
Abstract
Parkinson’s disease (PD) is characterized by the degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc), resulting in motor and non-motor dysfunction. Physical exercise improves these symptoms in PD patients. To explore the molecular mechanisms underlying the beneficial effects of physical exercise, we exposed 1-methyl-4-phenyl-1,2,3,6-tetrahydropyrimidine (MPTP)-treated mice to a four-week physical exercise regimen, and subsequently explored their motor performance and the transcriptome of multiple PD-linked brain areas. MPTP reduced the number of DA neurons in the SNpc, whereas physical exercise improved beam walking, rotarod performance, and motor behavior in the open field. Further, enrichment analyses of the RNA-sequencing data revealed that in the MPTP-treated mice physical exercise predominantly modulated signaling cascades that are regulated by the top upstream regulators L-DOPA, RICTOR, CREB1, or bicuculline/dalfampridine, associated with movement disorders, mitochondrial dysfunction, and epilepsy-related processes. To elucidate the molecular pathways underlying these cascades, we integrated the proteins encoded by the exercise-induced differentially expressed mRNAs for each of the upstream regulators into a molecular landscape, for multiple key brain areas. Most notable was the opposite effect of physical exercise compared to previously reported effects of L-DOPA on the expression of mRNAs in the SN and the ventromedial striatum that are involved in—among other processes—circadian rhythm and signaling involving DA, neuropeptides, and endocannabinoids. Altogether, our findings suggest that physical exercise can improve motor function in PD and may, at the same time, counteract L-DOPA-mediated molecular mechanisms. Further, we hypothesize that physical exercise has the potential to improve non-motor symptoms of PD, some of which may be the result of (chronic) L-DOPA use.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Parkinson’s disease (PD) is characterized by the degeneration of dopaminergic (DA) neurons in the substantia nigra pars compacta (SNpc). The clinical phenotype encompasses motor symptoms—including bradykinesia, rigidity, tremor, gait dysfunction, and postural instability—and non-motor symptoms such as sleeping disturbances, pain, or cognitive deficits that affect executive functions, attention, mood, and working memory [1,2,3]. Levodopa (L-DOPA), a precursor of DA, has been used since the 1960s to treat PD motor symptoms and is still considered the gold standard of therapy [4, 5]. In recent years, physical exercise—including intervention strategies such as aerobic exercise (e.g., treadmill exercise, cycling, or dancing) or strength training (e.g., using a modified fitness counts program or progressive resistance exercising)—has been reported to improve DA signaling [6, 7] and motor dysfunction [8,9,10,11], including bradykinesia [12, 13], rigidity [14], and tremor [12]. Physical exercise has also been reported to improve less dopamine-dependent symptoms involving postural control such as turning performance [6] and instability [15], as well as cognitive function [2, 16, 17] in PD patients. Although these beneficial clinical effects of exercise on PD symptoms are evident, the underlying molecular mechanisms are not well understood. A better understanding of these processes may ultimately lead to a more efficient treatment of these symptoms, through directly targeting the underlying pathways.
Systemic administration of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) in mice results in the loss of nigrostriatal DA neurons, and is widely used to study the pathophysiological mechanisms underlying DA neuron degeneration in PD [18]. Moreover, similar to human PD, physical exercise improves motor behavior and reduces cognitive impairment in MPTP-treated mice [19,20,21,22,23]. To our knowledge, in this study, we elucidate for the first time the molecular pathways underlying the beneficial effects of exercise in PD, using the MPTP mouse model of PD.
Methods
Animals
Six-month-old male C57BL/6J mice were housed, five to a cage, with ad libitum access to food and water and at a constant 12/12 h light/dark cycle (lights on between 07:00 and 19:00 h). Room temperature was controlled at 21 °C and rooms were homogenously lighted by 60 LUX with controlled humidity. Following arrival, the mice were acclimatized to their new housing for 1 week, after which they were randomly assigned to one of four treatment groups: (1) saline-treated; (2) saline-treated with physical exercise; (3) MPTP-treated; and (4) MPTP-treated with physical exercise. MPTP-HCl (Sigma-Aldrich) dissolved in saline was administered via four intraperitoneal injections at 2 h intervals, amounting to a total administered dose of 70 mg/kg (free-base), which in a dose-response pilot experiment was found as the highest tolerable dose, with a survival rate of 75%. The intended concentration of 80 mg/kg, based on similar previous experiments [7, 24, 25], resulted in a higher toxicity and death rate that could not be justified. The control mice underwent the same protocol using saline injections. Mice were allowed to recover from the injections for 2 weeks. All saline-treated mice survived the protocol, n = 14 for group (1) and n = 14 for group (2), whereas, due to MPTP treatment, group (3) and group (4) eventually consisted of n = 10 and n = 13 mice, respectively.
Physical Exercise
A forced chronic and aerobic physical exercise regimen was initiated 3 weeks following MPTP or saline treatment and was performed daily. Mice ran 30 min twice a day during a training period of 28 consecutive days in individual, horizontal lanes on a five-lane treadmill (Panlab Harvard Apparatus) at a speed of 20 cm/s (as used before in comparable experimental setups [7, 24, 26, 27]). Automated short air puffs were used to stimulate the mice to keep running when drifting too far to the back of the lane. All mice were able to perform the physical exercise without any noticeable problems. Mice assigned to the groups without physical exercise were placed in their housing cage in the same experimental room, adjacent to the treadmill.
Behavioral Testing
Behavioral testing commenced 1 week before the physical exercise regimen started (week 0), and was repeated each week (similar to comparable experiments by others [7, 28,29,30]) during the exercise regimen (weeks 1–4): beam walk on the first, rotarod on the third, and open field on the fifth day of each week, in each case performed between 08:00 and 13:00 h. Prior to all behavioral tests, the animals of all four treatment groups were habituated to the experimental room for 1 h. Mice from different treatment groups were tested concurrently on the rotarod and in the open field.
Open Field
The mice were placed in a white plexiglass box (50×50×40cm) and video recorded from above for 30 min using EthoVision XT 7.0 software (Noldus Information Technology B.V., Wageningen, The Netherlands). Afterwards, the parameters “total walking distance”, “total movement time”, “mean velocity”, and “mean angular velocity” were calculated by the software, as described previously [31].
Rotarod
Mice were placed on the rotarod apparatus (IITC Inc., Woodland Hills, CA, USA) with a rod diameter of 32 mm and an increasing speed of 4 to 38 rpm in 300 s. Five mice were tested simultaneously on the rotarod and their latency to fall was measured (similar to [7, 30, 32]). On each testing day, each mouse performed one pre-trial and three trials, each with a maximum duration of 300 s and with a minimum of 1 h of rest between the trials. The pre-trial enabled the mice to habituate (again) to the rotarod and was not included in the results. For each testing day, the latency times of the three trials were averaged per mouse.
Beam Walk
We assessed the motor coordination and balance by measuring the ability of the mice to transverse a narrow beam [28, 33]. The mice were placed on a white plasticized iron rod (full length 80 cm, diameter 10 mm) suspended at 40 cm height and were trained to cross the beam to their home cage. Training of the mice occurred on the first day. During the training, the distance to cross was increased each time they successfully reached their cage, until they were able to reach their home cage over the full length of the beam. Each week, the mice were habituated again to the experimental setup by a pre-trial, which was followed by three trials in which the time was recorded how long it took for the mice to cross the full beam to reach their home cage; inter-trial interval was in all cases at least 1 h. For each testing day, the times of the three trials were averaged per mouse.
Immunohistochemistry
We performed immunohistochemistry to establish TH protein expression [24, 34] in the DL, VM, SNpc, and VTA. Twenty-four hours following their last exercise training, mice were sacrificed by cervical dislocation, and brains were dissected and fixated in 4% paraformaldehyde in PBS solution for 3 h and subsequently cryoprotected by immersion in 30% sucrose for 24 h. After cryosectioning, DAB staining was performed on 20-μm-thick coronal slices, placed on gelatinized glass slides. For this, the sections were washed with PBS (3×10min), non-specific sites blocked with blocking buffer (2.5% normal donkey serum, 2.5% normal goat serum, 1% BSA, 1% glycine, 0.1% lysine, and 0.4% Triton X-100 in PBS) for 30 min and incubated with rabbit anti-tyrosine hydroxylase (TH, 1:1000; Pel-Freez Biologicals #P40101-0; lot no. 19335) for 16 h at 4 °C. This was followed by 1 h incubations with biotinylated goat-anti-rabbit (1:200; Jackson ImmunoResearch; 711-065-152; lot no.117858) and avidin-biotin-peroxidase complex (A and B 1:800; Vectastain Elite ABC kit, PK-6100 Standard), with PBS washing steps in between. To visualize antibody binding, the sections with SNpc and ventral tegmental areas (VTA) were incubated for 30 min, and those with dorsolateral striatum (DL) and ventromedial striatum (VM) areas were incubated for 20 min in a DAB/H2O2-solution potentiated by ammonium nickel sulfate. The sections were subsequently dehydrated and cover slipped. For each mouse, every sixth section throughout the DL, VM, SNpc, and VTA was included in the counting procedure, and for optimal comparison between groups, sections of different treatment groups were stained concurrently.
Images were captured by a Leica DM6000 B microscope. TH-positive (TH+) cells were counted in the sections of the SNpc (−2.54 to −3.88 mm to Bregma [35]) and VTA (−2.92 to −3.88 mm to Bregma [35]), using a 20× magnification. The number of TH+ cells in each section (both the left and right side) was counted by a blinded assessor and averaged over the total number of sections per animal. DA fiber density was estimated in the DL (1.18 to −0.10 mm to Bregma [35]) and VM (1.54 to 0.62 mm to Bregma [35]) by quantifying the optic density (OD) with FIJI [36], using a 5× magnification. In both areas, the OD per section was determined by averaging the OD of ten separate areas within the striatal matrix (i.e., in-between the striosomes). Subsequently, the OD was normalized by subtracting the OD of the corpus callosum (CC) or anterior commissure (AC) for the DL and VM respectively, in the same section, and all sections were averaged per animal.
RNA Isolation and Sample Preparation
Twenty-four hours following the last physical exercise training, brains of 8–10 mice per group—that were sacrificed by cervical dislocation—were dissected, immediately frozen on dry ice, and stored at −80 °C until further preparation. Specific brain areas, i.e., prefrontal cortex (PFC), DL, VM, VTA, SN, and pedunculopontine nucleus (PPN), were then cryopunched based on the stereotaxic atlas of the mouse brain [35] from 200-μm-thick coronal slices, using punch needles with a diameter of 0.5 and 0.75 mm (see Online Resource 1 for the estimated punching locations per area). All specimens were kept at −20 °C during processing. For RNA isolation, punched samples were homogenized with a TissueLyser (Retsch GmbH) in 800 μL TRIzol reagent and RNA isolation was performed according to the manufacturer’s instructions (Invitrogen). Total RNA concentration was determined with a NanodropTM ND-1000 spectrophotometer (Thermo Fisher Scientific Inc.), and RNA quality was visually assessed by 1% agarose gel electrophoresis. Genomic DNA was removed by treatment with DNase I in the presence of RNAsin (Thermo Fisher) in 5× FSB buffer and RNAse-free water. Subsequently, total RNA samples were stored at −80 °C until further use. For each treatment group and brain area, RNA samples of six mice were pooled for RNAseq analysis.
RNA Sequencing and Data Processing
All RNA samples were subjected to RNA sequencing (RNAseq; HudsonAlpha Genomic Services Lab, Huntsville, AL) as performed before [37]. In short, total RNA concentration was estimated by Qubit 2.0 Fluorometer (Invitrogen, Carlsbad, CA, USA) and RNA integrity by using the Agilent 2100 Bioanalyzer (Applied Biosystems, Carlsbad, CA, USA). RNAseq libraries were formed from approximately 500 ng total RNA of each pooled sample, followed by poly(A) enrichment. RNAseq was performed using paired-end sequencing on Illumina HiSeqH2000 (Illumina, San Diego, CA, USA), at 50 base pairs, generating over 25 million paired reads per sample. Raw RNAseq FASTQ files were demultiplexed by bcl2fastq conversion software v1.8.3 (Illumina, Inc., San Diego, CA, USA) using default settings.
RNAseq data was analyzed using GeneSifter software (VizX Labs, Seattle, WA). RNAseq reads were mapped to the Mus musculus reference genome build 37.2, and for this, the reads were trimmed by 15 base pairs at the five-prime end. Subsequently, transcript abundance was calculated by estimating the reads per kilobase of exon per million mapped reads (RPKM), and normalization to the number of mapped reads was used for comparison of two mRNA sets. A t test was used for pairwise comparison and a likelihood ratio test to adjust for distribution probability.
qPCR Validation
The RNAseq results were validated by comparing expression levels of at least eight mRNAs/genes per area with their expression as established by qPCR. These genes were chosen randomly, although there was one requirement, namely that genes from all three comparisons of interest, i.e., the comparisons to assess the effect of MPTP (group 3 vs. group 1), physical exercise (group 2 vs. group 1), and physical exercise in the MPTP model of PD (group 4 vs. group 3), should be included. RNA from the same samples used for the RNAseq pools was reverse-transcribed to cDNA with random primers using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Scientific, #K1632 lot no 00167909) according to the manufacturer’s protocol. Three-step qPCR (95 °C for 10 min, followed by 45 three-step cycles of 95 °C for 5 s, 65 °C for 10 s, and 72 °C for 20 s, and the generation of melting curves from 70 °C to 95 °C; Rotor-Gene 6000 Series, Corbett Life Science Pty. Ltd.) was performed using the 2× SensiFAST SYBR No-ROX mix (Bioline lot no SF582-313209) and primers designed with NCBI Primer-Blast (www.ncbi.nlm.nih.gov/tools/primerblast/) and synthesized at Sigma Life Sciences (The Netherlands) (For a complete overview of used primers, see Online Resource 10, Supp. Table 1). The housekeeping genes ACTB and YWHAZ were used as reference for normalization of gene expression. Based on the qPCR results, the minimum requirements to be included in the enrichment analysis—regarding fold change (FC) cut-off, maximum likelihood ratio value, and minimal RPKM value—were adjusted so that at least 90% of the expression changes could be validated by qPCR. As there was insufficient remaining RNA available to perform the complete qPCR validation for the PPN RNAseq data, the same cut-off values were used as for the other brain areas.
Overlap of MPTP- and Exercise-Regulated Genes
To determine the direct effect of exercise on MPTP-regulated genes, we looked at the overlap between the genes regulated by MPTP (group 3 vs. group 1) and the genes regulated by exercise in the MPTP model (group 4 vs. group 3). To quantify this overlap, we used the hypergeometric distribution test [38]:
and determined the chance of observing exactly x overlapping genes from a total of n differentially expressed genes by exercise in the MPTP model, with a total of M genes that were differentially expressed by MPTP and a total of N genes detected with RNAseq. The number of unique genes detected with RNAseq in each brain area (N), consists of genes detected in both comparisons (group 3 vs. group 1 and group 4 vs. group 3), irrespective of their FC or expression p value. Of note, for all comparisons only protein-coding genes were considered.
Enrichment Analysis and Building of Molecular Landscapes
The Ingenuity pathway analysis software package (www.ingenuity.com) was used to identify enriched categories in the lists of differentially expressed protein-coding mRNAs in each of the brain areas [39]. Again, we focused on the three main comparisons of interest (see above)—i.e., the comparisons that assess the effect of MPTP, physical exercise, and physical exercise in the MPTP model of PD—in the six brain areas. Ingenuity assigns genes or rather their corresponding mRNAs/proteins to functional (sub)-categories, i.e., “canonical pathways” and “biofunctions”, with the latter including “diseases and disorders” and “molecular and cellular functions”. In addition, Ingenuity generates a list of “upstream regulators”, i.e., proteins or compounds that regulate multiple proteins/mRNAs from the input list. When possible, the program also calculates a z score that is based on the expression changes of the input mRNAs and that is a measure for the directionality of the upstream regulator, canonical pathway, or biofunction. A z score < −2 or > 2 is considered significant. For all analyses, only functional categories and upstream regulators with significant enrichment (i.e., Benjamini-Hochberg corrected p < 0.05) and containing at least two genes were taken into account.
Proteins/mRNAs regulated by the top upstream regulators were analyzed in more depth to identify their relation to physical exercise-induced processes in the MPTP model of PD (i.e., the comparison of group 4 with group 3). Guided by the results of the Ingenuity enrichment analyses, an extensive literature search was performed for the (putative) roles of all proteins encoded by the differentially expressed mRNAs as well as their functional interactions, using the UniProt Protein Knowledge Base (http://www.uniprot.org) and PubMed (http://www.ncbi.nlm.nih.gov/pubmed). Based on these findings and applying an approach similar to the one we used previously for genome-wide association and expression data [39,40,41,42], we then built molecular landscapes containing interacting proteins encoded by the mRNAs that are differentially expressed by physical exercise and are known to be regulated by the top regulators for each brain area. To complement these protein interaction cascades, we added a number of proteins that were not encoded by the differentially expressed mRNAs but that have been implicated in PD etiology through other lines of (genetic) evidence. In this respect, proteins encoded by familial PD candidate genes were included if they have at least one functional interaction with one or more other landscape proteins. Additional proteins were included when having at least two interactions with other landscape proteins. The molecular landscapes were drawn with Serif DrawPlus 4.0.
Statistics
Statistical comparisons of values between multiple treatment groups were carried out using a two-way ANOVA. For behavioral test data, with data at multiple time points, a linear mixed model was applied using SPSS (IBM, version 23), with “week”, “physical exercise”, and “MPTP” as fixed factors to calculate the main effects of the training period, physical exercise, and the interaction between physical exercise and MPTP. The main effect of MPTP in the behavioral tests was assessed using a pairwise comparison of saline-treated and MPTP-treated mice before the start of the exercise regimen. For pairwise comparison, an F test was used to determine if the distributions of the compared two groups have the same variance. Based on the F test, a Student’s t test for equal or unequal variance was then used to evaluate the significance of the expression differences. For all comparisons, data are represented as mean with the standard error of the mean (SEM), and a p value < 0.05 was considered statistically significant.
The p values calculated with the hypergeometric distribution test were adjusted for multiple testing using the Bonferroni correction.
Results
In this study, we assessed the effects of physical exercise in the MPTP-treated mouse model of PD at the behavioral and molecular levels.
Physical Exercise Affects the Motor Function of MPTP-Treated Mice
At baseline, i.e., following recovery from MPTP treatment but before the exercise regimen started, MPTP-treated mice showed an increased total walking distance (p < 0.01), total movement time (p < 0.005), and mean velocity (p < 0.005), and a decreased mean angular velocity (p < 0.005) in the open field compared to saline-treated control mice. In contrast, their performance on rotarod and beam walk tests was not significantly different from controls (Fig. 1).

Effect of MPTP. Results of the behavioral tests in week 0 (set as the baseline (100%) for the effect of physical exercise, which is shown in Fig. 2). No effect of MPTP was shown for the beam walk (a), or rotarod (b) tests, but MPTP significantly affects the parameters in the open field (c–f). **p < 0.01; ***p < 0.005, mean + SEM, n = 28 for controls (saline-treated), and n = 23 for MPTP-treated mice
In Fig. 2, the effects of physical exercise during the course of the training period relative to baseline are shown for each of the four treatment groups. The beam walk task showed a clear training effect over time in all groups (main effect of “week” p < 0.001), and the test performance was improved by physical exercise in both the MPTP-treated and saline-treated mice, without significant differences between the groups (main effect of physical exercise p < 0.05) and no significant interaction between physical exercise and MPTP treatment (Fig. 2a). Rotarod performance was also significantly improved by physical exercise (p < 0.01), but no improvement over time or interaction with MPTP treatment was found (Fig. 2b). Of the tested parameters in the open field (total walking distance, total movement time, mean velocity, and mean angular velocity), the mean angular velocity was increased (p < 0.001), and the total movement time showed a decreasing trend (p = 0.051) for all treatment groups over time during the exercise regimen (i.e., main effect of “week”). There was no significant (main) effect of physical exercise on any of the four tested open field parameters, only a trend towards a higher “mean velocity” (p = 0.082). However, for all four open field parameters, significant interactions between physical exercise and MPTP treatment were found (p < 0.05). Physical exercise increased the walking distance and mean velocity of saline-treated mice, but not of MPTP-treated mice. Moreover, physical exercise increased the total movement time of saline-treated mice and decreased that of MPTP-treated mice. This opposite effect was also observed for mean angular velocity, i.e., a decrease by physical exercise in saline-treated mice and an increase in MPTP-treated mice (Fig. 2c–f).

Effect of physical exercise and interaction with MPTP compared to the baseline (week 0); results of the behavioral tests in week 0–4. Measurements in week 1–4 were normalized to week 0 (100%). Dark-orange arrows indicate the main effect of physical exercise (a, b), and light orange arrows show the effect of the interaction between physical exercise and MPTP treatment (c–f) mean ± SEM n = 14 for both CNR and CR, n = 10 for MNR and n = 13 for MR. CNR, control not running; CR control running, MNR MPTP-treated but not running, MR MPTP-treated and running
TH Depletion in the SNpc and Striatum Following MPTP Treatment
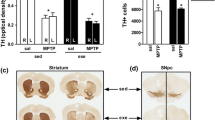
The number of DA neurons in the SNpc and VTA of each treatment group, as well as an estimate of DA fiber density in striatal target areas (DL and VM, respectively) was determined by immunohistochemistry for TH—the rate-limiting enzyme in DA synthesis. These measures were primarily taken to confirm and estimate the degree of neuronal loss due to MPTP treatment, but they may also provide some insight into whether exercise could affect these structural changes. MPTP significantly reduced the number of TH+ cells in the SNpc (p < 0.005), but not in the VTA. Pairwise comparison between the treatment groups revealed that the number of TH+ cells in the SNpc of MPTP-treated mice without and with physical exercise was reduced by 29 and 20%, respectively, compared to the saline-treated group without exercise (both p < 0.05; Fig. 3). There was no significant effect of physical exercise on the number of TH+ cells in either the SNpc or the VTA, and no interaction between MPTP and physical exercise.

TH+ neurons in the SNpc and VTA. The upper panel shows a representative picture for each of the four treatment groups and the lower panel shows the average number of TH+ neurons in the SNpc and the VTA per treatment group. *p < 0.05, mean + SEM, n = 5 for CNR and MR and n = 4 for CR, and MNR for both brain areas. CNR control not running, CR control running, MNR MPTP-treated but not running, MR MPTP-treated and running, SNpc substantia nigra pars compacta, VTA ventral tegmental area
In Online Resource 2, the relative OD of TH+ fibers in the DL, the primary striatal target area of the SNpc, is shown. The OD of TH+ fibers was reduced by MPTP (p < 0.05), without a main effect of physical exercise or an interaction between MPTP and physical exercise. Pairwise comparison showed that MPTP decreased the density of TH+ fibers in MPTP-treated mice without exercise by 33% (p < 0.005) compared to saline-treated mice without physical exercise. There was a trend towards an increased TH+ OD by physical exercise in MPTP-treated mice, but this increase was not significant.
Online Resource 3 shows the OD of TH+ fibers in the VM, the primary striatal target area of the VTA. Although all treatment groups (physical exercise, MPTP, and MPTP + physical exercise) showed a reduced OD of TH+ fibers, no significant effects of MPTP, physical exercise, or their interaction were found.
qPCR Validation of the RNAseq Data
The RNAseq data were obtained from pooled samples, and in order to validate these data, the mRNA expression levels in each of the investigated brain areas were determined in individual samples by qPCR. The results of the qPCR experiments (Online Resource 4) led us to adopt the following requirements for the inclusion of differentially expressed protein-coding mRNAs in the subsequent analyses: FC > 1.2, likelihood ratio < 0.05, RPKM > 5.
A Direct Effect of Physical Exercise on MPTP-Regulated Genes
The overlap between the protein-coding mRNAs that are differentially expressed due to MPTP alone and due to exercise in MPTP-treated mice is represented in Online Resource 5. In all brain areas, the probability of this overlap was calculated by using the hypergeometric distribution test, which showed that for all areas, the overlap is greater than would be expected based on random gene selection (p < 0.05). Further, in all areas, 82–99% of the overlapping mRNAs are regulated in opposite directions by MPTP and exercise. Enrichment analyses of mRNAs that overlap but are regulated in opposite directions are summarized in Online Resource 10, Supp. Table 2. The VTA and PFC show the most significant results, and are also the brain areas with the biggest absolute and relative overlap (i.e., the overlap in number and proportion of mRNAs). The analysis of the VTA displays a downregulation of the top regulator “inosine”, whereas the PFC and to a lesser extent also the DL show an increase in effect of dalfampridine and bicuculline.
RNAseq Data Analysis: Enriched Regulators, Pathways, and Biofunctions
Enrichment analysis of the differentially expressed mRNAs was performed for each of the brain areas examined to investigate the effects of MPTP (i.e., comparing the MPTP-treated group without exercise to the saline-treated mice without exercise), physical exercise (i.e., comparing saline-treated mice with exercise to saline-treated mice without exercise), and the effects of physical exercise in MPTP-treated mice (i.e., comparing the MPTP-treated mice with exercise to MPTP-treated mice without exercise). In Tables 1, 2, and 3, a short overview of the main effects—the top regulator(s), canonical pathway(s), and biofunction(s)—of MPTP, physical exercise, and physical exercise in MPTP-treated mice is provided for each brain area separately. A more elaborate overview of these enrichment analyses per brain area can be found in Online Resource 10, Supp. Tables 3–8.
In all brain areas examined, MPTP treatment affected a set of mRNAs that is involved in epilepsy, which is reflected by the presence of the epilepsy-regulating transcription factor CREB1, the convulsants bicuculline and dalfampridine, and the biofunction “epilepsy”. Other regulators and related functional themes enriched within the mRNAs affected by MPTP are RICTOR and its regulation of ribosomal and mitochondrial proteins, as well as L-DOPA and DA receptor signaling (Table 1). Of note, in line with the MPTP-mediated decrease in TH expression in the DL and VM (not significant; see above), L-DOPA is a significant upstream regulator in both the DL (p = 2.85E-06; z = −2.635) and the VM (p = 9.97E-04; z = −1.234), but was not among the top 10 upstream regulators and is therefore not included in the (Supplementary) Tables.
Furthermore, in the various brain areas examined, physical exercise affected sets of mRNAs that are regulated by the upstream regulators CREB1, RICTOR, L-DOPA, and dexamethasone. These regulators overlap to some extent with the upstream regulators for the MPTP-regulated mRNAs as mentioned above. However, the top canonical pathways and biofunctions due to physical exercise are not epilepsy-related, but rather associated with “mitochondrial dysfunction” and “movement disorder” (Table 2).
The top regulators of the mRNAs differentially expressed due to physical exercise in MPTP-treated mice are L-DOPA, RICTOR, bicuculline/dalfampridine, and CREB1. The top canonical pathways and biofunctions enriched in exercised MPTP-treated mice are “mitochondrial dysfunction” and “protein synthesis” in the VTA and DL, “G-protein signaling”, “movement disorder”, “seizures and cytoskeleton dynamics” in the VM and are related to (cell) death in the PFC (Table 3).
Of note, the predicted direction of effect of the top regulators RICTOR and L-DOPA is changed in the VTA, DL, and VM of exercised MPTP-treated mice compared to exercised saline-treated mice. More specifically, the predicted direction of effect of RICTOR is (strongly) decreased in the VTA and DL after exercise in saline-treated mice, but is strongly increased and has no significant predicted direction in the VTA and DL of exercised MPTP-treated mice, respectively. Further, L-DOPA shows a strongly decreased predicted direction of effect in the VM of exercised MPTP-treated mice, whereas this direction of effect was absent after exercise alone.
The Main Molecular Pathways Regulated by Physical Exercise
To elucidate the main molecular pathways regulated by physical exercise in MPTP-treated mice, the mRNA sets regulated by the top upstream regulators L-DOPA (in the SN and VM, Online Resource 10, Supp. Tables 9 and 10), RICTOR (in the DL and VTA, Online Resource 10, Supp. Tables 11 and 12), bicuculline/dalfampridine (in the PFC, Online Resource 10, Supp. Table 13), and CREB1 (in the PPN, Online Resource 10, Supp. Table 14) were studied in greater detail and used to build molecular landscapes for each top upstream regulator in the various brain areas. Here, we provide a short description of each of these molecular landscapes. In Online Resource 10, all landscapes are described in full detail. Of note, in the PPN, L-DOPA is the top upstream regulator following physical exercise or MPTP treatment, but L-DOPA (p = 1.97E-02; z score = −1.964), although significant, was not among the top 10 upstream regulators following physical exercise in MPTP-treated mice.
The molecular landscapes of interacting proteins encoded by the L-DOPA-regulated mRNAs that are differentially expressed in the SN and the VM due to physical exercise in MPTP-treated mice, are shown in Figs. 4 and 5, respectively. In the SN landscape, G-coupled receptor signaling (involving the proteins ARRB2 and GRP39), glucose uptake and signaling (SLC2A1), DA signaling (PPP1R1B), and reactive oxygen species (ROS) regulation (HSPB6, FTL, ROMO1) converge on the activation of ERK1/2 (ACKR1, EDNRB, GPR39, IER3, TP53), apoptotic pathways (CASP3, TP53), CREB1, and circadian clock regulation (PER1, DBP, CIART) (Fig. 4). In the VM landscape, the main molecular pathways are (interneuron-mediated) DA release (involving the proteins CHAT, DOC2B, SYN1, and TH) and signaling (DRD2, PPP1R1B), cannabinoid signaling (CNR1, FAAH), and neuropeptide signaling (PDYN, PENK, TAC1) that subsequently regulate/activate ERK1/2, CREB1, and CCND1 signaling. The latter is a cell cycle regulator that may also be involved in synaptic plasticity and learning [43] (Fig. 5). Of note, almost all proteins in this landscape are regulated by physical exercise and L-DOPA in opposite directions.

Landscape of proteins encoded by the mRNAs regulated by physical exercise and the upstream regulator L-DOPA in the SN. mRNAs differentially expressed in the SN due to physical exercise in MPTP-treated mice are shown in gray. Blue proteins are additional genes/proteins that are associated with PD through genetic and/or expression studies, whereas white proteins have no known link with PD. The direction of effect of physical exercise (measured) and L-DOPA (from literature) on the expression of these mRNAs is depicted through colored borders. L-DOPA-activated proteins are shown with purple writing for the protein name, and familial PD proteins are shown with a blue border

Landscape of proteins encoded by the mRNAs regulated by physical exercise and the upstream regulator L-DOPA in the VM. mRNAs differentially expressed in the VM due to physical exercise in MPTP-treated mice are shown in gray. Blue proteins are additional genes/proteins that are associated with PD through genetic and/or expression studies, whereas white proteins have no known link with PD. The direction of effect of physical exercise (measured) and L-DOPA (from literature) on the expression of these mRNAs is depicted through colored borders. L-DOPA-activated proteins are shown with purple writing for the protein name, and familial PD proteins are shown with a blue border
The RICTOR-regulated mRNAs that are differentially expressed in the DL and VTA due to physical exercise in the MPTP-treated mice encode proteins that are specifically involved in three cellular systems: the complex I-V of the electron transport chain, the 40S and 60S ribosomal subunits, and the proteasome (see Online Resource 10, Supp. Tables 11 and 12). These are complexes that regulate cellular energy, protein translation, and protein degradation, respectively (Online Resources 6 and 7). Of note, physical exercise and RICTOR have an opposite effect on the expression of all differentially expressed mRNAs in the mitochondrial electron transport chain in the DL, whereas physical exercise and RICTOR exert the same direction of effect (i.e., a decreasing effect) on the expression of electron transport chain mRNAs in the VTA.
In the PFC, 8 out of 9 mRNAs differentially expressed due to physical exercise in MPTP-treated mice and regulated by bicuculline/dalfampridine have been linked to epilepsy (Online Resource 10, Supp. Table 13). Immediate-early gene activation is one of the main processes regulated by these mRNAs e.g., via the early response genes/proteins FOS, FOSB, and NR4A1, which in turn are regulated by insulin and low-density lipoprotein. In Online Resource 8, an overview of the interactions of the proteins encoded by these mRNAs and their regulation by bicuculline/dalfampridine and physical exercise is shown in a molecular landscape.
In the PPN, the proteins encoded by the mRNAs that were differentially expressed due to physical exercise in MPTP-treated mice and regulated by CREB1 have only a limited number of interactions in the built landscape (Online Resource 9). Nevertheless, a few functional themes such as vascular remodeling, neuropeptide signaling, lipid metabolism, epilepsy/immediate-early gene regulation, and calcium signaling were identified, with CREB1 as their central regulator (Online Resource 10, Supp. Table 14).
Discussion
This study aimed to explore the molecular mechanisms underlying the beneficial effects of physical exercise on motor functioning in the MPTP-treated mouse model of PD. After validation of the model, through demonstrating significant nigral neuronal loss following MPTP treatment, the effects of a four-week physical exercise regimen on motor performance, and the accompanying molecular changes in multiple brain areas were assessed using behavioral tests and RNAseq analysis, respectively. The behavioral tests showed that physical exercise improved beam walk and rotarod performance in both MPTP-treated and control mice, but had a different and often opposite effect on the four tested open field parameters in these groups. Our RNAseq findings demonstrated that physical exercise in MPTP-treated mice mainly affects the expression of mRNAs involved in L-DOPA-mediated pathways in the SN and VM that regulate DA signaling, RICTOR-mediated pathways in the VTA and DL involved in energy metabolism and cellular stress [44, 45], and bicuculline/dalfampridine-mediated pathways in the PFC and CREB1-mediated pathways in the PPN that are both a measure of neuronal activity [46, 47]. To further elucidate the specific molecular mechanisms underlying the effects of physical exercise in MPTP-treated mice, the differentially expressed mRNAs regulated by these top regulators were integrated into molecular landscapes, depicting the main biological processes and signaling cascades affected.
Our animal model was validated by demonstrating a significant nigral DA neuronal loss following MPTP treatment. The observed moderate neuronal loss in the midbrain due to MPTP treatment, i.e., a 29% reduction of TH-positive neurons in the SNpc without a statistical significant loss in the VTA, is in keeping with earlier studies using a similar MPTP treatment regimen in 5-month-old mice showing 33% loss in the SNpc and no significant loss in the VTA [48]. Other studies, on 8–10-week-old mice, have reported a neuronal loss of 29–45% [49, 50], but also of more than 50% loss in the SNpc [7, 24, 51]. Differences in level of neurodegeneration [52] and molecular effects [39] due to MPTP toxicity may be explained by MPTP dosing, age of the mice, and the duration between MPTP injection and sacrifice [48, 52]. We used aged (6-month-old) mice to better model age-dependent processes such as regulation of anti-oxidants [53], neuroplasticity, neurogenesis [54, 55], and the immune response in PD [56, 57]. To assess how exercise may boost any neuroplastic mechanisms of the injured basal ganglia, the physical exercise regime was performed within the recovery phase of striatal DA levels as reported in younger MPTP-treated mice [58], but after the acute neurotoxic (molecular) effects of MPTP [39, 59]. We did not find a significant effect of physical exercise on the number of surviving DA neurons, but noted a trend towards an increased number of TH-positive neurons in the SNpc and an increased TH-positive fiber density in the DL and VM in MPTP-treated mice with physical exercise compared to MPTP-treated mice without exercise. From previous studies, it remains unclear whether physical exercise can protect against cellular loss in the MPTP-mouse model. Preservation of SNpc neurons by physical exercise has been described before [27, 51], but the findings were inconsistent [7, 22].
Regarding motor function, forced exercise has more effect than voluntary exercise in both PD patients [60] and mice [61], and it activates the same brain areas as anti-PD medication does [62]. In this study, the mice were able to perform the physical exercise without any noticeable problems, suggesting that their physical exercise regimen is comparable to the forced moderate aerobic exercise that has been shown to improve both motor and non-motor functions in PD patients [11, 60, 63, 64]. MPTP treatment alone resulted in an increased activity in the open field, as reported before [48, 52, 65,66,67], but did not affect the performance on beam walk and rotarod. It should be noted that the training effect on the beam walk as seen in all four treatment groups, especially in week 1 compared to week 0, may implicate the necessity for more extensive training of the mice before the beam walk task in week 0. Further, the effects of exercise on the motor performance included an improvement on the beam walk and rotarod in both saline and MPTP-treated groups. However, the effects of physical exercise on the open field parameters in saline-treated mice was either absent or opposite in MPTP-treated animals. These findings suggest that some effects of physical exercise may be dependent on the “disease-state” (i.e., saline- or MPTP-treated). It could be argued, however, that the lack of effect of physical exercise in MPTP-treated mice on total walking distance and mean velocity (Fig. 2) may be due to their MPTP-induced hyperactivity (Fig. 1) that could have limited a further increase in motor performance due to physical exercise. This hyperactivity has been observed more often following MPTP treatment [48, 65, 66, 68,69,70] and may result from compensatory effects induced by e.g., brain areas of the mesolimbic pathway (see also below). Furthermore, the opposite effect of exercise on total movement time and mean angular velocity in MPTP-treated mice (Fig. 2) compared to the effect of MPTP alone (Fig. 1) suggests that physical exercise counteracts the effect of MPTP. This finding could have important translational value as axial symptoms in PD—such as hypokinetic rigidity which is reflected by reduced angular velocity [71,72,73]—are notoriously more difficult to treat by medication than appendicular symptoms.
The RNAseq analysis showed that the level of overlap between MPTP-regulated genes and physical exercise-regulated genes differed between the brain areas studied and was particularly high in the PFC and VTA. These data suggest that in the PFC and VTA, physical exercise influences the processes affected by MPTP more directly than in the other areas in which more indirect mechanisms may prevail. Nevertheless, in all brain areas examined, the majority of overlapping genes (82–99%) were regulated in opposite directions by physical exercise compared to MPTP, suggesting counteracting effects of physical exercise on MPTP-regulated mechanisms. For example, the enrichment analysis of the overlapping genes in the PFC and DL (see Online Resource 10, Supp. Table 2) shows a predicted activation of the top regulators dalfampridine, bicuculline, and CREB1—indicative for neuronal activation [46, 47]—whereas these are inactivated by MPTP.
The roles of the PD-related brain areas examined in this study can be summarized in a simplified basal ganglia circuitry model, wherein PPN, SN, and DL are mainly involved in motor control, and the VTA, VM, and PFC contribute particularly to the regulation of (complex) behavior and cognition (Fig. 6) [74,75,76,77,78,79]. The top regulators—and to a lesser extent also the canonical pathways and biofunctions—regulated by physical exercise in the cognition-associated brain areas of MPTP-treated mice, showed highly significant predicted directions of effect, whereas these effects were less prominent in the motor-related areas. This implicates that, although physical exercise is able to improve motor function (as supported by the behavioral tests), it may also have strong effects on cognition and behavior. This is interesting from a therapeutic point of view, because non-motor symptoms in PD patients—including cognitive impairment, depression, pain, and sleep disorders—are usually less responsive to dopamine replacement therapy and therefore treatment options are limited [80,81,82]. It remains unclear, however, to what extent these motor and non-motor features of PD have truly discernible neuroanatomical or molecular substrates, as effects of changes in mRNA expression in the “behavioral areas” VTA, VM, and PFC on motor function of our animals cannot be excluded. For example, a recent paper reported that VTA-specific knockout of RICTOR in mice may affect cognition and mood, but also results in hyperactivity in the open field [83]. In addition, it has been suggested that during exercise, the mesolimbic pathway (including the VTA and VM) may provide a compensatory functional activation of the motor loop [84]. Furthermore, whereas L-DOPA is known to improve DL-mediated motor symptoms, it may impair VM function in PD patients [85, 86]. Therefore, exercise may counteract L-DOPA-mediated pathways in the VM and as such improve VM functionality, which could in turn result in increased compensatory motor-loop activation. Finally, inhibition of GABAergic interneurons in the PFC by bicuculline increases the release of DA in the DL through the glutamatergic corticostriatal pathway [87,88,89] and may increase the locomotor activity of mice [88, 90]. This is in line with the reduced TH expression we observed in the DL and the inactivation of bicuculline/dalfampridine-regulated pathways in the PFC following MPTP treatment, as predicted on the basis of the RNAseq analysis. Moreover, we found no significantly reduced TH expression in the DL of exercised MPTP-treated mice that, in contrast to MPTP-treated mice without exercise, showed a predicted activation of the bicucullin/dalfampridine-regulated pathways in the PFC.

Overview of the brain areas analyzed, and the top upstream regulators, and processes per area. The brain areas are shown in a simplified model of the basal ganglia circuitry. Green, red, and gray triangles depict positive (> 2), negative (< −2) or non-significant z scores, respectively, from the enrichment analyses of the physical exercise-regulated mRNAs in MPTP-treated mice. DL dorsolateral striatum, GPe globus pallidus external, GPi globus pallidus internal, PFC prefrontal cortex, PPN pedunculopontine nucleus, SNpc substantia nigra pars compacta, SNr substantia nigra reticularis, STN subthalamic nucleus, VM ventromedial striatum, VTA ventral tegmental area
Almost five decades after its introduction [4], the DA precursor L-DOPA is still the gold standard for symptomatic treatment to alleviate the motor symptoms of PD [5]. It should be noted, however, that chronic high-dose L-DOPA use is associated with complications such as dyskinesias [91,92,93]. Moreover, the effects of L-DOPA on non-motor symptoms in PD are even less predictable and L-DOPA use may even lead to deterioration of these symptoms, e.g., impaired reversal learning or motor sequence learning deficits [94,95,96,97,98,99,100,101]. It has been suggested that these adverse cognitive effects of L-DOPA may be due to a higher L-DOPA demand in the motor systems compared to cognitive areas, resulting in a relative L-DOPA overdose in cognitive areas [102,103,104] e.g., the VM (see also above). Therefore, novel “add-on” treatments that can enable low-dose L-DOPA use and/or reduce the adverse effects of (long-term) L-DOPA use are desirable. In this respect, our study suggests that physical exercise is an attractive add-on treatment for PD, and that exercise combined with L-DOPA treatment may be more beneficial than treatment of PD patients with L-DOPA alone [9, 105]. Other findings that support this hypothesis include the reports indicating that physical exercise not only improves the motor symptoms of PD patients [8, 9], but also L-DOPA-induced dyskinesias in PD patients [106] and animal models [107], and cognitive function in PD patients [2, 16, 17]. In this light, it is of note that L-DOPA use may result in alpha-synuclein-induced neuroinflammation [108] that very recently has been shown to be reduced by physical exercise [109,110,111]. Although the major pathways regulated in our study are not directly related to inflammation, L-DOPA-mediated pathways may affect alpha-synuclein regulation [108], and the RICTOR-regulated pathways may improve mitochondrial function and protein turn-over, i.e., processes that have been suggested to reduce alpha-synuclein-induced neuroinflammation [111].
Considering the above, it is worth noting that our landscapes revealed that physical exercise and L-DOPA regulate similar pathways in the SN and VM—often in an opposite direction—and that most of these pathways have been linked to sleeping problems (SN) and cognitive and/or motor dysfunctioning (VM) in PD. For example, the expression of clock proteins was affected by physical exercise and L-DOPA in the SN, a brain region known to be involved in the regulation of REM sleep [112, 113] and causing circadian rhythm irregularities when damaged by MPTP [114, 115]. Further, the use of L-DOPA can disturb REM sleep [116] and result in a delayed sleep onset in PD patients, which suggests an uncoupling of sleep and circadian regulation [117]. On the other hand, physical exercise can improve circadian rhythm regulation [118,119,120] and may therefore serve as a complementary therapy to strengthen circadian function in PD, as suggested earlier [121].
In the VM, both physical exercise and L-DOPA regulate DA, neuropeptide, and endocannabinoid signaling, but in opposite directions. L-DOPA treatment results in sustained DA signaling in the striatum and can disrupt DA and (endo)cannabinoid receptor crosstalk [122, 123]. In contrast, physical exercise may rebalance DA signaling after sustained L-DOPA treatment (by reducing PPP1R1B activation) [107], attenuates depression-like behavior by decreasing the expression of neuropeptides [124] and activates the endocannabinoid system [125,126,127]. In turn, the endocannabinoid system modulates synaptic (DA) transmission in the striatum of PD patients [128,129,130], restores homeostasis following DA depletion [131, 132] and exerts beneficial effects on cognition, mood, and nociception [126]. Therefore, physical exercise seems to exert a positive effect on the regulation of DA, neuropeptide, and endocannabinoid signaling. Moreover, these three signaling pathways are not only associated with L-DOPA-induced dyskinesia [133,134,135,136,137,138], a process that is mainly due to dysregulation in the DL, but are also involved in regulating VM-associated cognitive functions and behaviors [124, 139,140,141,142,143,144], supporting the notion that the anatomical and neurophysiological boundaries of the striatal domains regulating control of movement (DL) and (more) cognition-related processes (VM) may functionally overlap [145, 146].
In summary, the molecular pathways that are regulated in the SN and VM by both physical exercise and L-DOPA can be directly linked to clinical features of PD. Interestingly, the overall effects of physical exercise on these pathways seem to particularly improve the motor and behavioral clinical phenotype, whereas (chronic) L-DOPA-treatment can also cause adverse effects. Moreover, to our knowledge, physical exercise exerts—although it may counteract some L-DOPA-regulated pathways—no adverse effects on PD patients. To confirm the positive effects of physical exercise on cognitive function, future physical exercise studies in PD animal models and patients should include cognitive tests, e.g., the Y-maze, the water maze, or reversal learning tasks. Furthermore, these studies should aim at further elucidating the molecular pathways underlying physical exercise in relation to (chronic) L-DOPA treatment in animal models.
Taken together, our findings provide further evidence that physical exercise improves motor function in PD, while it also affects the regulation of non-motor brain areas of MPTP-treated mice. We found that physical exercise and L-DOPA exert opposite effects on molecular pathways in several PD-associated brain areas, including those involved in sleeping and cognitive function. Overall, the present study suggests that physical exercise has therapeutic potential, not only to improve motor function but it may also improve non-motor symptoms of PD—and perhaps even alleviate detrimental effects associated with (chronic) L-DOPA use.
References
Goetz CG, Tilley BC, Shaftman SR, Stebbins GT, Fahn S, Martinez-Martin P et al (2008) Movement Disorder Society-sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS-UPDRS): scale presentation and clinimetric testing results. Mov Disord 23(15):2129–2170
David FJ, Robichaud JA, Leurgans SE, Poon C, Kohrt WM, Goldman JG et al (2015) Exercise improves cognition in Parkinson's disease: The PRET-PD randomized, clinical trial. Mov Disord 30(12):1657–1663
Sveinbjornsdottir S (2016) The clinical symptoms of Parkinson's disease. J Neurochem 139(Suppl 1):318–324
Cotzias GC, Van Woert MH, Schiffer LM (1967) Aromatic amino acids and modification of parkinsonism. N Engl J Med 276(7):374–379
Fox SH, Katzenschlager R, Lim SY, Ravina B, Seppi K, Coelho M et al (2011) The Movement Disorder Society evidence-based medicine review update: treatments for the motor symptoms of Parkinson's disease. Mov Disord 26(Suppl 3):S2–41
Fisher BE, Li Q, Nacca A, Salem GJ, Song J, Yip J et al (2013) Treadmill exercise elevates striatal dopamine D2 receptor binding potential in patients with early Parkinson's disease. Neuroreport 24(10):509–514
Petzinger GM, Walsh JP, Akopian G, Hogg E, Abernathy A, Arevalo P et al (2007) Effects of treadmill exercise on dopaminergic transmission in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse model of basal ganglia injury. J Neurosci Off J Soc Neurosci 27(20):5291–5300
Crizzle AM, Newhouse IJ (2006) Is physical exercise beneficial for persons with Parkinson's disease? Clin J Sport Med 16(5):422–425
Muller T, Muhlack S (2010) Effect of exercise on reactivity and motor behaviour in patients with Parkinson's disease. J Neurol Neurosurg Psychiatry 81(7):747–753
Corcos DM, Robichaud JA, David FJ, Leurgans SE, Vaillancourt DE, Poon C et al (2013) A two-year randomized controlled trial of progressive resistance exercise for Parkinson's disease. Mov Disord 28(9):1230–1240
Wu PL, Lee M, Huang TT (2017) Effectiveness of physical activity on patients with depression and Parkinson's disease: a systematic review. PLoS One 12(7):e0181515
Ridgel AL, Peacock CA, Fickes EJ, Kim CH (2012) Active-assisted cycling improves tremor and bradykinesia in Parkinson's disease. Arch Phys Med Rehabil 93(11):2049–2054
Uygur M, Bellumori M, LeNoir K, Poole K, Pretzer-Aboff I, Knight CA (2015) Immediate effects of high-speed cycling intervals on bradykinesia in Parkinson's disease. Physiother Theory Pract 31(2):77–82
Marusiak J, Zeligowska E, Mencel J, Kisiel-Sajewicz K, Majerczak J, Zoladz JA et al (2015) Interval training-induced alleviation of rigidity and hypertonia in patients with Parkinson's disease is accompanied by increased basal serum brain-derived neurotrophic factor. J Rehabil Med 47(4):372–375
Klamroth S, Steib S, Devan S, Pfeifer K (2016) Effects of exercise therapy on postural instability in Parkinson disease: a meta-analysis. J Neurol Phys Ther 40(1):3–14
Hashimoto H, Takabatake S, Miyaguchi H, Nakanishi H, Naitou Y (2015) Effects of dance on motor functions, cognitive functions, and mental symptoms of Parkinson's disease: a quasi-randomized pilot trial. Complement Ther Med 23(2):210–219
Reynolds GO, Otto MW, Ellis TD, Cronin-Golomb A (2016) The therapeutic potential of exercise to improve mood, cognition, and sleep in Parkinson's disease. Mov Disord 31(1):23–38
Meredith GE, Rademacher DJ (2011) MPTP mouse models of Parkinson's disease: an update. J Park Dis 1(1):19–33
Archer T, Fredriksson A (2010) Physical exercise attenuates MPTP-induced deficits in mice. Neurotox Res 18(3–4):313–327
Fredriksson A, Stigsdotter IM, Hurtig A, Ewalds-Kvist B, Archer T (2011) Running wheel activity restores MPTP-induced functional deficits. J Neural Transm 118(3):407–420
Lau YS, Patki G, Das-Panja K, Le WD, Ahmad SO (2011) Neuroprotective effects and mechanisms of exercise in a chronic mouse model of Parkinson's disease with moderate neurodegeneration. Eur J Neurosci 33(7):1264–1274
Aguiar AS Jr, Lopes SC, Tristao FS, Rial D, de Oliveira G, da Cunha C et al (2016) Exercise improves cognitive impairment and dopamine metabolism in MPTP-treated mice. Neurotox Res 29(1):118–125
Churchill MJ, Pflibsen L, Sconce MD, Moore C, Kim K, Meshul CK (2017) Exercise in an animal model of Parkinson's disease: Motor recovery but not restoration of the nigrostriatal pathway. Neuroscience 359:224–247
Fisher BE, Petzinger GM, Nixon K, Hogg E, Bremmer S, Meshul CK et al (2004) Exercise-induced behavioral recovery and neuroplasticity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse basal ganglia. J Neurosci Res 77(3):378–390
Jackson-Lewis V, Przedborski S (2007) Protocol for the MPTP mouse model of Parkinson's disease. Nat Protoc 2(1):141–151
Guillot TS, Asress SA, Richardson JR, Glass JD, Miller GW (2008) Treadmill gait analysis does not detect motor deficits in animal models of Parkinson's disease or amyotrophic lateral sclerosis. J Mot Behav 40(6):568–577
Smith BA, Goldberg NR, Meshul CK (2011) Effects of treadmill exercise on behavioral recovery and neural changes in the substantia nigra and striatum of the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned mouse. Brain Res 1386:70–80
Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP et al (1999) Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci Off J Soc Neurosci 19(8):3248–3257
Klivenyi P, Bende Z, Hartai Z, Penke Z, Nemeth H, Toldi J et al (2006) Behaviour changes in a transgenic model of Huntington's disease. Behav Brain Res 169(1):137–141
Pang TY, Stam NC, Nithianantharajah J, Howard ML, Hannan AJ (2006) Differential effects of voluntary physical exercise on behavioral and brain-derived neurotrophic factor expression deficits in Huntington's disease transgenic mice. Neuroscience 141(2):569–584
Jansen D, Zerbi V, Arnoldussen IA, Wiesmann M, Rijpma A, Fang XT et al (2013) Effects of specific multi-nutrient enriched diets on cerebral metabolism, cognition and neuropathology in AbetaPPswe-PS1dE9 mice. PLoS One 8(9):e75393
Janssen CI, Zerbi V, Mutsaers MP, de Jong BS, Wiesmann M, Arnoldussen IA et al (2015) Impact of dietary n-3 polyunsaturated fatty acids on cognition, motor skills and hippocampal neurogenesis in developing C57BL/6J mice. J Nutr Biochem 26(1):24–35
Carter RJ, Morton J, Dunnett SB (2001) Motor coordination and balance in rodents. Curr Protoc Neurosci 15:8.12:8.12.1–8.12.14
Chung YC, Kim SR, Park JY, Chung ES, Park KW, Won SY et al (2011) Fluoxetine prevents MPTP-induced loss of dopaminergic neurons by inhibiting microglial activation. Neuropharmacology 60(6):963–974
Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates. Academic Press, San Diego
Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T et al (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9(7):676–682
Olde Loohuis NF, Kole K, Glennon JC, Karel P, Van der Borg G, Van Gemert Y et al (2015) Elevated microRNA-181c and microRNA-30d levels in the enlarged amygdala of the valproic acid rat model of autism. Neurobiol Dis 80:42–53
Wu WS, Li WH (2008) Systematic identification of yeast cell cycle transcription factors using multiple data sources. BMC Bioinformatics 9:522
Klemann CJHM, Martens GJM, Poelmans G, Visser JE (2016) Validity of the MPTP-treated mouse as a model for Parkinson's disease. Mol Neurobiol 53(3):1625–1636
Poelmans G, Pauls DL, Buitelaar JK, Franke B (2011) Integrated genome-wide association study findings: identification of a neurodevelopmental network for attention deficit hyperactivity disorder. Am J Psychiatry 168(4):365–377
Poelmans G, Franke B, Pauls DL, Glennon JC, Buitelaar JK (2013) AKAPs integrate genetic findings for autism spectrum disorders. Transl Psychiatry 3:e270
Klemann C, Martens GJM, Sharma M, Martens MB, Isacson O, Gasser T et al (2017) Integrated molecular landscape of Parkinson's disease. NPJ Park Dis 3:14
Wu K, Li S, Bodhinathan K, Meyers C, Chen W, Campbell-Thompson M et al (2012) Enhanced expression of Pctk1, Tcf12 and Ccnd1 in hippocampus of rats: Impact on cognitive function, synaptic plasticity and pathology. Neurobiol Learn Mem 97(1):69–80
UniProt (2015) UniProt: a hub for protein information. Nucleic Acids Res 43(Database issue):D204–D212
Chen CH, Shaikenov T, Peterson TR, Aimbetov R, Bissenbaev AK, Lee SW et al (2011) ER stress inhibits mTORC2 and Akt signaling through GSK-3beta-mediated phosphorylation of rictor. Sci Signal 4(161):ra10
Moore AN, Waxham MN, Dash PK (1996) Neuronal activity increases the phosphorylation of the transcription factor cAMP response element-binding protein (CREB) in rat hippocampus and cortex. J Biol Chem 271(24):14214–14220
Beaumont TL, Yao B, Shah A, Kapatos G, Loeb JA (2012) Layer-specific CREB target gene induction in human neocortical epilepsy. J Neurosci Off J Soc Neurosci 32(41):14389–14401
Rousselet E, Joubert C, Callebert J, Parain K, Tremblay L, Orieux G et al (2003) Behavioral changes are not directly related to striatal monoamine levels, number of nigral neurons, or dose of parkinsonian toxin MPTP in mice. Neurobiol Dis 14(2):218–228
Muthane U, Ramsay KA, Jiang H, Jackson-Lewis V, Donaldson D, Fernando S et al (1994) Differences in nigral neuron number and sensitivity to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine in C57/bl and CD-1 mice. Exp Neurol 126(2):195–204
Guo Z, Xu S, Du N, Liu J, Huang Y, Han M (2016) Neuroprotective effects of stemazole in the MPTP-induced acute model of Parkinson's disease: involvement of the dopamine system. Neurosci Lett 616:152–159
Shin MS, Jeong HY, An DI, Lee HY, Sung YH (2016) Treadmill exercise facilitates synaptic plasticity on dopaminergic neurons and fibers in the mouse model with Parkinson's disease. Neurosci Lett 621:28–33
Schumm S, Sebban C, Cohen-Salmon C, Callebert J, Launay JM, Golmard JL et al (2012) Aging of the dopaminergic system and motor behavior in mice intoxicated with the parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J Neurochem 122(5):1032–1046
Patki G, Che Y, Lau YS (2009) Mitochondrial dysfunction in the striatum of aged chronic mouse model of Parkinson's disease. Front Aging Neurosci 1:3
L'Episcopo F, Tirolo C, Testa N, Caniglia S, Morale MC, Serapide MF et al (2014) Wnt/beta-catenin signaling is required to rescue midbrain dopaminergic progenitors and promote neurorepair in ageing mouse model of Parkinson's disease. Stem Cells 32(8):2147–2163
Hood RL, Liguore WA, Moore C, Pflibsen L, Meshul CK (1646) Exercise intervention increases spontaneous locomotion but fails to attenuate dopaminergic system loss in a progressive MPTP model in aged mice. Brain Res 2016:535–542
Sawada M, Sawada H, Nagatsu T (2008) Effects of aging on neuroprotective and neurotoxic properties of microglia in neurodegenerative diseases. Neurodegener Dis 5(3–4):254–256
Munoz-Manchado AB, Villadiego J, Romo-Madero S, Suarez-Luna N, Bermejo-Navas A, Rodriguez-Gomez JA et al (2016) Chronic and progressive Parkinson's disease MPTP model in adult and aged mice. J Neurochem 136(2):373–387
Jakowec MW, Nixon K, Hogg E, McNeill T, Petzinger GM (2004) Tyrosine hydroxylase and dopamine transporter expression following 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurodegeneration of the mouse nigrostriatal pathway. J Neurosci Res 76(4):539–550
Vila M, Jackson-Lewis V, Vukosavic S, Djaldetti R, Liberatore G, Offen D et al (2001) Bax ablation prevents dopaminergic neurodegeneration in the 1-methyl- 4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson's disease. Proc Natl Acad Sci U S A 98(5):2837–2842
Ridgel AL, Vitek JL, Alberts JL (2009) Forced, not voluntary, exercise improves motor function in Parkinson's disease patients. Neurorehabil Neural Repair 23(6):600–608
Gorton LM, Vuckovic MG, Vertelkina N, Petzinger GM, Jakowec MW, Wood RI (2010) Exercise effects on motor and affective behavior and catecholamine neurochemistry in the MPTP-lesioned mouse. Behav Brain Res 213(2):253–262
Alberts JL, Phillips M, Lowe MJ, Frankemolle A, Thota A, Beall EB et al (2016) Cortical and motor responses to acute forced exercise in Parkinson's disease. Parkinsonism Relat Disord 24:56–62
Shu HF, Yang T, Yu SX, Huang HD, Jiang LL, Gu JW et al (2014) Aerobic exercise for Parkinson's disease: a systematic review and meta-analysis of randomized controlled trials. PLoS One 9(7):e100503
Uc EY, Doerschug KC, Magnotta V, Dawson JD, Thomsen TR, Kline JN et al (2014) Phase I/II randomized trial of aerobic exercise in Parkinson disease in a community setting. Neurology 83(5):413–425
Luchtman DW, Meng Q, Song C (2012) Ethyl-eicosapentaenoate (E-EPA) attenuates motor impairments and inflammation in the MPTP-probenecid mouse model of Parkinson's disease. Behav Brain Res 226(2):386–396
Wang H, Liang X, Wang X, Luo D, Jia J, Wang X (2013) Electro-acupuncture stimulation improves spontaneous locomotor hyperactivity in MPTP intoxicated mice. PLoS One 8(5):e64403
Ferguson SA, Law CD, Sarkar S (2015) Chronic MPTP treatment produces hyperactivity in male mice which is not alleviated by concurrent trehalose treatment. Behav Brain Res 292:68–78
Colotla VA, Flores E, Oscos A, Meneses A, Tapia R (1990) Effects of MPTP on locomotor activity in mice. Neurotoxicol Teratol 12(4):405–407
Chia LG, Ni DR, Cheng FC, Ho YP, Kuo JS (1999) Intrastriatal injection of 5,7-dihydroxytryptamine decreased 5-HT levels in the striatum and suppressed locomotor activity in C57BL/6 mice. Neurochem Res 24(6):719–722
Luchtman DW, Shao D, Song C (2009) Behavior, neurotransmitters and inflammation in three regimens of the MPTP mouse model of Parkinson's disease. Physiol Behav 98(1–2):130–138
Visser JE, Voermans NC, Oude Nijhuis LB, van der Eijk M, Nijk R, Munneke M et al (2007) Quantification of trunk rotations during turning and walking in Parkinson's disease. Clin Neurophysiol 118(7):1602–1606
Tabbal SD, Ushe M, Mink JW, Revilla FJ, Wernle AR, Hong M et al (2008) Unilateral subthalamic nucleus stimulation has a measurable ipsilateral effect on rigidity and bradykinesia in Parkinson disease. Exp Neurol 211(1):234–242
Godoy R, Noble S, Yoon K, Anisman H, Ekker M (2015) Chemogenetic ablation of dopaminergic neurons leads to transient locomotor impairments in zebrafish larvae. J Neurochem 135(2):249–260
Alexander GE (1994) Basal ganglia-thalamocortical circuits: their role in control of movements. J Clin Neurophysiol 11(4):420–431
Herrero MT, Barcia C, Navarro JM (2002) Functional anatomy of thalamus and basal ganglia. Childs Nerv Syst 18(8):386–404
Cools R (2008) Role of dopamine in the motivational and cognitive control of behavior. Neuroscientist 14(4):381–395
Leisman G, Braun-Benjamin O, Melillo R (2014) Cognitive-motor interactions of the basal ganglia in development. Front Syst Neurosci 8:16
Haber SN (2014) The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 282:248–257
Morita H, Hass CJ, Moro E, Sudhyadhom A, Kumar R, Okun MS (2014) Pedunculopontine nucleus stimulation: where are we now and what needs to be done to move the field forward? Front Neurol 5:243
Chaudhuri KR, Schapira AH (2009) Non-motor symptoms of Parkinson's disease: dopaminergic pathophysiology and treatment. Lancet Neurol 8(5):464–474
Wood LD, Neumiller JJ, Setter SM, Dobbins EK (2010) Clinical review of treatment options for select nonmotor symptoms of Parkinson's disease. Am J Geriatr Pharmacother 8(4):294–315
Seppi K, Weintraub D, Coelho M, Perez-Lloret S, Fox SH, Katzenschlager R et al (2011) The Movement Disorder Society evidence-based medicine review update: treatments for the non-motor symptoms of Parkinson's disease. Mov Disord 26(Suppl 3):S42–S80
Kaska S, Brunk R, Bali V, Kechner M, Mazei-Robison MS (2017) Deletion of Rictor in catecholaminergic neurons alters locomotor activity and ingestive behavior. Neuropharmacology 117:158–170
Nozaki T, Sugiyama K, Yagi S, Yoshikawa E, Kanno T, Asakawa T et al (2013) Effect of subthalamic nucleus stimulation during exercise on the mesolimbocortical dopaminergic region in Parkinson's disease: a positron emission tomography study. J Cereb Blood Flow Metab 33(3):415–421
MacDonald PA, MacDonald AA, Seergobin KN, Tamjeedi R, Ganjavi H, Provost JS et al (2011) The effect of dopamine therapy on ventral and dorsal striatum-mediated cognition in Parkinson's disease: support from functional MRI. Brain 134(Pt 5):1447–1463
Yang W, Liu B, Huang B, Huang R, Wang L, Zhang Y et al (2016) Altered resting-state functional connectivity of the striatum in Parkinson's disease after levodopa administration. PLoS One 11(9):e0161935
Karler R, Calder LD, Thai DK, Bedingfield JB (1998) The role of dopamine and GABA in the frontal cortex of mice in modulating a motor-stimulant effect of amphetamine and cocaine. Pharmacol Biochem Behav 60(1):237–244
Matsumoto M, Kanno M, Togashi H, Ueno K, Otani H, Mano Y et al (2003) Involvement of GABAA receptors in the regulation of the prefrontal cortex on dopamine release in the rat dorsolateral striatum. Eur J Pharmacol 482(1–3):177–184
Jiao D, Liu Y, Li X, Liu J, Zhao M (2015) The role of the GABA system in amphetamine-type stimulant use disorders. Front Cell Neurosci 9:162
Bubser M, Feenstra MG, Erdtsieck-Ernste EB, Botterblom MH, Van Uum HF, Pool CW (1997) Modulatory role of catecholamines in the transsynaptic expression of c-fos in the rat medial prefrontal cortex induced by disinhibition of the mediodorsal thalamus: a study employing microdialysis and immunohistochemistry. Brain Res 749(2):214–225
Picconi B, Paille V, Ghiglieri V, Bagetta V, Barone I, Lindgren HS et al (2008) L-DOPA dosage is critically involved in dyskinesia via loss of synaptic depotentiation. Neurobiol Dis 29(2):327–335
Jenner P (2008) Molecular mechanisms of L-DOPA-induced dyskinesia. Nat Rev Neurosci 9(9):665–677
Calabresi P, Di Filippo M, Ghiglieri V, Tambasco N, Picconi B (2010) Levodopa-induced dyskinesias in patients with Parkinson's disease: filling the bench-to-bedside gap. Lancet Neurol 9(11):1106–1117
Gotham AM, Brown RG, Marsden CD (1988) ‘Frontal’ cognitive function in patients with Parkinson's disease 'on' and 'off' levodopa. Brain 111(Pt 2):299–321
Swainson R, Rogers RD, Sahakian BJ, Summers BA, Polkey CE, Robbins TW (2000) Probabilistic learning and reversal deficits in patients with Parkinson's disease or frontal or temporal lobe lesions: possible adverse effects of dopaminergic medication. Neuropsychologia 38(5):596–612
Cools R, Barker RA, Sahakian BJ, Robbins TW (2001) Enhanced or impaired cognitive function in Parkinson's disease as a function of dopaminergic medication and task demands. Cereb Cortex 11(12):1136–1143
Cools R, Lewis SJ, Clark L, Barker RA, Robbins TW (2007) L-DOPA disrupts activity in the nucleus accumbens during reversal learning in Parkinson's disease. Neuropsychopharmacology 32(1):180–189
Cools R, Barker RA, Sahakian BJ, Robbins TW (2003) L-Dopa medication remediates cognitive inflexibility, but increases impulsivity in patients with Parkinson's disease. Neuropsychologia 41(11):1431–1441
Kwak Y, Muller ML, Bohnen NI, Dayalu P, Seidler RD (2012) L-DOPA changes ventral striatum recruitment during motor sequence learning in Parkinson's disease. Behav Brain Res 230(1):116–124
Nombela C, Rittman T, Robbins TW, Rowe JB (2014) Multiple modes of impulsivity in Parkinson's disease. PLoS One 9(1):e85747
Calabresi P, Ghiglieri V, Mazzocchetti P, Corbelli I, Picconi B (2015) Levodopa-induced plasticity: a double-edged sword in Parkinson's disease? Philos Trans R Soc Lond B Biol Sci. 370(1672). pii: 20140184
Kehagia AA, Barker RA, Robbins TW (2013) Cognitive impairment in Parkinson's disease: the dual syndrome hypothesis. Neurodegener Dis 11(2):79–92
Vaillancourt DE, Schonfeld D, Kwak Y, Bohnen NI, Seidler R (2013) Dopamine overdose hypothesis: evidence and clinical implications. Mov Disord 28(14):1920–1929
Vo A, Seergobin KN, Morrow SA, MacDonald PA (2016) Levodopa impairs probabilistic reversal learning in healthy young adults. Psychopharmacology 233(14):2753–2763
Muhlack S, Welnic J, Woitalla D, Muller T (2007) Exercise improves efficacy of levodopa in patients with Parkinson's disease. Move Disord 22(3):427–430
Frazzitta G, Bertotti G, Morelli M, Riboldazzi G, Pelosin E, Balbi P et al (2012) Rehabilitation improves dyskinesias in Parkinsonian patients: a pilot study comparing two different rehabilitative treatments. NeuroRehabilitation 30(4):295–301
Aguiar AS Jr, Moreira EL, Hoeller AA, Oliveira PA, Cordova FM, Glaser V et al (2013) Exercise attenuates levodopa-induced dyskinesia in 6-hydroxydopamine-lesioned mice. Neuroscience 243:46–53
Lee HJ, Baek SM, Ho DH, Suk JE, Cho ED, Lee SJ (2011) Dopamine promotes formation and secretion of non-fibrillar alpha-synuclein oligomers. Exp Mol Med 43(4):216–222
Jang Y, Koo JH, Kwon I, Kang EB, Um HS, Soya H et al (1655) Neuroprotective effects of endurance exercise against neuroinflammation in MPTP-induced Parkinson's disease mice. Brain Res 2017:186–193
Koo JH, Jang YC, Hwang DJ, Um HS, Lee NH, Jung JH et al (2017) Treadmill exercise produces neuroprotective effects in a murine model of Parkinson's disease by regulating the TLR2/MyD88/NF-kappaB signaling pathway. Neuroscience 356:102–113
Koo JH, Cho JY (2017) Treadmill exercise attenuates alpha-synuclein levels by promoting mitochondrial function and autophagy possibly via SIRT1 in the chronic MPTP/P-induced mouse model of Parkinson's disease. Neurotox Res. https://doi.org/10.1007/s12640-017-9770-5
Lima MM, Andersen ML, Reksidler AB, Vital MA, Tufik S (2007) The role of the substantia nigra pars compacta in regulating sleep patterns in rats. PLoS One 2(6):e513
Lima MM (2013) Sleep disturbances in Parkinson's disease: the contribution of dopamine in REM sleep regulation. Sleep Med Rev 17(5):367–375
Tanaka M, Yamaguchi E, Takahashi M, Hashimura K, Shibata T, Nakamura W et al (2012) Effects of age-related dopaminergic neuron loss in the substantia nigra on the circadian rhythms of locomotor activity in mice. Neurosci Res 74(3–4):210–215
Hayashi A, Matsunaga N, Okazaki H, Kakimoto K, Kimura Y, Azuma H et al (2013) A disruption mechanism of the molecular clock in a MPTP mouse model of Parkinson's disease. NeuroMolecular Med 15(2):238–251
Alatriste-Booth V, Rodriguez-Violante M, Camacho-Ordonez A, Cervantes-Arriaga A (2015) Prevalence and correlates of sleep disorders in Parkinson's disease: a polysomnographic study. Arq Neuropsiquiatr 73(3):241–245
Bolitho SJ, Naismith SL, Rajaratnam SM, Grunstein RR, Hodges JR, Terpening Z et al (2014) Disturbances in melatonin secretion and circadian sleep-wake regulation in Parkinson disease. Sleep Med 15(3):342–347
Wolff G, Esser KA (2012) Scheduled exercise phase shifts the circadian clock in skeletal muscle. Med Sci Sports Exerc 44(9):1663–1670
Schroeder AM, Truong D, Loh DH, Jordan MC, Roos KP, Colwell CS (2012) Voluntary scheduled exercise alters diurnal rhythms of behaviour, physiology and gene expression in wild-type and vasoactive intestinal peptide-deficient mice. J Physiol 590(23):6213–6226
Harrington ME (2012) Exercise strengthens circadian clocks. J Physiol 590(23):5929
Videnovic A, Noble C, Reid KJ, Peng J, Turek FW, Marconi A et al (2014) Circadian melatonin rhythm and excessive daytime sleepiness in Parkinson disease. JAMA Neurol 71(4):463–469
Bonaventura J, Rico AJ, Moreno E, Sierra S, Sanchez M, Luquin N et al (2014) L-DOPA-treatment in primates disrupts the expression of A(2A) adenosine-CB(1) cannabinoid-D(2) dopamine receptor heteromers in the caudate nucleus. Neuropharmacology 79:90–100
Pinna A, Bonaventura J, Farre D, Sanchez M, Simola N, Mallol J et al (2014) L-DOPA disrupts adenosine A(2A)-cannabinoid CB(1)-dopamine D(2) receptor heteromer cross-talk in the striatum of hemiparkinsonian rats: biochemical and behavioral studies. Exp Neurol 253:180–191
Bilkei-Gorzo A, Racz I, Michel K, Zimmer A (2002) Diminished anxiety- and depression-related behaviors in mice with selective deletion of the Tac1 gene. J Neurosci Off J Soc Neurosci 22(22):10046–10052
Sparling PB, Giuffrida A, Piomelli D, Rosskopf L, Dietrich A (2003) Exercise activates the endocannabinoid system. Neuroreport 14(17):2209–2211
Tantimonaco M, Ceci R, Sabatini S, Catani MV, Rossi A, Gasperi V et al (2014) Physical activity and the endocannabinoid system: an overview. Cell Mol Life Sci 71(14):2681–2698
Brellenthin AG, Crombie KM, Hillard CJ, Koltyn KF (2016) Endocannabinoid responses to exercise in low, moderate, and high active individuals: 3765 board #204 June 4, 8: 00 AM - 9: 30 AM. Med Sci Sports Exerc 48(5 Suppl 1):1052–1053
Di Filippo M, Picconi B, Tozzi A, Ghiglieri V, Rossi A, Calabresi P (2008) The endocannabinoid system in Parkinson's disease. Curr Pharm Des 14(23):2337–2347
Tozzi A, de Iure A, Di Filippo M, Tantucci M, Costa C, Borsini F et al (2011) The distinct role of medium spiny neurons and cholinergic interneurons in the D(2)/A(2)A receptor interaction in the striatum: Implications for Parkinson's disease. J Neurosci Off J Soc Neurosci 31(5):1850–1862
Farkas S, Nagy K, Jia Z, Harkany T, Palkovits M, Donohou SR et al (2012) The decrease of dopamine D(2)/D(3) receptor densities in the putamen and nucleus caudatus goes parallel with maintained levels of CB(1) cannabinoid receptors in Parkinson's disease: a preliminary autoradiographic study with the selective dopamine D(2)/D(3) antagonist [(3)H]raclopride and the novel CB(1) inverse agonist [(1)(2)(5)I]SD7015. Brain Res Bull 87(6):504–510
Bisogno T, Di Marzo V (2010) Cannabinoid receptors and endocannabinoids: role in neuroinflammatory and neurodegenerative disorders. CNS Neurol Disord Drug Targets 9(5):564–573
Pisani V, Madeo G, Tassone A, Sciamanna G, Maccarrone M, Stanzione P et al (2011) Homeostatic changes of the endocannabinoid system in Parkinson's disease. Mov Disord 26(2):216–222
Nisbet AP, Foster OJ, Kingsbury A, Eve DJ, Daniel SE, Marsden CD et al (1995) Preproenkephalin and preprotachykinin messenger RNA expression in normal human basal ganglia and in Parkinson's disease. Neuroscience 66(2):361–376
Calon F, Birdi S, Rajput AH, Hornykiewicz O, Bedard PJ, Di Paolo T (2002) Increase of preproenkephalin mRNA levels in the putamen of Parkinson disease patients with levodopa-induced dyskinesias. J Neuropathol Exp Neurol 61(2):186–196
Hanrieder J, Ljungdahl A, Falth M, Mammo SE, Bergquist J, Andersson M (2011) L-DOPA-induced dyskinesia is associated with regional increase of striatal dynorphin peptides as elucidated by imaging mass spectrometry. Mol Cell Proteomics 10(10):M111.009308
Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA et al (2007) Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci Off J Soc Neurosci 27(26):6995–7005
Huot P, Johnston TH, Koprich JB, Fox SH, Brotchie JM (2013) The pharmacology of L-DOPA-induced dyskinesia in Parkinson's disease. Pharmacol Rev 65(1):171–222
Wang Y, Zhang QJ, Wang HS, Wang T, Liu J (2014) Genome-wide microarray analysis identifies a potential role for striatal retrograde endocannabinoid signaling in the pathogenesis of experimental L-DOPA-induced dyskinesia. Synapse 68(8):332–343
Berrendero F, Mendizabal V, Robledo P, Galeote L, Bilkei-Gorzo A, Zimmer A et al (2005) Nicotine-induced antinociception, rewarding effects, and physical dependence are decreased in mice lacking the preproenkephalin gene. J Neurosci Off J Soc Neurosci 25(5):1103–1112
Meyer-Lindenberg A, Straub RE, Lipska BK, Verchinski BA, Goldberg T, Callicott JH et al (2007) Genetic evidence implicating DARPP-32 in human frontostriatal structure, function, and cognition. J Clin Invest 117(3):672–682
Kolsch H, Wagner M, Bilkei-Gorzo A, Toliat MR, Pentzek M, Fuchs A et al (2009) Gene polymorphisms in prodynorphin (PDYN) are associated with episodic memory in the elderly. J Neural Transm 116(7):897–903
Frank MJ, Fossella JA (2011) Neurogenetics and pharmacology of learning, motivation, and cognition. Neuropsychopharmacology 36(1):133–152
Zanettini C, Panlilio LV, Alicki M, Goldberg SR, Haller J, Yasar S (2011) Effects of endocannabinoid system modulation on cognitive and emotional behavior. Front Behav Neurosci 5:57
Votinov M, Pripfl J, Windischberger C, Moser E, Sailer U, Lamm C (2015) A functional polymorphism in the prodynorphin gene affects cognitive flexibility and brain activation during reversal learning. Front Behav Neurosci 9:172
Voorn P, Vanderschuren LJ, Groenewegen HJ, Robbins TW, Pennartz CM (2004) Putting a spin on the dorsal-ventral divide of the striatum. Trends Neurosci 27(8):468–474
Haber SN (2016) Corticostriatal circuitry. Dialogues Clin Neurosci 18(1):7–21
Funding
Author J.E. Visser was supported by Stichting Parkinsonfonds, The Netherlands Organization for Scientific Research (NWO/ZonMw, VENI 916.12.167) and The Netherlands Brain Foundation (F2014(1)-16).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
All animal experiments were approved by the Animal Care Committee of the Radboud University Nijmegen Medical Center, The Netherlands, and performed according to the guidelines of the Dutch Council for Animal Care and the European Communities Council Directive 2010/63/EU.
Conflict of Interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
Punching locations per brain area. For each brain area—PFC (A), DL (B), VM (C), SN (D), VTA (E), and PPN (F)—the punching locations are visualized in a cross section adapted from the Paxinos mouse brain atlas [35]. Punching locations with a punching needle of 0.5 mm are shown with red circles, and blue circles indicate the location with 0.75-mm punch needles. (TIFF 2554 kb)
ESM 2
Optical density of fibers in the DL. The upper panel shows a representative picture for each of the four treatment groups and the lower panel shows the optic density in the DL per treatment group. *p < 0.05, means ± SEM, n = 5 for CNR, and MR and n = 4 for CR and MNR. CC corpus callosum, DL dorsolateral striatum. (TIFF 9852 kb)
ESM 3
Optical density of fibers in the VM. The upper panel shows a representative picture for each of the four treatment groups and the lower panel shows the optic density in the VM per treatment group. Means ± SEM, n = 5 for CNR and MR, and n = 4 for CR and MNR. AC anterior commissure, VM ventromedial striatum. (TIFF 9563 kb)
ESM 4
Validation of the RNAseq data using qPCR. The fold change of each mRNA in the RNAseq dataset and the brain area (PFC, DL, VM, SN, VTA) is shown above each graph, with the expression levels as measured by qPCR shown underneath. Expression levels are normalized to ACTB and YWHAZ and shown in arbitrary units (AU). Mean + SEM. The p values are indicated in each graph (Student’s t test). (PDF 1160 kb)
ESM 5
The effect of physical exercise on MPTP-mediated genes. Per brain area, the number of differentially expressed genes due to MPTP alone (in blue) or due to exercise in MPTP-treated mice (in gray), and their overlap are shown. The chance of observing this overlap is calculated with the hypergeometric distribution test and shown next to the overlapping area. Below the blue and gray circles, the total number of unique genes detected by RNAseq for each brain area is shown and also the number and percentage of overlapping genes that is regulated in opposite direction by MPTP and exercise in MPTP-treated mice. (TIFF 313 kb)
ESM 6
mRNAs differentially expressed in the DL due to physical exercise in MPTP-treated mice and regulated by RICTOR. The expression of the purple mRNAs is decreased by both physical exercise and RICTOR. The expression of orange mRNAs is increased by physical exercise and decreased by RICTOR. (TIFF 1309 kb)
ESM 7
mRNAs differentially expressed in the VTA due to physical exercise in MPTP-treated mice and regulated by RICTOR. The expression of the purple mRNAs is decreased by both physical exercise and RICTOR. (TIFF 1511 kb)
ESM 8
mRNAs differentially expressed in the PFC due to physical exercise in MPTP-treated mice and regulated by bicuculline/dalfampridine. mRNAs differentially expressed in the PFC due to physical exercise in MPTP-treated mice are shown in gray. Blue proteins are additional genes/proteins that are associated with PD through genetic and/or expression studies, whereas white proteins have no known link with PD. The direction of effect of physical exercise (measured) and bicuculline/dalfampridine (from literature) on the expression of these mRNAs is depicted through colored borders. Bicuculline-regulated proteins are shown with purple writing for the protein name, and familial PD proteins are shown with a blue border. (TIFF 1075 kb)
ESM 9
mRNAs differentially expressed in the PPN due to physical exercise in MPTP-treated mice and regulated by CREB1. mRNAs differentially expressed in the PPN due to physical exercise in MPTP-treated mice are shown in gray. Blue proteins are additional genes/proteins that are associated with PD through genetic and/or expression studies, whereas white proteins have no known link with PD. The direction of effect of physical exercise (measured) on the expression of these mRNAs is depicted through orange (increase) or purple (decrease) borders. Familial PD proteins are shown with a blue border. (TIFF 1180 kb)
ESM 10
Supplementary Tables 1–14, the detailed landscape descriptions, the references, and a list of abbreviations. (PDF 1852 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Klemann, C.J.H.M., Xicoy, H., Poelmans, G. et al. Physical Exercise Modulates L-DOPA-Regulated Molecular Pathways in the MPTP Mouse Model of Parkinson’s Disease. Mol Neurobiol 55, 5639–5657 (2018). https://doi.org/10.1007/s12035-017-0775-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0775-0