
Abstract
Autoantibodies represent a hallmark of rheumatoid arthritis (RA), with the rheumatoid factor (RF) and antibodies against citrullinated proteins (ACPA) being the most acknowledged ones. RA patients who are positive for RF and/or ACPA (“seropositive”) in general display a different etiology and disease course compared to so-called “seronegative” patients. Still, the seronegative patient population is very heterogeneous and not well characterized. Due to the identification of new autoantibodies and advancements in the diagnosis of rheumatic diseases in the last years, the group of seronegative patients is constantly shrinking. Aside from antibodies towards various post-translational modifications, recent studies describe autoantibodies targeting some native proteins, further broadening the spectrum of recognized antigens. Next to the detection of new autoantibody groups, much research has been done to answer the question if and how autoantibodies contribute to the pathogenesis of RA. Since autoantibodies can be detected years prior to RA onset, it is a matter of debate whether their presence alone is sufficient to trigger the disease. Nevertheless, there is gathering evidence of direct autoantibody effector functions, such as stimulation of osteoclastogenesis and synovial fibroblast migration in in vitro experiments. In addition, autoantibody positive patients display a worse clinical course and stronger radiographic progression. In this review, we discuss current findings regarding different autoantibody types, the underlying disease-driving mechanisms, the role of Fab and Fc glycosylation and clinical implications.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory joint disease, which is considered to be of predominantly autoimmune origin. The disease was first described in 1800 by Dr. Landré-Beauvais and was initially regarded as a form of gout. Only in the mid-nineteenth century was it distinguished from actual gout based on the uric acid measurement [1, 2]. RA is characterized by a progressive course, which in the absence of treatment is ultimately leading to pannus formation and joint destruction. Since its first description, the understanding of RA pathophysiology has evolved immensely. As is the case with most rheumatic diseases, the pathogenesis of RA is very complex and influenced by genetic and environmental factors, microbiota, barrier layers, and hormones. The main event in the pathogenesis is still considered to be the breach of tolerance followed by one or multiple “second hits” [3].
Although autoantibodies in RA are a topic of extensive research, the understanding of their precise role in RA development is still far from being complete. In the last decades, many self-antibodies have been identified as hallmarks of RA, starting with rheumatoid factor (RF) first described in 1957 in the sera of RA patients [4]. Although RF has proven to be not specific enough for RA and is present in a variety of other conditions, its discovery was the first step towards recognition of the important role played by autoimmunity in the pathogenesis of arthritis. In 1964 the so-called antiperinuclear factor was described in the serum of patients with RA and several other diseases, including systemic lupus erythematosus and ankylosing spondylitis [5, 6]. The nature of these antibodies was not perceived, and until 1978, when keratin-specific antibodies were described, there were no major discoveries in the field of autoantibodies in RA. Finally, in the 1990s, the crucial understanding of citrullination and its importance for autoimmunity in RA was acknowledged [7]. The success of B-cell-depleting agents in RA, which was first observed with rituximab approved for the treatment of RA in 2007, further confirmed the driving role of adaptive immunity in the pathogenesis of the disease.
Despite the generally accepted concept of RA as a disease of autoimmune origin, in the regular clinical practice, there is a significant proportion of patients (up to 40% [8]) classified as “seronegative RA” — assuming they do not possess autoantibodies. Seronegativity is usually defined by the absence of anti-citrullinated protein antibodies (ACPA) and/or IgM-RF. However, research in recent years has also shown that the presence of fine-specificities [9] or newly discovered autoantibodies [10], as well as re-diagnosis to other rheumatic diseases [11] are making this group highly heterogenic and its place in the classification of musculoskeletal diseases remains to be clarified.
The autoantibody response in RA is an intensively researched field of high importance and interest. In this review, we discuss the latest findings concerning autoantibodies in RA, their clinical significance and the current consensus on the place of autoantibodies in the pathogenetic cascade.
Various Autoantibodies in Patients with RA
Sensitivity and specificity of various autoantibodies identified in RA are displayed in Table 1.
ACPA
The concept of ACPA was born when citrullinated epitopes were discovered to be “the common ground” of recognition by both the “perinuclear factor” and anti-keratin antibodies. It was noticed that these antibodies both reacted with fillagrin but not with pro-fillagrin, and it was hypothesized that a posttranslational modification could take place [7, 12]. Schellekens et al. also noticed already at that time that ACPA could be detected very early in the disease course [12]. Since then, extensive research has been conducted on the role and diagnostic importance of ACPA. ACPA display high sensitivity (60–78%) and specificity (86–99%) for RA [5, 13]. Today, various ACPA tests are available (reviewed in detail in [5]). Currently, the most widely used test for the measurement of ACPA is the cyclic citrullinated peptide (CCP) 2 test, first introduced in 2000. In the CCP3 test, additional citrullinated epitopes are introduced to the same protein structure. Both tests are similar in their performance [5] and the additional advantage of the CCP3 test (which is also more expensive than the standard CCP2) is not completely apparent. However, CCP3.1 showed a slightly higher specificity compared to CCP2 and anti-modified citrullinated vimentin (MCV) tests [13], and CCP3-positivity was shown to add predictive value to the CCP2 test for the development of RA in individuals-at-risk [14]. The anti-MCV antibody test is also universally used and has comparable sensitivity and specificity profiles to CCP tests.
In the process of citrullination, peptidyl arginine is being transformed to citrulline by the calcium-dependent enzyme peptidylarginine deiminase (PAD). There are multiple forms of PADs, which differ in their localization and function. PAD2 and PAD4 play a prominent role in RA not only due to direct production of citrullinated antigens but also by contributing to neutrophil extracellular trap (NET) formation via citrullination of nuclear proteins [15, 16].
ACPA recognize citrullinated parts of various targets and are therefore somewhat “promiscuous” in their recognition profile, binding to proteins like vimentin, α-enolase, type II collagen (CII), fibrinogen, fillagrin, fibronectin, immunoglobulin-binding protein (BiP), and potentially various others, provided they contain accessible citrullinated epitopes [5, 17,18,19,20,21]. Consequently, oftentimes these antibodies are showing extreme cross-reactivity. In case of collagen II (CII), it seems that citrullination is crucial for the recognition of epitopes by autoantibodies in humans, in contrast to mouse antibodies in the collagen-induced arthritis model, where citrullination prevents the binding to CII [22, 23].
New epitopes for the recognition by ACPA are being constantly discovered and antigen arrays are getting more attention as a means to investigate the complete “citrullinome” and to identify other potential targets [10, 24, 25]. It has been demonstrated in the last years that glycine motifs next to the cit-position are essential for ACPA binding [24, 26,27,28]. Hydrogen bonds are responsible for the interaction of complementarity-determining regions with the antigen main chain and ACPA show limited interaction with protein side chains. Both factors are considered to be the basis for their wide cross-reactivity [27]. However, a specific three-dimensional fold structure of proteins also interferes with binding, and the cross-reactivity has therefore its limits.
The cross-reactivity profile is not uniform and can differ between ACPA, as was shown for monoclonal antibodies and polyclonal ACPA from the serum of RA patients [26, 27, 29, 30]. The concept of “promiscuous” and “private” ACPA was suggested recently based on studies demonstrating the presence of more selective (“private”) antibodies alongside classical cross-reactive (“promiscuous”) ACPA [19]. It has been shown for instance that some ACPA recognize not both, but only either one of citrullinated residues in CII. Interestingly, only non-cross-reactive antibodies were shown to bind to cartilage proteins [22].
AMPA
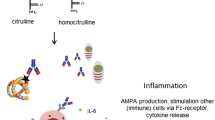
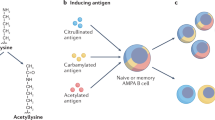
In the recent years, autoantibodies targeting various other posttranslational modifications have been discovered. While citrulline is a product of enzymatic conversion of arginine, other modifications happen mostly to lysine residues. This amino acid can be transformed non-enzymatically into homocitrulline via addition of isocyanic groups (process known as carbamylation) or into malondialdehyde (MDA)-acetaldehyde (MAA)-lysine. MDA is a product of lipid peroxidation by reactive oxygen species and can be attached to a variety of aminoacidic residues, such as glutamine, asparagine, arginine, and histidine [31]. Another frequent enzymatic modification of lysine is acetylation. It has been demonstrated that ACPA are cross-reactive not only towards various citrullinated peptides, but also towards other posttranslational modifications [28, 32,33,34,35]. Therefore, the concept of anti-modified protein antibodies (AMPA) was suggested. AMPA are extensively reviewed elsewhere [34, 36]; hence, here we focus only on the milestones and most recent findings.
It is worth mentioning that AMPA are present mostly in ACPA-positive patients, suggesting a cross-reactive nature. Despite that, the recognition profile of anti-carbamylated protein antibodies (anti-CarP) for example only partially overlaps with that of ACPA [32,33,34, 37, 38]. It is noteworthy that homocitrulline, the product of carbamylation, highly resembles citrulline and only contains one additional CH2 group, which might be partially responsible for the cross-reactivity. Another reason is suggested to be the Cit-Gly “consensus motif” [26, 39]. Interestingly, isocyanic acid for the process of carbamylation can be generated by myeloperoxidase from thiocyanate, which is abundant in the blood of smokers [40]. This might provide an additional link between the risk of RA and smoking. Importantly, anti-CarP antibodies were found to be associated with interstitial lung disease (ILD) in RA patients even after adjusting for other factors, including smoking; though anti-CarP were more prevalent among smokers [41].
Similarly, anti-MAA antibodies are present mostly in ACPA-positive patients as well. The expression of MAA-modified-proteins is found in the synovium of RA patients, but not in osteoarthritis (OA) patients, and co-localizes with citrullinated proteins [31, 42]. However, the data on MAA cross-reactivity with citrullinated proteins are controversial. In another study, monoclonal anti-MAA antibodies did not cross-react with either CCP, carbamylated, or 4-hydroxynonenal (HNE)-carbonylated proteins [43]. Interestingly, just like ACPA, anti-MAA can bind various peptides containing MAA modification. Similarly to carbamylation, MAA overexpression was identified in the lung tissue of RA patients with ILD, and anti-MAA antibodies were associated with ILD in these patients [44].
Anti-acetylated protein antibodies are mostly reserved to ACPA-positive patients as well [45, 46]. Anti-acetylated ornithine antibodies slightly improved the specificity of EULAR/ACR 2010 diagnostic criteria in one study, but not in case of seronegative (ACPA- and RF-negative) RA [46]. By contrast, the addition of anti-CarP antibodies to the classification criteria was favoring sensitivity, while the specificity was reduced; although both effects were very minor and not cost-effective [47].
There is still lack in comprehensive understanding of how AMPA responses develop. Recent studies also show that the process is most likely to be highly individual and heterogeneous, with various monoclonal ACPA showing different specificity profiles and exhibiting distinct effector functions [28, 39].
RF
The discovery of RF served as a first evidence of autoimmunity in RA. It encompasses immunoglobulins of IgM, IgG, and IgA classes targeting the Fc-fragment of IgG. However, RF can be present in a variety of other rheumatologic and non-rheumatologic conditions, including a broad spectrum of infectious diseases — most importantly hepatitis C-virus infection and cryoglobulinemia [48], as well as cancer [49,50,51,52]. The specificity of RF in RA is estimated to be 85% at best, and the sensitivity to be in the range between 60 and 90% [52]. Compared to ACPA, RF is slightly more sensitive; however, it certainly lacks specificity. Still, both tests are included in the current EULAR/ACR diagnostic criteria. As will be mentioned later in one of the sections, RF of IgA class is probably playing a more significant role compared to other immunoglobulin classes.
Antibodies to Native Proteins
Recently, several novel candidates unrelated to posttranslational modifications were suggested as promising markers in RA. The concept of seronegative RA is problematic, both from diagnostic and pathogenetic perspectives. Some novel candidate autoantibodies present in this subset of patients have been suggested, including anti-pentraxin 3 (PTX3) and anti-dual specificity phosphatase 11 (DUSP11) antibodies. These antibodies were present in about 30–40% of both ACPA-positive and ACPA-negative patients in one study [10]. In the same study, additional candidates were suggested. However, these novel antibodies are lacking sensitivity (which was below 30%). Moreover, in another study, anti-pentraxin-related protein 3 (PTX3) antibodies were found in about 50% of systemic lupus erythematosus patients and in none of RA patients [53]. These antibodies have also been reported in anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (about 40% of patients in one cohort were positive) [54]. Therefore, the data on the specificity of these antibodies for RA need to be considered with caution.
In another array-based study, antibodies against a total of 102 native proteins (76 in ACPA-negative RA patients and 86 recognized by the serum of ACPA-positive patients) were found to be specific for RA [55]. Some of the antigens were found in synovial fluid [56], but this still needs to be validated in other studies.
The better known and studied anti-PAD-antibodies are slightly more sensitive (20–70%) and specific (77–97%) for RA compared to other antibodies against native proteins [15]. These antibodies are mostly associated with ACPA but can also be identified in up to 19% of ACPA-negative RA patients [15].
Considering all this, it seems that signs of autoimmunity can be found in at least a subset of “seronegative” patients. The autoantibody spectrum might be broader than we currently know and is likely not reserved to citrullinated proteins. However, it is hard to implicate these findings into the daily clinical practice, since the benefit of testing for other autoantibodies remains to be proven.
Factors Contributing to Autoantibody Production in RA
The presence of ACPA is highly associated with the human leukocyte antigen (HLA)-shared epitope (SE) — the susceptibility loci encoding a particular amino acid sequence in the third hypervariable region in the HLA-DR beta-chain 1 (HLA-DR4, HLA-DR1, or HLA-DR10) [57, 58]. HLA shared epitope is linked to both the risk of developing RA and the risk of having ACPA-positive RA, but not to the risk of having ACPA alone [59], which indicates its role in both ACPA production and the effector phase of arthritis. There also seems to be a ‘gene-dose’-dependent effect, with patients harboring two alleles or HLA-DRB1.04 having higher ACPA titers [57, 58].
Since the role of HLA is to present antigens to T cells and to aid B cells in getting T cell-help, it was logically proposed that SE alleles are contributing to the risk of having ACPA and arthritis due to a higher ability of the MHC to bind citrullinated epitopes. However, this hypothesis, though very plausible, has its flaws. While the higher binding affinity of citrullinated vimentin as opposed to its native form has been shown for three HLA-SE alleles (HLA-DRB1*01:01, *04:01, and *04:04) [60, 61], at the same time, citrullination of fibrinogen was demonstrated to have no influence on its binding to HLA, and there were no differences in T cell proliferation upon stimulation with citrullinated or non-citrullinated proteins in HLA-DRB1* patients [62]. Overall, the data on this topic are controversial [57]. Moreover, a novel hypothesis, which has been supported by some recent studies, has emerged. It is suggested that the HLA-DR alleles might contribute to preferable binding of PAD4 peptides, and not citrullinated or native fibrinogen [63, 64]. The authors propose an intriguing hypothesis that PAD is serving as a hapten for citrullinated proteins, evoking T cell immune response and triggering the production of ACPA. This is, however, somewhat challenged by the evidence of ACPA response usually preceding the anti-PAD-antibody response [65].
Additional susceptibility gene variants for ACPA-positivity, such as in PTPN22 [66] and in HLA-DQ and HLA-DP [67] have been described.
Other well-known risk factors for ACPA-positivity are environmental stressors. Smoking is estimated to dose-dependently contribute to at least 35% of ACPA-positive RA cases and is associated synergistically with the HLA-DRB1 shared epitope [68, 69]. In a large cohort of ACPA-positive individuals at-risk and ACPA-positive RA patients, smoking has been found to be associated with both ACPA- and anti-carbamylated protein antibody (anti-CarP)-positivity [70]. Interestingly, the presence of secretory IgA ACPA in serum of RA patients was found to be associated with smoking [71]. Smoking is considered to contribute to ACPA production via excessive citrullination in lung tissue. This is backed up by immunohistochemistry showing citrullinated proteins and increased PAD expression in the bronchoalveolar lavage from smokers [67, 72]. If an individual in addition has one or more risk-alleles, which provide the needed T-cell help, this combination is considered to increase the risk of RA up to 21-fold [73].
Periodontitis is another suggested risk factor for autoantibody production in RA and individuals at-risk. First, the incidence of RA was found to be higher among patients referred for the treatment of periodontal disease than in the general population [74]. Periodontitis was more frequently found among RA patients in some studies [75, 76] and meta-analysis [77], while equal prevalence among RA and non-RA individuals was shown in others [77, 78]. These associations, if found, were also correlating with ACPA-positivity. Likewise, ACPA-positivity was found to be associated with the periodontal health in first-degree relatives of RA patients, with a higher prevalence of periodontitis if ACPA-positive [79]. Higher titers of anti-Porphyromonas gingivalis antibodies were also found in ACPA- or RF-positive subjects at-risk for RA [80]. P. gingivalis produces PAD, which could account for its possible contribution to citrullination and subsequent autoantibody production in the context of RA [77, 78]. Though data on the topic are controversial, mostly, the results point towards a possible role of periodontal disease in autoantibody production. This topic is discussed in more detail in other reviews, like [81].
Finally, the so-called leukotoxic hypercitrullination via the pore-forming leukotoxin A (LtxA) has been described for neutrophils in Aggregatibacter actinomycetemcomitans (Aa) infection. Antibodies to both Aa and LtxA were present in patients with RA and periodontitis [82]. Recently, the presence of anti-Aa- and anti-LtxA-antibodies was found to be associated with the measures of atherosclerosis in patients with RA in one study, suggesting periodontal disease prevention to be important, though these findings do not allow to conclude if these associations are specific for RA, since the study did not include a non-RA control group [83].
Pathogenetic Role of Autoantibodies in RA
As ACPA are the main autoantibodies in RA, there has been interest in identifying their target antigens in the RA synovium. One study found a 1.4-fold increase in the proportion of ACPA in total IgG, IgA, and IgM in the RA synovial fluid compared to serum [84] and a 7.5-fold increase of IgG in synovial lining compared to serum [85]. Interestingly, immunohistological staining with citrulline-specific antibodies, including CCP1-specific antibodies isolated from RA serum, showed the presence of citrullinated proteins not only in RA synovium, but also in the inflamed synovium from patients with other inflammatory joint diseases [84]. Other studies identified citrullinated proteins in a wide spectrum of inflamed tissues, but only in a minority of healthy specimens, confirming the notion that citrullination is the result of an ongoing inflammation [86, 87]. Synovial tissue is infiltrated by neutrophils that might serve as major contributors to citrullination through the release of PAD4 [88]. In a more recent study, authors found higher levels of citrulline and homocitrulline in the synovial tissues of seropositive RA patients than in seronegative RA and OA (citrulline = 0,019 µg/mg in seropositive RA versus 0,007 µg/ml in both OA and seronegative RA) [87]. In the same study, the majority of citrullinated and homocitrullinated proteins were recognized by a synthetic antibody in the area of necrotic tissue in the synovium and rheumatic nodules, as well as in the synovial lining and endothelium. The expression of PAD2, PAD3, and PAD4 could be localized in the synovial lining and the expression of myeloperoxidase was identified around necrotic areas [87, 89].
The notion that ACPA could have a direct pathogenetic role has its back up in the clinical setting. ACPA-positivity is associated with bone loss in individuals at-risk for RA [90] and in the early disease [72, 91]. Overall, seropositive RA is characterized by a more aggressive inflammation and destruction compared to seronegative RA [72, 87, 92, 93], though one study showed contrasting results [94]. The presence of autoantibodies has been associated with bone loss and erosion formation (radiographic progression) [90, 95,96,97]. Interestingly, newborns of women with RA, although having no long-term developmental consequences, were shown to be smaller for their gestational age [98]. On the other hand, not every ACPA-positive individual progresses to RA even after several years. In a study by Hensvold et al., only 8.5% (21 of 247) of ACPA-positive individuals from a large prospective twin cohort developed RA in 3 years of follow-up [99]. In some RA patients, ACPA can be detected up to 10 years prior to diagnosis [100]. These observations challenge the concept of the pathogenicity of autoantibodies in RA.
Still, there is gathering evidence that ACPA might be exhibiting direct effector functions in RA. Studies have demonstrated ACPA to stimulate bone resorption through enhanced osteoclast differentiation, while in addition inducing TNF-α production by osteoclast precursors [101]. In a recent study, the presence of ACPA was associated with the elevation of osteoclast activation and bone resorption markers like tartrate-resistant acid phosphatase 5b (TRAP5), cathepsin K and C-terminal telopeptide of type I collagen (CTX-I) in the bone marrow of RA patients [102].
ACPA were also shown to stimulate cytokine production, specifically of TNF-α by macrophages via Fcγ receptors in vitro, and this effect was amplified by the presence of IgM RF through its conjugation with IgG [103]. Recently, Dong et al. showed that ACPA are able to induce the production of interleukin (IL)-1β by macrophages in vitro via NLRP3 inflammasome-activation through CD147, integrin β1 and Akt/NFkB signaling [104].
In another recent study, ACPA have been shown to promote migration of fibroblast-like synoviocytes (FLS) under starving conditions and upon inflammatory stimuli, such as IL-8 [105]. The effects of ACPA on FLS were elicited via their binding to citrullinated residues and were dependent on the expression of PAD-2 and PAD-4 in FLS.
A potential direct involvement of ACPA in joint pain pattern is controversially discussed. It has been demonstrated with monoclonal ACPA; however, there was a following correction, since the monoclonal antibodies used in the study were proven to be unspecific. Still, those antibodies were also showing effector functions, which raises another interesting question of whether the properties of autoantibodies are dependent on their specificity profile or are exhibited through Fc-receptors, irrespective of the Fab-recognition [106]. ACPA-positivity was not associated with pain at the time of diagnosis in a Swedish cohort of newly diagnosed RA patients [72]. On the other hand, antibodies targeting cartilage proteins (collagen II and cartilage oligomeric matrix protein) were shown to induce pain in mice before the onset of inflammation in a neuronal Fcγ-receptor-dependent manner [107].
There is limited evidence on whether mice spontaneously develop ACPA upon the induction of experimental arthritis. This might be strain-dependent [108,109,110]. In addition, ACPA alone are usually not sufficient to induce arthritis in mice [22, 110]. However, upon immunization with citrullinated collagen II without adjuvant, mice developed ACPA, which was associated with increased bone resorption and cartilage loss [109]. Additionally, pre-immunization with citrullinated antigen BiP has been shown to exacerbate experimental arthritis [21].
Influence of Glycosylation on the Properties of Autoantibodies in RA
Glycosylation has been shown to considerably modulate effector functions of immunoglobulins. Particularly important is the presence of sialic acid at the glycan attached to asparagine at the position 297 — known as sialylation. It has been shown to skew the functional profile of the antibodies towards anti-inflammatory [111]. Desialylated immune complexes were shown to be potent inducers of osteoclastogenesis and treatment with the sialic acid precursor N-acetylmannosamine (ManNAc) significantly reduced the susceptibility of mice to CIA [112, 113]. It was also observed that patients with less sialylated IgG have decreased bone volume. However, in the context of strong inflammation, the role of glycosylation in the osteoclastic activity was much less significant than under physiological conditions [113].
It has been shown that peripheral blood plasmablasts of ACPA-positive RA patients have much less sialic acid residues on their surface compared to plasmablasts from healthy individuals. The same was observed for IgG [114]. Moreover, it has been demonstrated that IgG from RA patients acts less inflammatory and its pro-inflammatory potential could be boosted upon pre-treatment with the sialic acid cleaving enzyme neuraminidase. Importantly, as was shown in mouse models, the expression of the sialylating enzyme ST6GAL1 in antibody-secreting cells was impaired by cytokines from the IL-23-axis and this effect was reserved to autoantibodies [114]. In line with this study, the ACPA glycosylation profile has been shown to differ from that of total IgG in serum, as well as in synovial fluid [115].
Another important feature of ACPA is their Fab-glycosylation. This non-conserved glycosylation has been shown to negatively influence the avidity profile of ACPA and seems to be the result of the introduction of new N-glycosylation sites during the extensive somatic hypermutation process [116, 117]. In a recent study, the importance of Fab-glycosylation for the recognition of NET-antigens and citrullinated histones was demonstrated [118]. Interestingly, IgM ACPA do not appear to have this characteristic Fab-glycosylation [119]. Most recently, Fab-glycosylation was proposed to play a role in the affinity maturation and clonal selection of ACPA [120]. Interestingly, ACPA of lower avidity profile were associated with more advanced joint damage [121]. Common features of ACPA, such as their cross-reactivity, recognition of post-translational modifications and relatively low avidity (Kd predominantly in the micromolar range), are contradicting the evidence of extensive somatic hypermutations. The conflict between the extensive process of selection and the low-affinity output raises the question of how ACPA-expressing B cells avoid the survival requirement for higher affinity. Vergroesen et al. proposed that the favoring of N-glycosylation sites is required for the survival of autoreactive B cell clones. This notion is supported by their discovery that the number of N-glycosylation sites in ACPA-reactive B cells did not correlate with mutation frequency and hence is unlikely to be merely the consequence of the high ACPA mutation rate [120].
Clinical Relevance of Autoantibodies in RA
The prevalence of ACPA in the general population is estimated to be around 1% and is increased with older age, in smokers and individuals with joint complaints [122].
Multiple autoantibodies, including ACPA, anti-PAD- and anti-CarP-antibodies, precede the onset of RA [65, 123, 124], with ACPA still rendering the highest risk [125] and appearing before clinically apparent synovitis [126]. However, over 30% of ACPA- and RF-positive individuals with arthralgia do not develop RA within 2 years of follow-up [125], with the level of ACPA seeming to be of importance. In a recent study, 46% of at-risk persons with high ACPA levels developed RA in a 5 year-period, compared to only 21.5% among all ACPA-positive individuals [127]. Similarly, in another study, 32% of ACPA-positive first-degree relatives of RA patients with the values ≥ 2 cutoffs developed RA in a 5-year period, compared to 26% if the value was just above the cutoff [128].
The positive predictive value (PPV) of ACPA tests, which demonstrates the likelihood of an individual with a positive test to have or develop a condition, varies in different studied populations – from 9% in the general population to 55% when both environmental and genetic risk factors are present, 52% in clinically suspect arthralgia (CSA) and >80% in undifferentiated arthritis patients [129]. Multivariable analysis in one study showed ACPA-positivity to be an independent risk factor for RA development in individuals with CSA (hazard ratio (HR) = 5.1; 2.0–13.2) and to have a 50–67% PPV (RF-negative/RF-positive) for RA development within 2 years [125]. Anti-CarP-antibodies also showed an increased HR for RA of 3.9 (1.9–7.7) in univariable analysis [125]. PPVs vary between different studies due to the ambiguous criteria for CSA, the length of the observational follow-up period they have been calculated for, and other potential confounding factors. However, it can be concluded that ACPA and other autoantibodies mostly precede the arthritis phase and have quite a high predictive value in individuals with joint complaints.
During the timeline before the onset of arthritis, such events as epitope spreading [25, 124], Fc-glycosylation changes of IgG ACPA towards a more pro-inflammatory phenotype [130] and expansion of ACPA immunoglobulin isotype repertoire [131] are taking place.
ACPA and RF are included in the EULAR/ACR 2010 classification criteria for RA due to their specificity and high prevalence in patients. On the other hand, there is no advice for using autoantibody monitoring during treatment since their levels usually do not correlate with disease activity. Nevertheless, studies suggest that the presence of multiple AMPA and/or autoantibody isotypes is associated with significantly lower success rates of achieving drug-free remission [132, 133] but might be associated with better early treatment outcomes [132, 134]. Moreover, seropositivity, especially ACPA-positivity, is associated with a better response to some biological disease-modifying anti-rheumatic drugs: especially to rituximab, but also to abatacept, tocilizumab and tofacitinib but not to TNF-inhibitors [135,136,137,138].
Collectively, studies show that seropositivity for various autoantibodies in RA is associated with a more severe disease course, radiographic damage and bone loss, even prior to the disease onset [90, 139, 140]. In a recent observational study, various AMPA (to citrullinated, carbamylated, and acetylated peptides) were shown to contribute to the risk of radiographic progression only if present as a combination but not separately [95]. IgA RF was also found to be associated with erosive disease [140, 141].
About 40–30% of RA patients are still being classified as “seronegative RA.” Interestingly, the incidence of RF-negative RA has risen in Minnesota, USA, in the last decade compared to 1995–2004, while the incidence of ACPA-positive or ACPA-negative RA has not changed [142]. It has been discussed for decades, whether these patients truly represent RA or are being misclassified. As mentioned above in the section “Autoantibodies to native proteins”, novel autoantibodies, as well as fine-specificities are detected in a proportion of seronegative RA patients. For example, ACPA fine-specificities were found in 30% of seronegative RA patients in one study and anti-CarP antibodies—in 16% [9]. Taking into account other antibody classes than IgG and tests other than CCP-2, this group of patients will probably be shrinking further in the upcoming years. Moreover, another study showed that after 15 years of follow-up, 8.8% of patients initially diagnosed with seronegative RA have been re-diagnosed with spondyloarthritis [143]. In another cohort, 3% in a 10-year period were re-classified to various rheumatic conditions: polymyalgia rheumatica, psoriatic arthritis, osteoarthritis, spondyloarthritis and others [144]. This indicates the need for more careful diagnostics and follow-up in patients who are diagnosed with seronegative RA.
Conclusion
Autoimmunity in RA is the subject of intensive research, but despite that fact, multiple unanswered questions remain. A variety of autoantibodies has been described, though the mechanisms of the breach of tolerance and disease progression are still not completely understood. The role of autoantibodies in the pathogenetic cascade is likely to be significant, although many other factors, such as cells of the myeloid lineage, FLS, cytokines, T- and B- cells are also involved and the placing of autoantibodies among them is not an easy task. The expansion of the knowledge on autoimmunity in RA is crucial for better understanding of its development and for further treatment advancements. Some aspects remain to be investigated, such as the origin, role and interplay of different autoantibody isotypes, the importance of epigenetic changes in the autoantibody production and the physiological role and origins of ACPA under “normal” conditions. Expanding the range of clinically tested autoantibodies in seronegative RA and individuals at-risk is an important point for the future of patient care.
References
Landré Beauvais AJ (2001) The first description of rheumatoid arthritis. Unabridged text of the doctoral dissertation presented in 1800. Jt Bone Spine 68:130–143. https://doi.org/10.1016/S1297-319X(00)00247-5
Entezami P, Fox DA, Clapham PJ, Chung KC (2011) Historical perspective on the etiology of rheumatoid arthritis. Hand Clin 27:1–10. https://doi.org/10.1016/j.hcl.2010.09.006
Scherer HU, Huizinga TWJ, Krönke G, Schett G, Toes REM (2018) The B cell response to citrullinated antigens in the development of rheumatoid arthritis. Nat Rev Rheumatol 14:157–169. https://doi.org/10.1038/nrrheum.2018.10
Franklin EC, Holman HR, Müller-Eberhard HJ, Kunkel HG (1957) An unusual protein component of high molecular weight in the serum of certain patients with rheumatoid arthritis. J Exp Med 105:425–438. https://doi.org/10.1084/jem.105.5.425
Aggarwal R, Liao K, Nair R, Ringold S, Costenbader KH (2009) Anti-citrullinated peptide antibody assays and their role in the diagnosis of rheumatoid arthritis. Arthritis Care Res 61:1472–1483. https://doi.org/10.1002/art.24827
Nienhuis RLF, Mandema E, Smids C (1964) New serum factor in patients with rheumatoid arthritis: the antiperinuclear factor. Ann Rheum Dis 23:302–305. https://doi.org/10.1136/ard.23.4.302
Van Venrooij WJ, Pruijn GJM (2014) How citrullination invaded rheumatoid arthritis research. Arthritis Res Ther 16:1–5. https://doi.org/10.1186/ar4458
de Brito RS, Baldo DC, Andrade LEC (2019) Clinical and pathophysiologic relevance of autoantibodies in rheumatoid arthritis. Adv Rheumatol 59:2. https://doi.org/10.1186/s42358-018-0042-8
Reed E, Hedström AK, Hansson M, Mathsson-Alm L, Brynedal B, Saevarsdottir S, Cornillet M, Jakobsson PJ, Holmdahl R, Skriner K, Serre G, Alfredsson L, Rönnelid J, Lundberg K (2020) Presence of autoantibodies in “seronegative” rheumatoid arthritis associates with classical risk factors and high disease activity. Arthritis Res Ther 22:1–11. https://doi.org/10.1186/s13075-020-02191-2
Li K, Mo W, Wu L, Wu X, Luo C, Xiao X, Jia X, Yang H, Fei Y, Chen H, Zhang F, Li Y, Zhao L, Zhang X (2021) Novel autoantibodies identified in ACPA-negative rheumatoid arthritis. Ann Rheum Dis 80:739–747. https://doi.org/10.1136/annrheumdis-2020-218460
Paalanen K, Puolakka K, Nikiphorou E, Hannonen P, Sokka T (2020) Is seronegative rheumatoid arthritis true rheumatoid arthritis? A nationwide cohort study. Rheumatology (Oxford) 60:2391–2395. https://doi.org/10.1093/rheumatology/keaa623
Schellekens GA, De Jong BAW, Van Den Hoogen FHJ, Van De Putte LBA, Van Venrooij WJ (2015) Citrulline is an essential constituent of antigenic determinants recognized by rheumatoid arthritis-specific autoantibodies. J Immunol 195:8–16. https://doi.org/10.1172/JCI1316
Szekanecz Z, Szabó Z, Zeher M, Soós L, Dankó K, Horváth I, Lakos G (2013) Superior performance of the CCP3.1 test compared to CCP2 and MCV in the rheumatoid factor-negative RA population. Immunol Res 56:439–443. https://doi.org/10.1007/s12026-013-8425-8
Di Matteo A, Mankia K, Duquenne L, Mahler M, Corscadden D, Mbara K, Garcia-Montoya L, Nam JL, Emery P (2020) Third-generation anti–cyclic citrullinated peptide antibodies improve prediction of clinical arthritis in individuals at risk of rheumatoid arthritis. Arthritis Rheumatol 72:1820–1828. https://doi.org/10.1002/art.41402
Curran AM, Naik P, Giles JT, Darrah E (2020) PAD enzymes in rheumatoid arthritis: pathogenic effectors and autoimmune targets. Nat Rev Rheumatol 16:301–315. https://doi.org/10.1038/s41584-020-0409-1
Li P, Li M, Lindberg MR, Kennett MJ, Xiong N, Wang Y (2010) PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J Exp Med 207:1853–1862. https://doi.org/10.1084/jem.20100239
Liang B, Ge C, Lönnblom E, Lin X, Feng H, Xiao L, Bai J, Ayoglu B, Nilsson P, Nandakumar KS, Zhao M, Holmdahl R (2019) The autoantibody response to cyclic citrullinated collagen type II peptides in rheumatoid arthritis. Rheumatology (Oxford) 58:1623–1633. https://doi.org/10.1093/rheumatology/kez073
Joshua V, Schobers L, Titcombe PJ, Israelsson L, Rönnelid J, Hansson M, Catrina AI, Pruijn GJM, Malmström V (2016) Antibody responses to de novo identified citrullinated fibrinogen peptides in rheumatoid arthritis and visualization of the corresponding B cells. Arthritis Res Ther 18:1–9. https://doi.org/10.1186/s13075-016-1181-0
Ge C, Holmdahl R (2019) The structure, specificity and function of anti-citrullinated protein antibodies. Nature Rev Rheumatol 15:503–508. https://doi.org/10.1038/s41584-019-0244-4
Lundberg K, Kinloch A, Fisher BA, Wegner N, Wait R, Charles P, Mikuls TR, Venables PJ (2008) Antibodies to Citrullinated-Enolase Peptide 1 Are Specific for Rheumatoid Arthritis and Cross-React With Bacterial Enolase. Arthritis Rheum 58:3009–3019. https://doi.org/10.1002/art.23936
Shoda H, Fujio K, Shibuya M, Okamura T, Sumitomo S, Okamoto A, Sawada T, Yamamoto K (2011) Detection of autoantibodies to citrullinated BiP in rheumatoid arthritis patients and pro-inflammatory role of citrullinated BiP in collagen-induced arthritis. Arthritis Res Ther 13:191. https://doi.org/10.1186/ar3520
Haag S, Schneider N, Mason DE, Tuncel J, Andersson IE, Peters EC, Burkhardt H, Holmdahl R (2014) Identification of new citrulline-specific autoantibodies, which bind to human arthritic cartilage, by mass spectrometric analysis of citrullinated type II collagen. Arthritis Rheumatol 66:1440–1449. https://doi.org/10.1002/art.38383
Burkhardt H, Sehnert B, Bockermann R, Engström Å, Kalden JR, Holmdahl R (2005) Humoral immune response to citrullinated collagen type II determinants in early rheumatoid arthritis. Eur J Immunol 35:1643–1652. https://doi.org/10.1002/eji.200526000
Lo KC, Sullivan E, Bannen RM, Jin H, Rowe M, Li H, Pinapati RS, Cartwright AJ, Tan JC, Patel J, Keystone EC, Siminovitch KA (2020) Comprehensive profiling of the rheumatoid arthritis antibody repertoire. Arthritis Rheumatol 72:242–250. https://doi.org/10.1002/art.41089
Sokolove J, Bromberg R, Deane KD, Lahey LJ, Derber LA, Chandra PE, Edison JD, Gilliland WR, Tibshirani RJ, Norris JM, Holers VM, Robinson WH (2012) Autoantibody epitope spreading in the pre-clinical phase predicts progression to rheumatoid arthritis. PLoS One 7:1–9. https://doi.org/10.1371/journal.pone.0035296
Steen J, Forsström B, Sahlström P, Odowd V et al (2019) Recognition of amino acid motifs, rather than specific proteins, by human plasma cell-derived monoclonal antibodies to posttranslationally modified proteins in rheumatoid arthritis. Arthritis Rheumatol 71:196–209. https://doi.org/10.1002/art.40699
Ge C, Xu B, Liang B, Lönnblom E, Lundström SL, Zubarev RA, Ayoglu B, Nilsson P, Skogh T, Kastbom A, Malmström V, Klareskog L, Toes REM, Rispens T, Dobritzsch D, Holmdahl R (2019) Structural basis of Cross-Reactivity of Anti-Citrullinated protein antibodies. Arthritis Rheumatol 71:210–221. https://doi.org/10.1002/art.40698
Sahlström P, Hansson M, Steen J, Amara K, Titcombe PJ, Forsström B, Stålesen R, Israelsson L, Piccoli L, Lundberg K, Klareskog L, Mueller DL, Catrina AI, Skriner K, Malmström V, Grönwall C (2020) Different hierarchies of anti–modified protein autoantibody reactivities in rheumatoid arthritis. Arthritis Rheumatol 72:1643–1657. https://doi.org/10.1002/art.41385
Ioan-Facsinay A, El-Bannoudi H, Scherer HU, Van Der Woude D, Ménard HA, Lora M, Trouw LA, Huizinga TWJ, Toes REM (2011) Anti-cyclic citrullinated peptide antibodies are a collection of anti-citrullinated protein antibodies and contain overlapping and non-overlapping reactivities. Ann Rheum Dis 70:188–193. https://doi.org/10.1136/ard.2010.131102
van de Stadt LA, van Schouwenburg PA, Bryde S, Kruithof S, van Schaardenburg D, Hamann D, Wolbink G, Rispens T (2013) Monoclonal anti-citrullinated protein antibodies selected on citrullinated fibrinogen have distinct targets with different cross-reactivity patterns. Rheumatol (Oxford) 52:631–635. https://doi.org/10.1093/rheumatology/kes371
Thiele GM, Duryee MJ, Anderson DR et al (2015) Malondialdehyde-acetaldehyde adducts and anti-malondialdehyde-acetaldehyde antibodies in rheumatoid arthritis. Arthritis Rheumatol 67:645–655. https://doi.org/10.1002/art.38969
Kissel T, Reijm S, Slot LM et al (2020) Antibodies and B cells recognising citrullinated proteins display a broad cross- reactivity towards other post- translational modifications. Ann Rheum Dis 79:472–480. https://doi.org/10.1136/annrheumdis-2019-216499
Shi J, Willemze A, Janssen GMC, Van Veelen PA, Drijfhout JW, Cerami A, Huizinga TWJ, Trouw LA, Toes RE (2013) Recognition of citrullinated and carbamylated proteins by human antibodies: Specificity, cross-reactivity and the “AMC-Senshu” method. Ann Rheum Dis 72:148–150. https://doi.org/10.1136/annrheumdis-2012-201559
Trouw LA, Rispens T, Toes REM (2017) Beyond citrullination: other post-translational protein modifications in rheumatoid arthritis. Nat Rev Rheumatol 13:331–339. https://doi.org/10.1038/nrrheum.2017.15
Volkov M, van Schie KA, van der Woude D (2020) Autoantibodies and B cells: the ABC of rheumatoid arthritis pathophysiology. Immunol Rev 294:148–163. https://doi.org/10.1111/imr.12829
Shi J, van Veelen PA, Mahler M, Janssen GMC, Drijfhout JW, Huizinga TWJ, Toes REM, Trouw LA (2014) Carbamylation and antibodies against carbamylated proteins in autoimmunity and other pathologies. Autoimmun Rev 13:225–230. https://doi.org/10.1016/j.autrev.2013.10.008
Shi J, Knevel R, Suwannalai P, Van Der Linden MP, Janssen GMC, Van Veelen PA, Levarht NEW, Van Der Helm-van Mil AHM, Cerami A, Huizinga TWJ, Toes REM, Trouw LA (2011) Autoantibodies recognizing carbamylated proteins are present in sera of patients with rheumatoid arthritis and predict joint damage. Proc Natl Acad Sci USA 108:17372–17377. https://doi.org/10.1073/pnas.1114465108
Reed E, Jiang X, Kharlamova N, Ytterberg AJ, Catrina AI, Israelsson L, Mathsson-Alm L, Hansson M, Alfredsson L, Rönnelid J, Lundberg K (2016) Antibodies to carbamylated α-enolase epitopes in rheumatoid arthritis also bind citrullinated epitopes and are largely indistinct from anti-citrullinated protein antibodies. Arthritis Res Ther 18:1–9. https://doi.org/10.1186/s13075-016-1001-6
Lloyd KA, Wigerblad G, Sahlström P et al (2019) Differential ACPA binding to nuclear antigens reveals a PAD-independent pathway and a distinct subset of acetylation cross-reactive autoantibodies in rheumatoid arthritis. Front Immunol 9:3033. https://doi.org/10.3389/fimmu.2018.03033
Wang Z, Nicholls SJ, Rodriguez ER, Kummu O, Hörkkö S, Barnard J, Reynolds WF, Topol EJ, DiDonato JA, Hazen SL (2007) Protein carbamylation links inflammation, smoking, uremia and atherogenesis. Nat Med 13:1176–1184. https://doi.org/10.1038/nm1637
Castellanos-Moreira R, Rodríguez-García SC, Gomara MJ, Ruiz-Esquide V, Cuervo A, Casafont-Solé I, Ramírez J, Holgado S, Gómez-Puerta JA, Cañete JD, Haro I, Sanmarti R (2020) Anti-carbamylated proteins antibody repertoire in rheumatoid arthritis: Evidence of a new autoantibody linked to interstitial lung disease. Ann Rheum Dis 79:587–594. https://doi.org/10.1136/annrheumdis-2019-216709
Mikuls TR, Duryee MJ, Rahman R et al (2017) Enrichment of malondialdehyde-acetaldehyde antibody in the rheumatoid arthritis joint. Rheumatology (Oxford) 56:1794–1803. https://doi.org/10.1093/rheumatology/kex212
Grönwall C, Amara K, Hardt U et al (2017) Autoreactivity to malondialdehyde-modifications in rheumatoid arthritis is linked to disease activity and synovial pathogenesis. J Autoimmun 84:29–45. https://doi.org/10.1016/j.jaut.2017.06.004
England BR, Duryee MJ, Roul P, Mahajan TD, Singh N, Poole JA, Ascherman DP, Caplan L, Demoruelle MK, Deane KD, Klassen LW, Thiele GM, Mikuls TR (2019) Malondialdehyde-Acetaldehyde Adducts and Antibody Responses in Rheumatoid Arthritis-Associated Interstitial Lung Disease. Arthritis Rheumatol 71:1483–1493. https://doi.org/10.1002/art.40900
Juarez M, Bang H, Hammar F, Reimer U, Dyke B, Sahbudin I, Buckley CD, Fisher B, Filer A, Raza K (2016) Identification of novel antiacetylated vimentin antibodies in patients with early inflammatory arthritis. Ann Rheum Dis 75:1099–1107. https://doi.org/10.1136/annrheumdis-2014-206785
Rodriguez-Martínez L, Bang H, Regueiro C et al (2020) Improved classification of rheumatoid arthritis with a score including anti-acetylated ornithine antibodies. Sci Rep 10:1–10. https://doi.org/10.1038/s41598-020-73919-y
Regueiro C, Nuño L, Ortiz AM, Peiteado D, Villalba A, Pascual-Salcedo D, Martínez-Feito A, González-Alvaro I, Balsa A, González A (2017) Value of measuring anti-carbamylated protein antibodies for classification on early arthritis patients. Sci Rep 7:1–8. https://doi.org/10.1038/s41598-017-09657-5
Pietrogrande M, Corona M, Milani S, Rosti A, Ramella M, Tordato G (1995) Relationship between rheumatoid factor and the immune response against hepatitis C virus in essential mixed cryoglobulinemia. Clin Exp Rheumatol 13(Suppl 1):109–113
Janssen KMJ, Hop H, Vissink A, Dijkstra G, de Smit MJ, Brouwer E, Westra J (2020) Levels of anti-citrullinated protein antibodies and rheumatoid factor, including iga isotypes, and articular manifestations in ulcerative colitis and crohn’s disease. Int J Environ Res Public Health 17:1–10. https://doi.org/10.3390/ijerph17218054
Maślińska M, Mańczak M, Kwiatkowska B, Ramsperger V, Shen L, Suresh L (2021) IgA immunoglobulin isotype of rheumatoid factor in primary Sjögren’s syndrome. Rheumatol Int 41:643–649. https://doi.org/10.1007/s00296-020-04782-3
Ugolini A, Nuti M (2021) Rheumatoid factor: A novel determiner in cancer history. Cancers (Basel) 13:1–9. https://doi.org/10.3390/cancers13040591
Ingegnoli F, Castelli R, Gualtierotti R (2013) Rheumatoid factors: Clinical applications. Dis Markers 35:727–734. https://doi.org/10.1155/2013/726598
Bassi N, Ghirardello A, Blank M, Zampieri S, Sarzi-Puttini P, Mantovani A, Shoenfeld Y, Doria A (2010) IgG anti-pentraxin 3 antibodies in systemic lupus erythematosus. Ann Rheum Dis 69:1704–1710. https://doi.org/10.1136/ard.2009.117804
Augusto JF, Poli C, Beauvillain C, Subra JF, Jaillon S, Renier G, Chevailler A, Puéchal X, Delneste Y, Jeannin P (2017) Anti-pentraxin antibodies in autoimmune systemic diseases: Focus on anti-pentraxin-3 autoantibodies. Int Rev Immunol 36:145–153. https://doi.org/10.1080/08830185.2017.1284210
Poulsen TBG, Damgaard D, Jørgensen MM, Senolt L, Blackburn JM, Nielsen CH, Stensballe A (2020) Identification of novel native autoantigens in rheumatoid arthritis. Biomedicines 8:1–13. https://doi.org/10.3390/BIOMEDICINES8060141
Bhattacharjee M, Balakrishnan L, Renuse S, Advani J, Goel R, Sathe G, Keshava Prasad TS, Nair B, Jois R, Shankar S, Pandey A (2016) Synovial fluid proteome in rheumatoid arthritis. Clin Proteomics 13:1–11. https://doi.org/10.1186/s12014-016-9113-1
Pratesi F, Petit Teixeira E, Sidney J, Michou L, Puxeddu I, Sette A, Cornelis F, Migliorini P (2013) HLA shared epitope and ACPA: Just a marker or an active player? Autoimmun Rev 12:1182–1187. https://doi.org/10.1016/j.autrev.2013.08.002
Huizinga TWJ, Amos CI, Van Der Helm-Van Mil AHM et al (2005) Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum 52:3433–3438. https://doi.org/10.1002/art.21385
Terao C, Ohmura K, Ikari K, Kawaguchi T, Takahashi M, Setoh K, Nakayama T, Kosugi S, Sekine A, Tabara Y, Taniguchi A, Momohara S, Yamanaka H, Yamada R, Matsuda F, Mimori T (2014) Effects of smoking and shared epitope on the production of anti-citrullinated peptide antibody in a Japanese adult population. Arthritis Care Res 66:1818–1827. https://doi.org/10.1002/acr.22385
Hill JA, Southwood S, Sette A, Jevnikar AM, Bell DA, Cairns E (2003) Cutting edge: the conversion of arginine to citrulline allows for a high-affinity peptide interaction with the rheumatoid arthritis-associated HLA-DRB1*0401 MHC class II molecule. J Immunol 171:538–541. https://doi.org/10.4049/jimmunol.171.2.538
Ting YT, Petersen J, Ramarathinam SH, Scally SW, Loh KL, Thomas R, Suri A, Baker DG, Purcell AW, Reid HH, Rossjohn J (2018) The interplay between citrullination and HLA-DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J Biol Chem 293:3236–3251. https://doi.org/10.1074/jbc.RA117.001013
Auger I, Sebbag M, Vincent C, Balandraud N, Guis S, Nogueira L, Svensson B, Cantagrel A, Serre G, Roudier J (2005) Influence of HLA-DR genes on the production of rheumatoid arthritis-specific autoantibodies to citrullinated fibrinogen. Arthritis Rheum 52:3424–3432. https://doi.org/10.1002/art.21391
Balandraud N, Auger I, Roudier J (2021) Do RA associated HLA-DR molecules bind citrullinated peptides or peptides from PAD4 to help the development of RA specific antibodies to citrullinated proteins? J Autoimmun 116:102542. https://doi.org/10.1016/j.jaut.2020.102542
Auger I, Balandraud N, Massy E, Hemon MF, Peen E, Arnoux F, Mariot C, Martin M, Lafforgue P, Busnel JM, Roudier J (2020) Peptidylarginine deiminase autoimmunity and the development of anti–citrullinated protein antibody in rheumatoid arthritis: the Hapten-Carrier model. Arthritis Rheumatol 72:903–911. https://doi.org/10.1002/art.41189
Kolfenbach JR, Deane KD, Derber LA, O’Donnell CI, Gilliland WR, Edison JD, Rosen A, Darrah E, Norris JM, Holers VM (2010) Autoimmunity to peptidyl arginine deiminase type 4 precedes clinical onset of rheumatoid arthritis. Arthritis Rheum 62:2633–2639. https://doi.org/10.1002/art.27570
Begovich AB, Carlton VEH, Honigberg LA et al (2004) A missense single-nucleotide polymorphism in a gene encoding a proteintyrosine phosphatase (PTPN22) is associated with rheumatoid arthritis. Am J Hum Genet 75:330–337. https://doi.org/10.1086/422827
Klareskog L, Malmström V, Lundberg K, Padyukov L, Alfredsson L (2011) Smoking, citrullination and genetic variability in the immunopathogenesis of rheumatoid arthritis. Semin Immunol 23:92–98. https://doi.org/10.1016/j.smim.2011.01.014
Källberg H, Ding B, Padyukov L et al (2011) Smoking is a major preventable risk factor for rheumatoid arthritis: estimations of risks after various exposures to cigarette smoke. Ann Rheum Dis 70:508–511. https://doi.org/10.1136/ard.2009.120899
Linn-Rasker SP, Van Der Helm-Van Mil AHM, Van Gaalen FA, Kloppenburg M, De Vries RRP, Le Cessie S, Breedveld FC, Toes REM, Huizinga TWJ (2006) Smoking is a risk factor for anti-CCP antibodies only in rheumatoid arthritis patients who carry HLA-DRB1 shared epitope alleles. Ann Rheum Dis 65:366–371. https://doi.org/10.1136/ard.2005.041079
van Wesemael TJ, Ajeganova S, Humphreys J et al (2016) Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: A multicenter cohort study. Arthritis Res Ther 18:1–10. https://doi.org/10.1186/s13075-016-1177-9
Roos Ljungberg K, Börjesson E, Martinsson K, Wetterö J, Kastbom A, Svärd A (2020) Presence of salivary IgA anti-citrullinated protein antibodies associate with higher disease activity in patients with rheumatoid arthritis. Arthritis Res Ther 22:1–10. https://doi.org/10.1186/s13075-020-02363-0
Llorente I, Merino L, Ortiz AM, Escolano E, González-Ortega S, García-Vicuña R, García-Vadillo JA, Castañeda S, González-Álvaro I (2017) Anti-citrullinated protein antibodies are associated with decreased bone mineral density: baseline data from a register of early arthritis patients. Rheumatol Int 37:799–806. https://doi.org/10.1007/s00296-017-3674-9
Klareskog L, Stolt P, Lundberg K, Källberg H, Bengtsson C, Grunewald J, Rönnelid J, Harris HE, Ulfgren AK, Rantapää-Dahlqvist S, Eklund A, Padyukov L, Alfredsson L (2006) A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum 54:38–46. https://doi.org/10.1002/art.21575
Mercado F, Marshall RI, Klestov AC, Bartold PM (2000) Is there a relationship between rheumatoid arthritis and periodontal disease? J Clin Periodontol 27:267–272. https://doi.org/10.1034/j.1600-051x.2000.027004267.x
Mikuls TR, Payne JB, Yu F, Thiele GM, Reynolds RJ, Cannon GW, Markt J, McGowan D, Kerr GS, Redman RS, Reimold A, Griffiths G, Beatty M, Gonzalez SM, Bergman DA, Hamilton BC, Erickson AR, Sokolove J, Robinson WH, Walker C, Chandad F, O’Dell JR (2014) Periodontitis and porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol 66:1090–1100. https://doi.org/10.1002/art.38348
Hitchon CA, Chandad F, Ferucci ED, Willemze A, Ioan-Facsinay A, Van Der Woude D, Markland J, Robinson D, Elias B, Newkirk M, Toes RM, Huizinga TWJ, El-Gabalawy HS (2010) Antibodies to Porphyromonas gingivalis are associated with anticitrullinated protein antibodies in patients with rheumatoid arthritis and their relatives. J Rheumatol 37:1105–1112. https://doi.org/10.3899/jrheum.091323
Bae SC, Lee YH (2018) Association between anti-Porphyromonas gingivalis antibody, anti-citrullinated protein antibodies, and rheumatoid arthritis: a meta-analysis. Z Rheumatol 77:522–532. https://doi.org/10.1007/s00393-017-0328-y
Laugisch O, Wong A, Sroka A, Kantyka T, Koziel J, Neuhaus K, Sculean A, Venables PJ, Potempa J, Möller B, Eick S (2016) Citrullination in the periodontium—a possible link between periodontitis and rheumatoid arthritis. Clin Oral Investig 20:675–683. https://doi.org/10.1007/s00784-015-1556-7
Loutan L, Alpizar-Rodriguez D, Courvoisier DS, Finckh A, Mombelli A, Giannopoulou C (2019) Periodontal status correlates with anti-citrullinated protein antibodies in first-degree relatives of individuals with rheumatoid arthritis. J Clin Periodontol 46:690–698. https://doi.org/10.1111/jcpe.13117
Mikuls TR, Thiele GM, Deane KD, Payne JB, O’Dell JR, Yu F, Sayles H, Weisman MH, Gregersen PK, Buckner JH, Keating RM, Derber LA, Robinson WH, Holers VM, Norris JM (2012) Porphyromonas gingivalis and disease-related autoantibodies in individuals at increased risk of rheumatoid arthritis. Arthritis Rheum 64:3522–3530. https://doi.org/10.1002/art.34595
Sakkas LI, Daoussis D, Liossis SN, Bogdanos DP (2017) The infectious basis of ACPA-positive rheumatoid arthritis. Front Microbiol 8:1–9. https://doi.org/10.3389/fmicb.2017.01853
Konig MF, Abusleme L, Reinholdt J, Palmer RJ, Teles RP, Sampson K, Rosen A, Nigrovic PA, Sokolove J, Giles JT, Moutsopoulos NM, Andrade F (2016) Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci Transl Med 8:1–13. https://doi.org/10.1126/scitranslmed.aaj1921
Giles JT, Reinholdt J, Andrade F, Konig MF (2021) Associations of antibodies targeting periodontal pathogens with subclinical coronary, carotid, and peripheral arterial atherosclerosis in rheumatoid arthritis. Arthritis Rheumatol 73:568–575. https://doi.org/10.1002/art.41572
Vossenaar ER, Smeets TJM, Kraan MC, Raats JM, Van Venrooij WJ, Tak PP (2004) The presence of citrullinated proteins is not specific for rheumatoid synovial tissue. Arthritis Rheum 50:3485–3494. https://doi.org/10.1002/art.20584
Masson-Bessière C, Sebbag M, Durieux JJ, Nogueira L, Vincent C, Girbal-Neuhauser E, Durroux R, Cantagrel A, Serre G (2000) In the rheumatoid pannus, anti-filaggrin autoantibodies are produced by local plasma cells and constitute a higher proportion of IgG than in synovial fluid and serum. Clin Exp Immunol 119:544–552. https://doi.org/10.1046/j.1365-2249.2000.01171.x
Makrygiannakis D, Af Klint E, Lundberg IE, Löfberg R, Ulfgren AK, Klareskog L, Catrina AI (2006) Citrullination is an inflammation-dependent process. Ann Rheum Dis 65:1219–1222. https://doi.org/10.1136/ard.2005.049403
Turunen S, Huhtakangas J, Nousiainen T, Valkealahti M, Melkko J, Risteli J, Lehenkari P (2016) Rheumatoid arthritis antigens homocitrulline and citrulline are generated by local myeloperoxidase and peptidyl arginine deiminases 2, 3 and 4 in rheumatoid nodule and synovial tissue. Arthritis Res Ther 18:1–15. https://doi.org/10.1186/s13075-016-1140-9
Vossenaar ER, Nijenhuis S, Helsen MMA, Van der Heijden A, Senshu T, Van den Berg WB, Van Venrooij WJ, Joosten LAB (2003) Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum 48:2489–2500. https://doi.org/10.1002/art.11229
Foulquier C, Sebbag M, Clavel C, Chapuy-Regaud S, Al Badine R, Méchin MC, Vincent C, Nachat R, Yamada M, Takahara H, Simon M, Guerrin M, Serre G (2007) Peptidyl arginine deiminase type 2 (PAD-2) and PAD-4 but not PAD-1, PAD-3, and PAD-6 are expressed in rheumatoid arthritis synovium in close association with tissue inflammation. Arthritis Rheum 56:3541–3553. https://doi.org/10.1002/art.22983
Kleyer A, Finzel S, Rech J, Manger B, Krieter M, Faustini F, Araujo E, Hueber AJ, Harre U, Engelke K, Schett G (2014) Bone loss before the clinical onset of rheumatoid arthritis in subjects with anticitrullinated protein antibodies. Ann Rheum Dis 73:854–860. https://doi.org/10.1136/annrheumdis-2012-202958
Hafström I, Ajeganova S, Forslind K, Svensson B (2019) Anti-citrullinated protein antibodies are associated with osteopenia but not with pain at diagnosis of rheumatoid arthritis: Data from the BARFOT cohort. Arthritis Res Ther 21:1–9. https://doi.org/10.1186/s13075-019-1833-y
De Punder YMR, Hendrikx J, Den Broeder AA, Pascual EV, Van Riel PL, Fransen J (2013) Should we redefine treatment targets in rheumatoid arthritis? Low disease activity is sufficiently strict for patients who are anticitrullinated protein antibody-negative. J Rheumatol 40:1268–1274. https://doi.org/10.3899/jrheum.121438
Ajeganova S, Huizinga TWJ (2015) Seronegative and seropositive RA: alike but different? Nat Rev Rheumatol 11:8–9. https://doi.org/10.1038/nrrheum.2014.194
Barra L, Pope JE, Orav JE, Boire G, Haraoui B, Hitchon C, Keystone EC, Thorne JC, Tin D, Bykerk VP (2014) Prognosis of seronegative patients in a large prospective cohort of patients with early inflammatory arthritis. J Rheumatol 41:2361–2369. https://doi.org/10.3899/jrheum.140082
Nijjar JS, Morton FR, Bang H et al (2021) The impact of autoantibodies against citrullinated, carbamylated, and acetylated peptides on radiographic progression in patients with new-onset rheumatoid arthritis: an observational cohort study. Lancet Rheumatol 3:284–293. https://doi.org/10.1016/S2665-9913(20)30381-7
Hecht C, Englbrecht M, Rech J, Schmidt S, Araujo E, Engelke K, Finzel S, Schett G (2015) Additive effect of anti-citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann Rheum Dis 74:2151–2156. https://doi.org/10.1136/annrheumdis-2014-205428
Bugatti S, Bogliolo L, Vitolo B, Manzo A, Montecucco C, Caporali R (2016) Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res Ther 18:1–8. https://doi.org/10.1186/s13075-016-1116-9
Ince-Askan H, Dolhain RJEM (2015) Pregnancy and rheumatoid arthritis. Best Pract Res Clin Rheumatol 29:580–596. https://doi.org/10.1016/j.berh.2015.07.001
Hensvold AH, Frisell T, Magnusson PKE, Holmdahl R, Askling J, Catrina AI (2017) How well do ACPA discriminate and predict RA in the general population: a study based on 12 590 population-representative Swedish twins. Ann Rheum Dis 76:119–125. https://doi.org/10.1136/annrheumdis-2015-208980
Nielen MMJ, Van Schaardenburg D, Reesink HW, Van De Stadt RJ, Van Der Horst-Bruinsma IE, De Koning MHMT, Habibuw MR, Vandenbroucke JP, Dijkmans BAC (2004) Specific autoantibodies precede the symptoms of rheumatoid arthritis: a study of serial measurements in blood donors. Arthritis Rheum 50:380–386. https://doi.org/10.1002/art.20018
Harre U, Georgess D, Bang H, Bozec A, Axmann R, Ossipova E, Jakobsson PJ, Baum W, Nimmerjahn F, Szarka E, Sarmay G, Krumbholz G, Neumann E, Toes R, Scherer HU, Catrina AI, Klareskog L, Jurdic P, Schett G (2012) Induction of osteoclastogenesis and bone loss by human autoantibodies against citrullinated vimentin. J Clin Invest 122:1791–1802. https://doi.org/10.1172/JCI60975
Kurowska W, Slowinska I, Krogulec Z, Syrowka P, Maslinski W (2021) Antibodies to citrullinated Proteins ( ACPA ) associate with markers of osteoclast activation and bone destruction in the bone marrow of patients with rheumatoid arthritis. J Clin Med 10:1778. https://doi.org/10.3390/jcm10081778
Laurent L, Anquetil F, Clavel C, Ndongo-Thiam N, Offer G, Miossec P, Pasquali JL, Sebbag M, Serre G (2015) IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann Rheum Dis 74:1425–1431. https://doi.org/10.1136/annrheumdis-2013-204543
Dong X, Zheng Z, Lin P, Fu X, Li F, Jiang J, Zhu P (2020) ACPAs promote IL-1β production in rheumatoid arthritis by activating the NLRP3 inflammasome. Cell Mol Immunol 17:261–271. https://doi.org/10.1038/s41423-019-0201-9
Sun M, Rethi B, Krishnamurthy A, Joshua V, Circiumaru A, Hensvold AH, Ossipova E, Grönwall C, Liu Y, Engstrom M, Catrina SB, Steen J, Malmstrom V, Klareskog L, Svensson C, Ospelt C, Wähämaa H, Catrina AI (2019) Anticitrullinated protein antibodies facilitate migration of synovial tissue-derived fibroblasts. Ann Rheum Dis 78:1621–1631. https://doi.org/10.1136/annrheumdis-2018-214967
Wigerblad G, Bas DB, Fernandes-Cerqueira C (2019) Correction: Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann Rheum Dis (2016) 75(730). https://doi.org/10.1136/annrheumdis-2015-208094. Ann Rheum Dis 78:865. https://doi.org/10.1136/annrheumdis-2015-208094corr1
O’Neil LJ, Barrera-Vargas A, Sandoval-Heglund D, Merayo-Chalico J, Aguirre-Aguilar E, Aponte AM, Ruiz-Perdomo Y, Gucek M, El-Gabalawy H, Fox DA, Katz JD, Kaplan MJ, Carmona-Rivera C (2020) Neutrophil-mediated carbamylation promotes articular damage in rheumatoid arthritis. Sci Adv 6:1–16. https://doi.org/10.1126/sciadv.abd2688
Kuhn KA, Kulik L, Tomooka B, Braschler KJ, Arend WP, Robinson WH, Holers VM (2006) Antibodies against citrullinated proteins enhance tissue injury in experimental autoimmune arthritis. J Clin Invest 116:961–973. https://doi.org/10.1172/JCI25422
Dusad A, Duryee MJ, Shaw AT, Klassen LW, Anderson DR, Wang D, Ren K, Gravallese EM, O’Dell JR, Mikuls TR, Thiele GM (2014) Induction of bone loss in DBA/1J mice immunized with citrullinated autologous mouse type II collagen in the absence of adjuvant. Immunol Res 58:51–60. https://doi.org/10.1007/s12026-013-8479-7
Cantaert T, Teitsma C, Tak PP, Baeten D (2013) Presence and role of anti-citrullinated protein antibodies in experimental arthritis models. Arthritis Rheum 65:939–948. https://doi.org/10.1002/art.37839
Böhm S, Schwab I, Lux A, Nimmerjahn F (2012) The role of sialic acid as a modulator of the anti-inflammatory activity of IgG. Semin Immunopathol 34:443–453. https://doi.org/10.1007/s00281-012-0308-x
Sehic E, Westerlund A, Lagerquist MK, Lerner UH, Carlsten H, Henning P, Engdahl C (2021) Immunoglobulin G complexes without sialic acids enhance osteoclastogenesis but do not affect arthritis-mediated bone loss. Scand J Immunol 93:1–11. https://doi.org/10.1111/sji.13009
Harre U, Lang SC, Pfeifle R, Rombouts Y, Frühbeißer S, Amara K, Bang H, Lux A, Koeleman CA, Baum W, Dietel K, Gröhn F, Malmström V, Klareskog L, Krönke G, Kocijan R, Nimmerjahn F, Toes REM, Herrmann M, Scherer HU, Schett G (2015) Glycosylation of immunoglobulin G determines osteoclast differentiation and bone loss. Nat Commun 6:6651. https://doi.org/10.1038/ncomms7651
Pfeifle R, Rothe T, Ipseiz N et al (2017) Regulation of autoantibody activity by the IL-23-T H 17 axis determines the onset of autoimmune disease. Nat Immunol 18:104–113. https://doi.org/10.1038/ni.3579
Scherer HU, Van Der Woude D, Ioan-Facsinay A, El Bannoudi H, Trouw LA, Wang J, Häupl T, Burmester GR, Deelder AM, Huizinga TWJ, Wuhrer M, Toes REM (2010) Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum 62:1620–1629. https://doi.org/10.1002/art.27414
Vergroesen RD, Slot LM, Hafkenscheid L, Koning MT, Van Der Voort EIH, Grooff CA, Zervakis G, Veelken H, Huizinga TWJ, Rispens T, Scherer HU, Toes REM (2018) B-cell receptor sequencing of anti-citrullinated protein antibody (ACPA) IgG-expressing B cells indicates a selective advantage for the introduction of N-glycosylation sites during somatic hypermutation. Ann Rheum Dis 77:955–957. https://doi.org/10.1136/annrheumdis-2017-212052
Rombouts Y, Willemze A, Van Beers JJBC et al (2016) Extensive glycosylation of ACPA-IgG variable domains modulates binding to citrullinated antigens in rheumatoid arthritis. Ann Rheum Dis 75:578–585. https://doi.org/10.1136/annrheumdis-2014-206598
Corsiero E, Carlotti E, Jagemann L, Perretti M, Pitzalis C, Bombardieri M (2020) H and L chain affinity maturation and/or Fab N-glycosylation influence immunoreactivity toward neutrophil extracellular trap antigens in rheumatoid arthritis synovial B cell clones. J Immunol 204:2374–2379. https://doi.org/10.4049/jimmunol.1901457
Kempers AC, Hafkenscheid L, Dorjée AL, Moutousidou E, Van De Bovenkamp FS, Rispens T, Trouw LA, Van Oosterhout M, Huizinga TWJ, Toes R, Scherer HU (2018) The extensive glycosylation of the ACPA variable domain observed for ACPA-IgG is absent from ACPA-IgM. Ann Rheum Dis 77:1087–1088. https://doi.org/10.1136/annrheumdis-2017-211533
Vergroesen RD, Slot LM, van Schaik BDC, Koning MT, Rispens T, van Kampen AHC, Toes REM, Scherer HU (2019) N-glycosylation site analysis of citrullinated antigen-specific B-cell receptors indicates alternative selection pathways during autoreactive B-cell development. Front Immunol 10:2092. https://doi.org/10.3389/fimmu.2019.02092
Suwannalai P, Britsemmer K, Knevel R, Scherer HU, Levarht EWN, Van Der Helm-Van Mil AH, Van Schaardenburg D, Huizinga TWJ, Toes REM, Trouw LA (2014) Low-avidity anticitrullinated protein antibodies (ACPA) are associated with a higher rate of joint destruction in rheumatoid arthritis. Ann Rheum Dis 73:270–276. https://doi.org/10.1136/annrheumdis-2012-202615
Van Zanten A, Arends S, Roozendaal C, Limburg PC, Maas F, Trouw LA, Toes REM, Huizinga TWJ, Bootsma H, Brouwer E (2017) Presence of anticitrullinated protein antibodies in a large population-based cohort from the Netherlands. Ann Rheum Dis 76:1184–1190. https://doi.org/10.1136/annrheumdis-2016-209991
Shi J, van de Stadt LA, Levarht EWN, Huizinga TWJ, Hamann D, van Schaardenburg D, Toes REM, Trouw LA (2014) Anti-carbamylated protein (anti-CarP) antibodies precede the onset of rheumatoid arthritis. Ann Rheum Dis 73:780–783. https://doi.org/10.1136/annrheumdis-2013-204154
Van Der Woude D, Rantapää-Dahlqvist S, Ioan-Facsinay A, Onnekink C, Schwarte CM, Verpoort KN, Drijfhout JW, Huizinga TWJ, Toes REM, Pruijn GJM (2010) Epitope spreading of the anti-citrullinated protein antibody response occurs before disease onset and is associated with the disease course of early arthritis. Ann Rheum Dis 69:1554–1561. https://doi.org/10.1136/ard.2009.124537
ten Brinck RM, van Steenbergen HW, van Delft MAM, Verheul MK, Toes REM, Trouw LA, van der Helm-van Mil AHM (2017) The risk of individual autoantibodies, autoantibody combinations and levels for arthritis development in clinically suspect arthralgia. Rheumatology (Oxford) 56:2145–2153. https://doi.org/10.1093/rheumatology/kex340
Mankia K, D’Agostino MA, Rowbotham E, Hensor EMA, Hunt L, Möller I, Miguel M, Mérida-Velasco JR, Murillo-González J, Naredo E, Nam JL, Tan AL, Freeston JE, Grainger A, Emery P (2019) MRI inflammation of the hand interosseous tendons occurs in anti-CCP-positive at-risk individuals and may precede the development of clinical synovitis. Ann Rheum Dis 78:781–786. https://doi.org/10.1136/annrheumdis-2018-214331
Ford JA, Liu X, Marshall AA, Zaccardelli A, Prado MG, Wiyarand C, Lu B, Karlson EW, Schur PH, Deane KD, Sparks JA (2019) Impact of cyclic citrullinated peptide antibody level on progression to rheumatoid arthritis in clinically tested cyclic citrullinated peptide antibody–positive patients without rheumatoid arthritis. Arthritis Care Res 71:1583–1592. https://doi.org/10.1002/acr.23820
Bemis EA, Demoruelle MK, Seifert JA, Polinski KJ, Weisman MH, Buckner JH, Gregersen PK, Mikuls TR, Odell JR, Keating RM, Deane KD, Holers VM, Norris JM (2021) Factors associated with progression to inflammatory arthritis in first-degree relatives of individuals with RA following autoantibody positive screening in a non-clinical setting. Ann Rheum Dis 80:154–161. https://doi.org/10.1136/annrheumdis-2020-217066
Finckh A, Courvoisier D, Lamacchia C (2020) Measuring ACPA in the general population or primary care: Is it useful? RMD Open 6:1–7. https://doi.org/10.1136/rmdopen-2019-001085
Rombouts Y, Ewing E, Van De Stadt LA, Selman MHJ, Trouw LA, Deelder AM, Huizinga TWJ, Wuhrer M, Van Schaardenburg D, Toes REM, Scherer HU (2015) Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann Rheum Dis 74:234–241. https://doi.org/10.1136/annrheumdis-2013-203565
Verpoort KN, Jol-Van Der Zijde CM, Papendrecht-Van Der Voort EAM, Ioan-Facsinay A, Drijfhout JW, Van Tol MJD, Breedveld FC, Huizinga TWJ, Toes REM (2006) Isotype distribution of anti-cyclic citrullinated peptide antibodies in undifferentiated arthritis and rheumatoid arthritis reflects an ongoing immune response. Arthritis Rheum 54:3799–3808. https://doi.org/10.1002/art.22279
de Moel EC, Derksen VFAM, Stoeken G, Trouw LA, Bang H, Goekoop RJ, Speyer I, Huizinga TWJ, Allaart CF, Toes REM, van der Woude D (2018) Baseline autoantibody profile in rheumatoid arthritis is associated with early treatment response but not long-term outcomes. Arthritis Res Ther 20:1–12. https://doi.org/10.1186/s13075-018-1520-4
Figueiredo CP, Bang H, Cobra JF et al (2017) Antimodified protein antibody response pattern influences the risk for disease relapse in patients with rheumatoid arthritis tapering disease modifying antirheumatic drugs. Ann Rheum Dis 76:399–407. https://doi.org/10.1136/annrheumdis-2016-209297
De Moel EC, Derksen VFAM, Trouw LA, Bang H, Collée G, Lard LR, Ramiro S, Huizinga TWJ, Allaart CF, Toes REM, Van Der Woude D (2019) In rheumatoid arthritis, changes in autoantibody levels reflect intensity of immunosuppression, not subsequent treatment response. Arthritis Res Ther 21:1–8. https://doi.org/10.1186/s13075-019-1815-0
Pyrpasopoulou A, Douma S, Triantafyllou A, Simoulidou E, Samara M, Parapanisiou E, Aslanidis S (2010) Response to Rituximab and Timeframe to Relapse in Rheumatoid Arthritis Patients. Mol Diagn Ther 14:43–48. https://doi.org/10.1007/BF03256352
Alemao E, Postema R, Elbez Y, Mamane C, Finckh A (2020) Presence of anti-cyclic citrullinated peptide antibodies is associated with better treatment response to abatacept but not to TNF inhibitors in patients with rheumatoid arthritis: a meta-analysis. Clin Exp Rheumatol 38:455–466
Courvoisier DS, Chatzidionysiou K, Mongin D et al (2021) The impact of seropositivity on the effectiveness of biologic anti-rheumatic agents: results from a collaboration of 16 registries. Rheumatology (Oxford) 60:820–828. https://doi.org/10.1093/rheumatology/keaa393
Bird P, Hall S, Nash P, Connell CA, Kwok K, Witcombe D, Thirunavukkarasu K (2019) Treatment outcomes in patients with seropositive versus seronegative rheumatoid arthritis in Phase III randomised clinical trials of tofacitinib. RMD Open 5:e000742. https://doi.org/10.1136/rmdopen-2018-000742
Lamacchia C, Courvoisier DS, Jarlborg M, Bas S, Roux-Lombard P, Möller B, Ciurea A, Finckh A, Bentow C, Martinez-Prat L, Mahler M, Gabay C, Nissen MJ (2021) Predictive value of anti-CarP and anti-PAD3 antibodies alone or in combination with RF and ACPA on the severity of rheumatoid arthritis. Rheumatology (Oxford) keab050. https://doi.org/10.1093/rheumatology/keab050
Berglin E, Johansson T, Sundin U, Jidell E, Wadell G, Hallmans G, Rantapää-Dahlqvist S (2006) Radiological outcome in rheumatoid arthritis is predicted by presence of antibodies against cyclic citrullinated peptide before and at disease onset, and by IgA-RF at disease onset. Ann Rheum Dis 65:453–458. https://doi.org/10.1136/ard.2005.041376
Fedele AL, Petricca L, Tolusso B, Alivernini S, Canestri S, Di Mario C, Bosello SL, Ferraccioli G, Gremese E (2018) Interleukin-6 and IgA-rheumatoid factor are crucial for baseline erosiveness, and anti-citrullinated peptide antibodies for radiographic progression in early rheumatoid arthritis treated according to a treat-to-target strategy. Scand J Rheumatol 47:351–359. https://doi.org/10.1080/03009742.2017.1416668
Myasoedova E, Davis J, Matteson EL, Crowson CS (2020) Is the epidemiology of rheumatoid arthritis changing? Results from a population-based incidence study, 1985–2014. Ann Rheum Dis 79:440–444. https://doi.org/10.1136/annrheumdis-2019-216694
Paalanen K, Puolakka K, Nikiphorou E, Hannonen P, Sokka T (2021) Is seronegative rheumatoid arthritis true rheumatoid arthritis? A nationwide cohort study. Rheumatology (Oxford) 60:2391–2395. https://doi.org/10.1093/rheumatology/keaa623
Paalanen K, Rannio K, Rannio T, Asikainen J, Hannonen P, Sokka T (2019) Does early seronegative arthritis develop into rheumatoid arthritis? A 10-year observational study. Clin Exp Rheumatol 37:37–43
Nishimura K, Sugiyama D, Kogata Y, Tsuji G, Nakazawa T, Kawano S, Saigo K, Morinobu A, Koshiba M, Kuntz KM, Kamae I, Kumagai S (2007) Meta-analysis: diagnostic accuracy of anti-cyclic citrullinated peptide antibody and rheumatoid factor for rheumatoid arthritis. Ann Intern Med 146:797–808. https://doi.org/10.7326/0003-4819-146-11-200706050-00008
Pecani A, Alessandri C, Spinelli FR, Priori R, Riccieri V, Di Franco M, Ceccarelli F, Colasanti T, Pendolino M, Mancini R, Truglia S, Barbati C, Vomero M, Sabatinelli D, Morello F, Valesini G, Conti F (2016) Prevalence, sensitivity and specificity of antibodies against carbamylated proteins in a monocentric cohort of patients with rheumatoid arthritis and other autoimmune rheumatic diseases. Arthritis Res Ther 18:1–8. https://doi.org/10.1186/s13075-016-1173-0
Sidiras P, Spruyt D, Gangji V, Imbault V, Sokolova T, Durez P, Communi D, Rasschaert J, Badot V (2021) Antibodies against carbamylated proteins: prevalence and associated disease characteristics in Belgian patients with rheumatoid arthritis or other rheumatic diseases. Scand J Rheumatol 50:118–123. https://doi.org/10.1080/03009742.2020.1798500
Kolarz B, Ciesla M, Rosenthal AK, Dryglewska M, Majdan M (2021) The value of anti-CarP and anti-PAD4 as markers of rheumatoid arthritis in ACPA/RF negative rheumatoid arthritis patients. Ther Adv Musculoskelet Dis 13:1–10. https://doi.org/10.1177/1759720X21989868
Funding
Open Access funding enabled and organized by Projekt DEAL. Deutsche Forschungsgemeinschaft (FOR2886 PANDORA TP03 and TP04; SPP1468-IMMUNOBONE), the EU/EFPIA Innovative Medicines Initiative 2 Joint Undertaking RTCure grant (no. 777357), and the Bundesministerium für Bildung und Forschung (BMBF; project MASCARA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Ethics Approval
Non-applicable.
Consent to Participate
Non-applicable.
Consent for Publication
Non-applicable.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sokolova, M.V., Schett, G. & Steffen, U. Autoantibodies in Rheumatoid Arthritis: Historical Background and Novel Findings. Clinic Rev Allerg Immunol 63, 138–151 (2022). https://doi.org/10.1007/s12016-021-08890-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08890-1