behavior and western blot studies, AMB with microsensor studies, and remaining co-authors with immunohistochemistry. All authors contributed to and have approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Ethical Approval
All applicable international, national, and institutional guidelines for the care and use of animals were followed.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional Information
Supplementary Information accompanies this paper.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shortall, S.E., Brown, A.M., Newton-Mann, E. et al. Calbindin Deficits May Underlie Dissociable Effects of 5-HT6 and mGlu7 Antagonists on Glutamate and Cognition in a Dual-Hit Neurodevelopmental Model for Schizophrenia. Mol Neurobiol 57, 3439–3457 (2020). https://doi.org/10.1007/s12035-020-01938-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-020-01938-x