
Abstract
Hereditary angioedema (HAE) is a rare genetic disease, characterized by recurrent and unexpected potentially life-threatening mucosal swelling. HAE may be further classified into HAE with C1‐inhibitor deficiency (C1‐INH‐HAE) and HAE with normal C1‐INH activity (nlC1‐INH‐HAE), mostly due to mutations leading to increased vascular permeability. Recent evidence implicates also the innate and adaptive immune responses in several aspects of angioedema pathophysiology. Monocytes/macrophages, granulocytes, lymphocytes, and mast cells contribute directly or indirectly to the pathophysiology of angioedema. Immune cells are a source of vasoactive mediators, including bradykinin, histamine, complement components, or vasoactive mediators, whose concentrations or activities are altered in both attacks and remissions of HAE. In turn, through the expression of various receptors, these cells are also activated by a plethora of molecules. Thereby, activated immune cells are the source of molecules in the context of HAE, and on the other hand, increased levels of certain mediators can, in turn, activate immune cells through the engagement of specific surface receptors and contribute to vascular endothelial processes that lead to hyperpemeability and tissue edema. In this review, we summarize recent developments in the putative involvement of the innate and adaptive immune system of angioedema.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Angioedema is a self‐limiting tissue swelling due to periodic increase in vascular permeability caused by the release of bradykinin (BK) and/or other cell–derived mediators. Recurrent swellings are localized to the skin and/or to the upper respiratory, gastrointestinal, and genitourinary tracts [1]. Angioedema can be hereditary or acquired. The most common form of hereditary angioedema (HAE) is caused by deficiency of C1 esterase inhibitor (C1‐INH‐HAE), but HAE can also occur with normal levels of C1‐INH (nl‐C1‐INH‐HAE) [1].
C1-INH is a protein of the complement system which is a critical component of both the innate and adaptive immunity [2,3,4]. The immune system is typically divided in two branches: innate and adaptive, although these distinctions are not completely exclusive [5]. The fundamentals of HAE (or angioedema) have been extensively reviewed previously [1, 6]. In this paper, we focus our discussion on the roles played by the immune system in the pathophysiology of angioedema.
Innate Immune System
Monocytes
Monocytes (MO) originate from myeloid progenitors in the bone marrow (BM). These cells are rapidly recruited to tissues during infections and inflammation, where they differentiate into macrophages or dendritic cells [7]. There are three subsets of human MO: classical (~90%), intermediate, and non-classical (~10%) [8]. These subpopulations can be further characterized by different functions and expression of surface markers and chemokine receptors [9]. They display phagocytic and microbial activity and produce pro-inflammatory cytokines. Intravital microscopy studies have revealed that non-classical MO continuously monitor the vasculature under physiological conditions through an LFA/ICAM-dependent crawling mechanism on resting endothelial cells (EC) [10, 11]. The role of MO has been poorly studied in HAE. It would be interesting to evaluate whether the surveillance of EC integrity driven by non-classical MO is altered in HAE patients which have an abnormal basal vascular permeability [12].
As an example, endothelial permeability is mediated by vasoactive mediator release, including vascular endothelial growth factors (VEGFs) [13] that were found increased in C1-INH-HAE patients and correlate with disease severity [14, 15]. VEGFs are produced by various cells including EC. They signal through the tyrosine kinase receptors, VEGFR‐1, VEGFR‐2, and VEGFR‐3 [16]. Indeed, MO express low levels of VEGFR‐1 and VEGFR-3 but do not express VEGFR‐2. MO produce high amounts of VEGF (in response to M‐CSF) or the antagonistic soluble VEGFR‐1 (in response to GM‐CSF) [8]. Increased VEGF-A in C1-INH-HAE could be caused by MO activation or vice versa circulating VEGF through binding to VEGFR-1 could attract and activate circulating MO (Table 1; Fig. 1).

Schematic representation of the effects of mediators increased in C1-INH-HAE on different immune cells
Additionally, MO secrete a wide spectrum of mediators including complement components such as C1-INH [17]. Hepatocytes are the major cellular source of plasma C1-INH [18]. MO contribute to C1-INH production, particularly at the site of inflammation where INF-γ is a potent inducer [19, 20]. Understanding the regulation of C1-INH synthesis by MO is essential to evaluate their potential role in C1-INH-HAE.
Lipopolysaccharide (LPS) does not trigger an increase in C1-INH levels in MO cultures, but it induces high levels of IL-1. LPS may play a role in regulation of C1-INH synthesis through the induction of IL-1, which is essential for T cell activation to yield IFN-γ through the induction of C1-INH in MO and hepatocytes [21]. LPS raised also C3 production by MO [22] but did not stimulate C1q and C1s secretion. Moreover, C2 was increased by IFN-γ to a similar extent as C1-INH [23, 24], whereas it did not affect C3 synthesis [23]. MO do not produce C4 in MO culture supernatants [17].
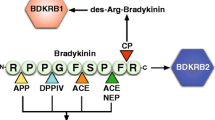
Another link to the pathophysiology of C1-INH-HAE might be the effect of BK on MO; BK is formed downstream the kallikrein-kinin system (KKS) and is unquestionably the most important mediator in C1-INH-HAE [25]. The vasoactive effects of BK are mediated by the cell surface BKR1 and BKR2 receptors expressed on several cell types. The existence and modulation of these receptors in MO are still limited. Activation of BKR1 promotes MO chemotaxis and arteriogenesis, whereas BKR2 signaling governs MO recruitment (Fig. 2) [26,27,28]. Therefore, it can be hypothesized that the BK increase in HAE could cause an activation of circulating MO.

Schematic representation of bradykinin effects on different immune cells
Expression of tissue factor (TF) by MO may represent another link between these cells and angioedema. TF may play a role in angioedema attacks by activating the coagulation pathway in association with reduced functions of C1‐INH [29]. TF, predominantly expressed in vessel wall, forms a complex with FVIIa and initiates the extrinsic coagulation pathway. TF is also present in the cytoplasm and on the surface of MO and can be released by various exogenous/endogenous inflammatory stimuli. The TF expression is up-regulated by LPS, immune complexes, C5a, cytokines, and oral contraceptives, which have been suggested as risk factor for angioedema [29]. TF is expressed in urticarial skin lesions with evidence of activation of the extrinsic pathway [30]. An increase of TF was demonstrated on MO of patients with C1-INH-HAE during remission compared with healthy controls [29]. C1‐INH efficiently inhibited LPS‐induced TF on MO, suggesting that MO of HAE due to C1‐INH deficiency are susceptible to induction TF expression. This is supported by four‐fold increase in TF expression a day after the infusion of C1‐INH [31]. However, no difference in TF expression was shown between remission and attack of HAE [31]. Taken together, in HAE the combination of low C1‐INH activity, subclinical activation of extrinsic coagulation pathways triggered by TF, may be important to the pathophysiology of angioedema. Further studies are needed to confirm whether the increase of TF expression on MO and the coagulation system activation observed is the cause or just an epiphenomenon in HAE attacks [29]. The role of MO in the pathophysiology of angioedema involves several mediators that can modulate MO activation (Table 1; Figs. 1 and 2).
Macrophages
The barrier properties of EC are critical for the maintenance of fluid and protein balance between the intravascular and extravascular compartments [32]. Imbalance of these barriers is implicated in the genesis or progression of angioedema [12]. The altered barrier function is linked to the release of a variety of soluble mediators acting on EC [32, 33] produced by resident cells, including macrophages [34].
Macrophages (MA) are innate immune cells that are localized in various tissues [35]. Most MA are derived from MO that migrate into connective tissues [36]. MA exert a variety of functions like phagocyting foreign agents, digesting dead cells, and regulation of innate immune response by releasing of several molecules [37]. Mediator secretion by MA is implicated in several disease states ranging, from chronic inflammation to allergy [36]. MA release upon inflammatory stimulation a plethora of inflammatory (e.g., TNF-α, IL-1, IL-6, CXCL8, IL-12) and anti-inflammatory cytokines (IL-10 and TGF-β) [36]. Several MA-derived cytokines are altered in serum of HAE patients [38] (Table 1). Concentrations of IL-1, IL-6, and TGF-β are significantly higher in HAE patients in remission compared with healthy controls [38]. These cytokines, plus IL-10, are further increased in HAE patients during attacks compared with asymptomatic period. In addition, IL-1 and TNF-α have been reported to stimulate EC and augment activation of the prekallikrein (PK)–high molecular weight kininogen (HK) complex, suggesting a possible role in the pathophysiology of HAE [39].
MA are a major source of VEGFs and angiopoietins (ANGPTs) [40, 41]. Their concentrations are increased in C1-INH-HAE patients and correlate with disease severity [14, 15] (Table 1).
Extracellular or secreted phospholipases A2 (sPLA2s) also modulate EC and vascular permeability. PLA2s enzymes hydrolyze membrane glycerol-phospholipids to release arachidonic acid and lyso-phospholipid [42, 43]. sPLA2s can modulate vascular permeability either by directly activating EC or by catalyzing the production/degradation of vasoactive molecules [43]. MA are a target for sPLA2 (Fig. 1). These enzymes activate MA and induce the production of cytokines, chemokines, VEGFs, and ANGPTs [40, 44, 45]. We found that sPLA2 activity is increased in biological fluid of C1-INH-HAE patients during symptom-free period compared with healthy controls [46]. sPLA2 group IIA (PLA2G2A) in C1-INH-HAE increases endothelial permeability and impairs C1-INH functional activity in vitro [46]. ANGPT1, a unique vascular stabilizer, is further increased during angioedema attack, whereas sPLA2 activity is decreased [46, 47] (Table 1).
Since BK is a potent vasodilator, promoter of vascular permeability [25]. It hypothesized that activated BKR2 on EC and/or on MA and/or mast cells may account for the altered levels of cytokines, angiogenic/lymphangiogenic factors, and sPLA2 in C1-INH-HAE patients [16, 48, 49]. MA express both BKR1 and BKR2 [50]. BK also induces TNF-α and IL-1 release from murine MA cell lines [52, 53] and stimulates prostaglandin E2 production from rat peritoneal MA [54] (Fig. 2). The activation of BKRs increases intracellular free calcium, which activates sPLA2 and consequently induces arachidonic acid release and its metabolites [51]. BK is more potent to activate intermediate-size MA compared with smaller peritoneal and alveolar MA [50]. Collectively, these findings indicate that overproduction of BK in HAE patients may affect the MA activation and their inflammatory responses in vivo.
MA are well known for their phagocytic activity and are highly specialized in removal of dying or dead cells [37]. Phagocytosis is facilitated by opsonization, a process by which serum components tag pathogens for recognition by MA and neutrophils. Opsonization is mediated by C1, C3, and C4 which are components of the complement classical pathway [3].
In C1-INH-HAE patients, low concentration/activity of C1-INH causes a gradual consumption of complement proteins in serum. Sera from HEA patients reduced the ability of MA to phagocyte apoptotic cells compared with sera from healthy donors [55]. Therefore, C1-INH-HAE patients can have immunological abnormalities due to decreased levels of complement components, which give rise to a lower capacity for opsonization from phagocyte cells including MA [55].
In conclusion, the role of MA in the pathophysiology of HAE is not yet completely understood. However, the ability of MA (1) to modulate vascular permeability by catalyzing the production of vasoactive molecules; (2) to be activated by key mediators of HAE, such as BK and sPLA2; and (3) to modify opsonization capacity suggests that these cells play an important role in both asymptomatic and symptomatic phases of HAE.
Neutrophils
Neutrophils, or polymorphonuclear leukocytes (PMN), are major effectors in innate immunity and acute inflammation [56]. They are circulating cells that must be lured into inflamed tissue by crossing the endothelial barrier. Sequential adhesive interactions between PMN and ECs are required for PMN extravasation. Adhesion molecules (i.e., ICAM-1, VCAM) lead to adhesion and arrest onto the endothelium and a subsequent PMN transmigration in the tissue where they play a critical role in pathogen elimination and tissue repair by releasing several cytotoxic products and reactive oxygen species (ROS) [57]. New evidences have highlighted our knowledge on PMN as cells playing a role beyond the acute infection including HAE [58].
PMN count is increased in C1-INH-HAE patients during edematous episodes. This PMN imbalance was attributed to the hemoconcentration caused by plasma extravasation during angioedema attack [59,60,61,62]. Veszeli et al. demonstrated a higher PMN count also in C1-INH-HAE patients during symptom-free period compared with healthy controls [63]. These authors described an increased release of neutrophil granule-derived enzymes in plasma (i.e., myeloperoxidase (MPO), elastase (NE), and pentraxin 3 (PTX3)) during attacks but not during attack-free period and in healthy controls. Plasma concentration of these enzymes was correlated with neutrophil counts. The increased plasma levels of MPO, NE, and PTX3 were attributed to neutrophil extracellular trap (NETs) release. Interestingly, CXCL8 and TNF-α levels, both involved in PMN activation and/or released by neutrophils, were also altered in C1-INH-HAE patients during acute phase compared with symptom-free period [63] (Table 1). Grymova and colleagues confirmed PMN activation and dysregulation in C1-INH-HAE type I and II patients [64]. mRNA expression of 10 genes related to PMN activation (CD274, IL1β, IL1RN, CXCL8, MMP9, and TLR4) was increased in HAE patients in symptom-free periods compared with healthy donors in addition to increased CD11b, decreased CD16 plasma membrane deposition, and increased relative CD274+ and CD87+ neutrophil counts, but not by increased NE or MPO plasma levels (Table 1). In addition, a co-culture of PMN and T-cells/PBMC showed a suppressive function of patient’ PMN resulted from a decreased CD25+ and IFN-γ+ T-cell/PBMC ratio in patients [64].
PMN can interact with the contact system in order to boost neutrophil extravasation induced by BK-mediated vasodilatation [65]. Brower et al. reported that NE can inactivate C1-INH allowing to contact system activation [66]. In addition, in vitro studies showed that BKR1 on ECs regulates neutrophil trafficking [67, 68]. BK levels are increased in HAE patients compared with healthy controls leading to PMN activation. In fact, BK increased the PMN adhesion to ECs [69, 70] and induced only a moderate migration of human peripheral PMN in BKR2-manner [51]. In addition, BK mediated NE release by PMN [71] (Fig. 2).
Neutrophil-derived proteinase 3 can proteolyze HK and liberate proteinase 3-kinin, thereby initiating kallikrein-independent activation of the kinin pathway [72]. Wachtfogel et al. reported that kallikrein (PKa) and Factor XIIa (FXIIa) can induce PMN degranulation [73]. Finally, it has been shown that NETs can activate FXII through several mechanisms. The negative charge of DNA could contribute to auto-activation of FXII or can sequester FXII and present it for activating cleavage [74].
PMN are source and/or target of several mediators and play a role in different context (i.e., inflammation, angiogenesis) [75]. Different isoforms of sPLA2s (e.g., groups II, V, and X) can activate human PMN inducing NE, CXCL8, or angiogenic factors (VEGFs and ANGPTs) release [76,77,78,79,80]. Therefore, increase of PLA2G2A in C1-INH-HAE patients could affect the activation of PMN and be responsible of release for the VEGFs and ANGPTs [14, 46, 47] (Fig. 1). Taken together, these results confirm the involvement of PMNs in the pathophysiology of angioedema by releasing mediators and induce endothelial preconditioning state, thereby predisposing HAE patients to edema formation.
Eosinophils and Basophils
Eosinophils and basophils are immune cells activated in several pathological conditions (e.g., allergic diseases, infections, cancer) [81, 82]. These cells are characterized by different phenotypes and by the ability to respond to specific stimuli, activating and inhibiting surface receptors [83]. Eosinophils derive from CD34+CD117+ hematopoietic stem cells in the BM. After their maturation they enter the circulation [84]. Activated eosinophils release pro-inflammatory cationic proteins, cytokines and chemokines, angiogenic [49, 85], and lipid mediators [86]. They also migrate to the inflammatory sites through the adhesion to activated ECs [84, 87, 88]. The involvement of eosinophils in HAE has not been investigated. They could be the source of VEGFs, TNF-α, and sPLA2 found in plasma of HAE patients. Moreover, eosinophils can be activated by BK leading to elastase release [71] and chemotaxis [89] (Fig. 2).
Episodic angioedema with eosinophilia (EAE), also known as Gleich syndrome, is a rare disorder characterized by recurrent episodes of angioedema, urticaria, fever, and marked eosinophilia that occur at 3–4 week intervals [90]. A peak of marked eosinophilia is preceded by a rise in serum IL-5 and IL-13 in EAE patients [91]. These findings suggest that immune cell-derived Th2-like cytokines are involved in this form of angioedema. However, the etiology of the cycling angioedema and eosinophilia remains to be elucidated.
Basophils represent less than 1% of peripheral blood leukocytes, and their activation leads to histamine release [92]. They are rarely present in tissues unless inflammation occurs [93, 94]. Basophil activation induces the release of VEGF-A [95, 96] and ANGPT1 and ANGPT2 (Table 1). Human basophils also produce CXCL8 [97]. Some of those mediators (e.g., VEGFs, ANGPTs, sPLA2, and CXCL8) were altered in HAE contributing to alteration of vascular homeostasis. Collectively, these data may suggest a potential role of eosinophils and basophils in the pathophysiology of certain forms of HAE.
Mast Cells
Mast cells (MC) can be identified in blood vessels, within mucosal and epithelial tissues and in the terminal nerve endings [98]. These cells release several preformed pro-inflammatory mediators (e.g., histamine, tryptase, chymase) [99]. MC derive from CD34+CD117+ (KIT) hematopoietic stem cells in the BM [100] and migrate as immature progenitor cells through the bloodstream to peripheral tissues where they complete maturation [101].
In this paper, we discuss the role of MC in angioedema without wheals. MC-mediated angioedema is pathophysiologically similar to urticaria, although it occurs in deeper levels of the dermis and involves probably different mediators. Except for C1-INH-HAE, the pathophysiology of angioedema without wheals is not completely clear. MC release several vasoactive mediators (e.g., histamine, prostaglandins, cysteinyl leukotrienes) contributing to extravasation of fluid in the deeper layers of the skin/mucosa of angioedema patients [102]. The canonical mechanism of MC activation is IgE-mediated [101]. However, in most patients, angioedema develops without an interaction between IgE-antigen complex bound to MC. Several non-IgE-mediated stimuli (e.g., drugs, C5a, C3a) can induce human MC degranulation [103].
Histamine is a relevant vasoactive amine contained in MC granules. It binds to H1-receptors on ECs, inducing vasodilatation; increases blood flow; and causes vessel [104]. Histamine stimulates nitric oxide expression and increases blood flow and plasma extravasation causing angioedema [105]. Most cases of angioedema are attributable to the vasoactive mediator BK and histamine. MC express BKR2 through which BK induces histamine release [106,107,108] (Fig. 2). Angioedema attacks of HAE patients are unresponsive to antihistamines and glucocorticoids [109]. Histamine is presumably not the main mediator of angioedema. In this paper, we present original results indicating that 102 patients with C1-INH-HAE in remission have increased concentrations of histamine compared with 64 healthy controls (Fig. 3a). We also measured the tryptase concentrations in these patients. Tryptase is a specific marker of MC activation [110]. We found that tryptase levels in C1-INH-HAE patients in remission are not altered compared with controls (Fig. 3b). These results are of some interest because histamine is secreted by basophils and MC, whereas tryptase is essentially released by MC [92, 111]. Therefore, it is possible to hypothesize that the increase in blood histamine in patients with C1-INH-HAE in remission derives essentially from basophils rather than MC. It would be interesting to evaluate tryptase and histamine levels during attacks in order to better understand their roles and the cellular sources (MC and/or basophils) in acute phase of angioedema.

Plasma concentrations of histamine and tryptase in C1-INH-HAE patients. The graph depicts plasma histamine (a) and tryptase (b) in 64 controls (Healthy) and in 102 patients with C1-INH-HAE in remission. Histamine was measured by ELISA. Tryptase was measured by fluoro-enzyme immune assay using Uni-CAP100. Data are shown as the median (horizontal black line), the 25th and 75th percentiles (boxes) and the 5th and 95th percentiles (whiskers) of 64 controls and 102 patients
Tryptase releases BK mainly through plasma kallikrein (PKa) activation and enhanced vascular permeability [112]. Sala-Cunill et al. demonstrated that tryptase levels are correlated to plasma HK cleavage during anaphylaxis [113]. These findings could indicate that tryptase might contribute to kinin cleavage and consequently BK production in angioedema.
Stimulation of MC can release PLA2. Human lung MC express and release multiple sPLA2s when activated by anti-IgE [114]. The sPLA2s released by MC contribute to leukotriene C4 production by acting in an autocrine fashion. PLA2 plasma activity was increased in patients with C1-INH-HAE during remission and was decreased during acute attack perhaps because of their activation and internalization in EC [46]. The cellular sources of these mediators remain unclear but could be partially attributable to MC activation in C1-INH-HAE (Table 1).
The proteoglycan matrix in MC cytoplasmic granules is mostly composed by heparin [115]. Heparin can initiate in vivo the contact system cascade activating FXIIa [116] and in turn BK production. Heparin appears to have a dual function in FXII activation: it has the negatively charged surface for binding and activation of plasmatic FXII [117]. and it blocks FXII inhibition binding antithrombin III [118]. Oschatz et al. described a paracrine mechanism by which MC-released heparin induces contact system-driven edema in mice [119]. These results suggest that MC activation and heparin can contribute to attacks in HAE patients [119].
Another MC mediator potentially capable of activating the contact system is elastase. It cleaves the light chain of HK and seems to be a positive regulator of the contact system activation [120]. Taken together, these findings indicate that MC degranulation may trigger FXII activation and the generation of BK through the release of heparin, tryptase, and elastase or other mediators. The potential roles of other MC mediators in contact system activation need to be further explored.
Adaptive Immune System
Lymphocytes
Lymphocytes mediate adaptive immune responses by providing the lifelong immunity following exposure to antigens [4]. These cells originate in the BM and migrate to tissues by circulating in the blood and in the lymphatic system [121]. There are several different lymphocytes including B and T cells, natural killer cells (NK cells), and innate lymphoid cells [122, 123]. Activated B lymphocytes differentiate into plasma cells, which secrete antibodies. T cells include by two main classes: CD8+ cytotoxic T cells and CD4+ T cells [124]. T and B cells express different receptors to recognize a wide spectrum of antigens [124]. The antigen receptor of B lymphocytes is the membrane isoform of IgM [125]. The T cell receptor (TCR) on T cells recognizes protein antigens or metabolized by dendritic cells and MAs.
Adaptive immunity has been poorly studied in HAE. Few reports have reported abnormal T and B cell counts, abnormal distribution of T cell surface IgG-receptors, and reduced Langerhans cell numbers in HAE patients [126, 127].
Several studies examined the involvement of cytokines in HAE. Arcoleo et al. comparing C1-INH-HAE patients before and after the acute attack with matched control subjects observed several modifications of IL-17 lymphokine network [38]. IL-17 concentrations were increased, whereas IL-23 levels were unmodified and TGF-β3 concentrations were reduced [38]. Comparing healthy and HAE subjects in remission, they found a significant difference for IL-17, GM-CSF, IL-21, and TGF-β1/2 [38]. These suggests that in HAE subjects there is a cytokine milieu favoring expansion of Th17 or Th17-type subsets capable of producing cytokines associated with contact activation by BK leading to local angioedema formation responsible of increase in permeability and subcutaneous swelling [38, 128, 129]. Th17 expansion could down-modulate inflammatory response favoring the natural resolution of angioedema [38, 130,131,132,133].
The alterations of circulating cytokines suggest HAE is a complex disorder caused by generation of BK associated with increase in several cytokines (Fig. 3). Lopez-Lera et al. evaluated the expression of HAE by profiling the RNA expression of peripheral blood mononuclear cells (PBMC) from C1-INH-HAE families [134]. This study did not reveal alterations in the expression pattern of PBMC in association to frequency and severity of disease [134]. Castellano et al., using a different approach, explored the involvement of several putative genes by performing a microarray gene expression analysis on RNA isolated from PBMC of HAE patients during attacks and in remission. They demonstrated the up-regulation of adrenomedullin (ADM) and cellular receptor for urokinase plasminogen activator (uPAR), during the acute attack. These gene activations involved in vascular tone regulation and in inflammatory response might have a pathogenic role by amplifying BK production and edema formation in HAE patients [135]. uPAR is a glycosylphosphatidylinositol-anchored protein [136] that binds uPA [137]. The function of uPA is the conversion of plasminogen to plasmin. uPAR is expressed by resting granulocytes and monocytes and by activated lymphocytes [138]. uPAR interacts with components of the BK-forming cascade. The neutralization of uPAR expressed on T cells leads to a reduction of BK. This observation highlights a potential role for adaptive immunity to modulate the edema formation through regulation of BK production [135].
Patients with HAE tend to produce autoantibodies. Kessel et al. demonstrated that HAE patients have an increase of autoantibodies presumably due to the activation of B cells associated with over-expression of TLR9 which plays a role in the induction and maintenance of autoimmunity [139].
Infection/Inflammation
As previously mentioned, an interesting aspect is the possibility that infections trigger angioedema attacks [140]. Bacteriuria and Helicobacter pylori could represent triggers of angioedema attacks [141, 142]. The observations were attributed to the excessive consumption of complement by antibodies directed against bacteria. The antibody response and the formation of immune complexes may trigger the consumption of already reduced C1-INH in HAE patients [134, 143].
Neutrophil-lymphocyte ratio (NLR) is a simple and easily used parameter for the assessment of inflammation. It has been found a positive correlation between the angioedema attack and NLR [144] suggesting that the NLR could be useful as a predictive biomarker for prediction of the attack in HAE patients.
To conclude this section, the roles of different subsets of lymphocytes in the pathophysiology of angioedema have not been thoroughly studied. Cells of adaptive immunity could have a role in the regulation of the severity of this disease in different forms of angioedema. Further studies with RNA sequencing and proteomic technologies will clarify the possible roles of multiple cells involved in adaptative immunity in the pathophysiology of angioedema attack.
Conclusions
In this review, we have summarized the results of relatively few studies examining the roles played by immune cells presumably involved in HAE. We have tried to distill the contribution that each immune cell can exert directly or indirectly in the pathophysiology of angioedema. The genetics and the resulting protein alterations of the majority of HAE patients are well characterized.
Acute phase of a disease is characterized by a transient increase of vascular permeability followed by the formation of local edema. Current research is focusing on EC receptors and the mechanisms of their activation in different phenotypes of angioedema. There is compelling evidence that the endothelium actively participates in both innate and adaptive immune responses. EC are in a strategic location to activate the circulating immune cells and those that transmigrate across the endothelium into the tissues. The roles played by EC in the recruitment of immune cells into lymph nodes and tissues highlight an intimate relationship between EC and immune cells [145, 146]. Therefore, the altered vascular permeability in both remission and acute phase of HAE can affect the effector functions of several immune cells.
The circulating levels of several mediators are altered in remission and/or during attack in HAE patients (Table 1). Activated immune cells might be the source of these molecules in the context of HAE. Moreover, the increased levels of several mediators can, in turn, activate the immune cells through the engagement of specific surface receptors (Fig. 1). In this paper, we have also discussed the effects of a variety of mediators on immune cells (Fig. 2).
In summary, it appears that the role of the multiple cells of innate and adaptive immune system in the pathophysiology of angioedema has not been thoroughly investigated. A better knowledge of these mechanisms could open new diagnostic and therapeutic opportunities for the different forms of angioedema.
Abbreviations
- AAE:
-
Acquired angioedema
- ADM:
-
Adrenomedullin
- ANGPT:
-
Angiopoietin
- b-FGF:
-
Basic fibroblast growth factor
- BKR:
-
Bradykinin receptor
- BK:
-
Bradykinin
- C1-INH:
-
C1-inhibitor
- EAE:
-
Episodic angioedema with eosinophilia
- EC:
-
Endothelial cell
- ECP:
-
Eosinophil cationic protein
- EDN:
-
Eosinophil-derived neurotoxin
- EPX:
-
Eosinophil peroxidase
- fMLP:
-
N-formyl-methionine-leucyl-phenylalanine
- FXII:
-
Factor XII
- GM‐CSF:
-
Granulocyte–macrophage colony‐stimulating factor
- HAE:
-
Hereditary angioedema
- His:
-
Histamine
- HK:
-
High molecular weight kininogen
- ICAM:
-
Intercellular adhesion molecule
- INF:
-
Interferon
- KKS:
-
Kallikrein-kinin system
- LFA:
-
Lymphocyte function-associated antigen
- LPS:
-
Lipopolysaccharide
- MA:
-
Macrophage
- MBP:
-
Major basic protein
- MC:
-
Mast cell
- M‐CSF:
-
Macrophage colony‐stimulating factor
- MMP9:
-
Matrix metallopeptidase 9
- MO:
-
Monocyte
- MPO:
-
Mieloperoxydase
- NE:
-
Neutrophil elastase
- NET:
-
Neutrophil extracellular trap
- NGF:
-
Nerve growth factor
- NK:
-
Natural killer
- NLR:
-
Neutrophil-lymphocyte ratio
- PLA2G2A:
-
Secretory phospholipase A2 group IIA
- PMN:
-
Polymorphonuclear cells
- PTX3:
-
Pentraxin 3
- ROS:
-
Reactive oxygen species
- sPLA2 :
-
Secretory phospholipases A2
- TCR:
-
T cell receptor
- TF:
-
Tissue factor
- TGF:
-
Transforming growth factor
- TLR:
-
Toll-like receptor
- TNF:
-
Tumor necrosis factor
- uPAR:
-
Urokinase receptor
- VEGF:
-
Vascular endothelial growth factor
References
Maurer M, Magerl M, Ansotegui I, Aygoren-Pursun E, Betschel S, Bork K, Bowen T et al (2018) The international WAO/EAACI guideline for the management of hereditary angioedema-The 2017 revision and update. Allergy 73:1575–1596
Ling M, Murali M (2019) Analysis of the complement system in the clinical immunology laboratory. Clin Lab Med 39:579–590
Dunkelberger JR, Song WC (2010) Complement and its role in innate and adaptive immune responses. Cell Res 20:34–50
Nielsen CH, Leslie RG (2002) Complement’s participation in acquired immunity. J Leukoc Biol 72:249–261
Chaplin DD (2010) Overview of the immune response. J Allergy Clin Immunol 125:S3-23
Germenis AE, Speletas M (2016) Genetics of hereditary angioedema revisited. Clin Rev Allergy Immunol 51:170–182
Swirski FK, Nahrendorf M, Etzrodt M, Wildgruber M, Cortez-Retamozo V, Panizzi P, Figueiredo JL, Kohler RH, Chudnovskiy A, Waterman P, Aikawa E, Mempel TR, Libby P, Weissleder R, Pittet MJ (2009) Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 325:612–616
Guilliams M, Mildner A, Yona S (2018) Developmental and Functional Heterogeneity of Monocytes. Immunity 49:595–613
Hijdra D, Vorselaars AD, Grutters JC, Claessen AM, Rijkers GT (2012) Differential expression of TNFR1 (CD120a) and TNFR2 (CD120b) on subpopulations of human monocytes. J Inflamm 9:38
Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F (2007) Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317:666–670
Carlin LM, Stamatiades EG, Auffray C, Hanna RN, Glover L, Vizcay-Barrena G, Hedrick CC, Cook HT, Diebold S, Geissmann F (2013) Nr4a1-dependent Ly6C(low) monocytes monitor endothelial cells and orchestrate their disposal. Cell 153:362–375
Wu MA, Bova M, Berra S, Senter R, Parolin D, Caccia S, Cicardi M (2020) The central role of endothelium in hereditary angioedema due to C1 inhibitor deficiency. Int Immunopharmacol 82:106304
Dvorak HF, Nagy JA, Feng D, Brown LF, Dvorak AM (1999) Vascular permeability factor/vascular endothelial growth factor and the significance of microvascular hyperpermeability in angiogenesis. Curr Top Microbiol Immunol 237:97–132
Loffredo S, Bova M, Suffritti C, Borriello F, Zanichelli A, Petraroli A, Varricchi G, Triggiani M, Cicardi M, Marone G (2016) Elevated plasma levels of vascular permeability factors in C1 inhibitor-deficient hereditary angioedema. Allergy 71:989–996
Loffredo S, Marone G (2018) Hereditary angioedema: the plasma contact system out of control: comment. J Thromb Haemost 16:2347–2348
Loffredo S, Staiano RI, Granata F, Genovese A, Marone G (2014) Immune cells as a source and target of angiogenic and lymphangiogenic factors. Chem Immunol Allergy 99:15–36
Yeung Laiwah AC, Jones L, Hamilton AO, Whaley K (1985) Complement-subcomponent-C1-inhibitor synthesis by human monocytes. Biochem J 226:199–205
Johnson AM, Alper CA, Rosen FS, Craig JM (1971) C1 inhibitor: evidence for decreased hepatic synthesis in hereditary angioneurotic edema. Science 173:553–554
Werb ZBM, Takemura R, Gordon S (1986) Secreted proteins of resting and activated macrophages . In handbook of Experimental Immunology. In Cellular immunology, Volume 2 (D. M. W. E. B. Oxford, ed)
Lotz M, Zuraw BL (1987) Interferon-gamma is a major regulator of C1-inhibitor synthesis by human blood monocytes. J Immunol 139:3382–3387
Zuraw BL, Lotz M (1990) Regulation of the hepatic synthesis of C1 inhibitor by the hepatocyte stimulating factors interleukin 6 and interferon gamma. J Biol Chem 265:12664–12670
Strunk RC, Whitehead AS, Cole FS (1985) Pretranslational regulation of the synthesis of the third component of complement in human mononuclear phagocytes by the lipid A portion of lipopolysaccharide. J Clin Investig 76:985–990
Strunk RC, Cole FS, Perlmutter DH, Colten HR (1985) gamma-Interferon increases expression of class III complement genes C2 and factor B in human monocytes and in murine fibroblasts transfected with human C2 and factor B genes. J Biol Chem 260:15280–15285
Sanders KM, Littman BH (1986) Lymphokine stimulation of human macrophage C2 production is partially due to interferon-gamma. J Immunol 137:876–879
Marcelino-Rodriguez I, Callero A, Mendoza-Alvarez A, Perez-Rodriguez E, Barrios-Recio J, Garcia-Robaina JC, Flores C (2019) Bradykinin-mediated angioedema: an update of the genetic causes and the impact of genomics. Front Genet 10:900
Bertram CM, Baltic S, Misso NL, Bhoola KD, Foster PS, Thompson PJ, Fogel-Petrovic M (2007) Expression of kinin B1 and B2 receptors in immature, monocyte-derived dendritic cells and bradykinin-mediated increase in intracellular Ca2+ and cell migration. J Leukoc Biol 81:1445–1454
Hillmeister P, Gatzke N, Dulsner A, Bader M, Schadock I, Hoefer I, Hamann I, Infante-Duarte C, Jung G, Troidl K, Urban D, Stawowy P, Frentsch M, Li M, Nagorka S, Wang H, Shi Y, le Noble F, Buschmann I (2011) Arteriogenesis is modulated by bradykinin receptor signaling. Circ Res 109:524–533
Marketou ME, Kontaraki J, Zacharis E, Parthenakis F, Maragkoudakis S, Gavras I, Gavras H, Vardas PE (2014) Differential gene expression of bradykinin receptors 1 and 2 in peripheral monocytes from patients with essential hypertension. J Hum Hypertens 28:450–455
Iwamoto K, Morioke S, Yanase Y, Uchida K, Hide M (2014) Tissue factor expression on the surface of monocytes from a patient with hereditary angioedema. J Dermatol 41:929–932
Asero R, Tedeschi A, Coppola R, Griffini S, Paparella P, Riboldi P, Marzano AV, Fanoni D, Cugno M (2007) Activation of the tissue factor pathway of blood coagulation in patients with chronic urticaria. J Allergy Clin Immunol 119:705–710
Landsem A, Nielsen EW, Fure H, Christiansen D, Ludviksen JK, Lambris JD, Osterud B, Mollnes TE, Brekke OL (2013) C1-inhibitor efficiently inhibits Escherichia coli-induced tissue factor mRNA up-regulation, monocyte tissue factor expression and coagulation activation in human whole blood. Clin Exp Immunol 173:217–229
Rodrigues SF, Granger DN (2015) Blood cells and endothelial barrier function. Tissue Barriers 3:e978720
Yuan SY, Rigor RR (2010) Regulation of Endothelial Barrier Function. Morgan & Claypool Life Sciences
Kalucka J, Bierhansl L, Wielockx B, Carmeliet P, Eelen G (2017) Interaction of endothelial cells with macrophages-linking molecular and metabolic signaling. Pflugers Arch- Eur J Physiol 469:473–483
Varol C, Mildner A, Jung S (2015) Macrophages: development and tissue specialization. Annu Rev Immunol 33:643–675
Arango Duque G, Descoteaux A (2014) Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol 5:491
Gordon S, Pluddemann A (2017) Tissue macrophages: heterogeneity and functions. BMC Biol 15:53
Arcoleo F, Lo Pizzo M, Misiano G, Milano S, Romano GC, Muggeo V, Cillari E (2018) The complex alteration in the network of IL-17-type cytokines in patients with hereditary angioedema. Clin Exp Med 18:355–361
Joseph K, Tholanikunnel BG, Kaplan AP (2017) Cytokine and estrogen stimulation of endothelial cells augments activation of the prekallikrein-high molecular weight kininogen complex: Implications for hereditary angioedema. J Allergy Clin Immunol 140:170–176
Granata F, Frattini A, Loffredo S, Staiano RI, Petraroli A, Ribatti D, Oslund R, Gelb MH, Lambeau G, Marone G, Triggiani M (2010) Production of vascular endothelial growth factors from human lung macrophages induced by group IIA and group X secreted phospholipases A2. J Immunol 184:5232–5241
Staiano RI, Loffredo S, Borriello F, Iannotti FA, Piscitelli F, Orlando P, Secondo A, Granata F, Lepore MT, Fiorelli A, Varricchi G, Santini M, Triggiani M, Di Marzo V, Marone G (2016) Human lung-resident macrophages express CB1 and CB2 receptors whose activation inhibits the release of angiogenic and lymphangiogenic factors. J Leukoc Biol 99:531–540
Granata F, Nardicchi V, Loffredo S, Frattini A, Ilaria Staiano R, Agostini C, Triggiani M (2009) Secreted phospholipases A(2): A proinflammatory connection between macrophages and mast cells in the human lung. Immunobiology 214:811–821
Lambeau G, Gelb MH (2008) Biochemistry and physiology of mammalian secreted phospholipases A2. Annu Rev Biochem 77:495–520
Granata F, Frattini A, Loffredo S, Del Prete A, Sozzani S, Marone G, Triggiani M (2006) Signaling events involved in cytokine and chemokine production induced by secretory phospholipase A2 in human lung macrophages. Eur J Immunol 36:1938–1950
Triggiani M, Granata F, Petraroli A, Loffredo S, Frattini A, Staiano RI, Monaco G, Marone G (2009) Inhibition of secretory phospholipase A2-induced cytokine production in human lung macrophages by budesonide. Int Arch Allergy Immunol 150:144–155
Loffredo S, Ferrara AL, Bova M, Borriello F, Suffritti C, Veszeli N, Petraroli A, Galdiero MR, Varricchi G, Granata F, Zanichelli A, Farkas H, Cicardi M, Lambeau G, Marone G (2018) Secreted phospholipases A2 in hereditary angioedema with C1-inhibitor deficiency. Front Immunol 9:1721
Ferrara AL, Bova M, Petraroli A, Veszeli N, Galdiero MR, Braile M, Marone G, Cristinziano L, Marcella S, Modestino L, Farkas H, Loffredo S (2020) Hereditary angioedema attack: what happens to vasoactive mediators? Int Immunopharmacol 78:106079
Terzuoli E, Meini S, Cucchi P, Catalani C, Cialdai C, Maggi CA, Giachetti A, Ziche M, Donnini S (2014) Antagonism of bradykinin B2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-kB pathway activation. PLoS One 9:e84358
Varricchi G, Granata F, Loffredo S, Genovese A, Marone G (2015) Angiogenesis and lymphangiogenesis in inflammatory skin disorders. J Am Acad Dermatol 73:144–153
Bockmann S, Mohrdieck K, Schmidt H, Zundorf G, Paegelow I (1998) Differential sensitivity of macrophages to bradykinin. Naunyn Schmiedebergs Arch Pharmacol 357:151–158
Bockmann S, Paegelow I (2000) Kinins and kinin receptors: importance for the activation of leukocytes. J Leukoc Biol 68:587–592
Burch RM, Kyle DJ (1992) Recent developments in the understanding of bradykinin receptors. Life Sci 50:829–838
Bockmann S, Mohrdieck K, Paegelow I (1999) Influence of interleukin-1 beta on bradykinin-induced responses in guinea pig peritoneal macrophages. Inflammation research : official journal of the European Histamine Research Society ... [et al.] 48:56–62
Sakamoto W, Kaga M, Handa H, Gotoh K, Suzuki S, Fujie K, Inoue N, Nishihira J (1994) Bradykinin and Met-T-kinin-Leu stimulated PGE2 production by rat macrophage and fibroblast. Brazilian journal of medical and biological research = Revista brasileira de pesquisas medicas e biologicas 27:1923–1927
Honda D, Ohsawa I, Sato N, Inoshita H, Mano S, Tomino Y, Suzuki Y (2017) Diminished capacity of opsonization and immune complex solubilization, and detection of anti-C1q antibodies in sera from patients with hereditary angioedema. Allergology international : official journal of the Japanese Society of Allergology 66:603–609
Kolaczkowska E, Kubes P (2013) Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol 13:159–175
El-Benna J, Hurtado-Nedelec M, Marzaioli V, Marie JC, Gougerot-Pocidalo MA, Dang PM (2016) Priming of the neutrophil respiratory burst: role in host defense and inflammation. Immunol Rev 273:180–193
Silvestre-Roig C, Hidalgo A, Soehnlein O (2016) Neutrophil heterogeneity: implications for homeostasis and pathogenesis. Blood 127:2173–2181
Koruth JS, Eckardt AJ, Levey JM (2005) Hereditary angioedema involving the colon: endoscopic appearance and review of GI manifestations. Gastrointest Endosc 61:907–911
Kodama J, Uchida K, Yoshimura S, Katayama Y, Kushiro H, Yutani C, Funahashi S, Takamiya O, Matsumoto Y, Ando Y et al (1984) Studies of four Japanese families with hereditary angioneurotic edema: simultaneous activation of plasma protease systems and exogenous triggering stimuli. Blut 49:405–418
Cohen N, Sharon A, Golik A, Zaidenstein R, Modai D (1993) Hereditary angioneurotic edema with severe hypovolemic shock. J Clin Gastroenterol 16:237–239
Goti F, Melcher GA, Spath P, Wuthrich B (1998) Hereditary angioedema. A rare cause of acute abdominal pain with ascites. Dtsch Med Wochenschr 123:1166–1171
Veszeli N, Csuka D, Zotter Z, Imreh E, Jozsi M, Benedek S, Varga L, Farkas H (2015) Neutrophil activation during attacks in patients with hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis 10:156
Grymova T, Vlkova M, Soucek P, Hakl R, Nechvatalova J, Slanina P, Stichova J, Litzman J, Freiberger T (2019) Neutrophils are dysregulated in patients with hereditary angioedema types I and II in a symptom-free period. Mediators Inflamm 2019:9515628
Hofman Z, de Maat S, Hack CE, Maas C (2016) Bradykinin: inflammatory product of the coagulation system. Clin Rev Allergy Immunol 51:152–161
Brower MS, Harpel PC (1982) Proteolytic cleavage and inactivation of alpha 2-plasmin inhibitor and C1 inactivator by human polymorphonuclear leukocyte elastase. J Biol Chem 257:9849–9854
Stuardo M, Gonzalez CB, Nualart F, Boric M, Corthorn J, Bhoola KD, Figueroa CD (2004) Stimulated human neutrophils form biologically active kinin peptides from high and low molecular weight kininogens. J Leukoc Biol 75:631–640
Araujo RC, Kettritz R, Fichtner I, Paiva AC, Pesquero JB, Bader M (2001) Altered neutrophil homeostasis in kinin B1 receptor-deficient mice. Biol Chem 382:91–95
Schuschke DA, Saari JT, Miller FN (2001) Leukocyte-endothelial adhesion is impaired in the cremaster muscle microcirculation of the copper-deficient rat. Immunol Lett 76:139–144
Guevara-Lora I, Labedz A, Skrzeczynska-Moncznik J, Kozik A (2011) Bradykinin and des-Arg10-kallidin enhance the adhesion of polymorphonuclear leukocytes to extracellular matrix proteins and endothelial cells. Cell Commun Adhes 18:67–71
Carl VS, Moore EE, Moore FA, Whalley ET (1996) Involvement of bradykinin B1 and B2 receptors in human PMN elastase release and increase in endothelial cell monolayer permeability. Immunopharmacology 33:325–329
Kahn R, Hellmark T, Leeb-Lundberg LM, Akbari N, Todiras M, Olofsson T, Wieslander J, Christensson A, Westman K, Bader M, Muller-Esterl W, Karpman D (2009) Neutrophil-derived proteinase 3 induces kallikrein-independent release of a novel vasoactive kinin. J Immunol 182:7906–7915
Wachtfogel YT, Pixley RA, Kucich U, Abrams W, Weinbaum G, Schapira M, Colman RW (1986) Purified plasma factor XIIa aggregates human neutrophils and causes degranulation. Blood 67:1731–1737
Oehmcke S, Morgelin M, Herwald H (2009) Activation of the human contact system on neutrophil extracellular traps. J Innate Immun 1:225–230
Scapini P, Lapinet-Vera JA, Gasperini S, Calzetti F, Bazzoni F, Cassatella MA (2000) The neutrophil as a cellular source of chemokines. Immunol Rev 177:195–203
Kim KP, Rafter JD, Bittova L, Han SK, Snitko Y, Munoz NM, Leff AR, Cho W (2001) Mechanism of human group V phospholipase A2 (PLA2)-induced leukotriene biosynthesis in human neutrophils. A potential role of heparan sulfate binding in PLA2 internalization and degradation. J Biol Chem 276:11126–11134
Triggiani M, Granata F, Frattini A, Marone G (2006) Activation of human inflammatory cells by secreted phospholipases A2. Biochim Biophys Acta 1761:1289–1300
Jo EJ, Lee HY, Lee YN, Kim JI, Kang HK, Park DW, Baek SH, Kwak JY, Bae YS (2004) Group IB secretory phospholipase A2 stimulates CXC chemokine ligand 8 production via ERK and NF-kappa B in human neutrophils. J Immunol 173:6433–6439
Silliman CC, Moore EE, Zallen G, Gonzalez R, Johnson JL, Elzi DJ, Meng X, Hanasaki K, Ishizaki J, Arita H, Ao L, England KM, Banerjee A (2002) Presence of the M-type sPLA(2) receptor on neutrophils and its role in elastase release and adhesion 283: C1102– C1113
Loffredo S, Borriello F, Iannone R, Ferrara AL, Galdiero MR, Gigantino V, Esposito P, Varricchi G, Lambeau G, Cassatella MA, Granata F, Marone G (2017) Group V secreted phospholipase A2 induces the release of proangiogenic and antiangiogenic factors by human neutrophils. Front Immunol 8:443
Marone G, Gambardella AR, Mattei F, Mancini J, Schiavoni G, Varricchi G (2020) Basophils in tumor microenvironment and surroundings. Adv Exp Med Biol 1224:21–34
Varricchi G, Galdiero MR, Loffredo S, Lucarini V, Marone G, Mattei F, Marone G, Schiavoni G (2018) Eosinophils: the unsung heroes in cancer? Oncoimmunology 7:e1393134
Galdiero MR, Varricchi G, Seaf M, Marone G, Levi-Schaffer F, Marone G (2017) Bidirectional mast cell-eosinophil interactions in inflammatory disorders and cancer. Front Med 4:103
Johnston LK, Bryce PJ (2017) Understanding interleukin 33 and its roles in eosinophil development. Front Medicine 4:51
Efraim AHNB, Levi-Schaffer F (2014) Roles of eosinophils in the modulation of angiogenesis. Chem Immunol Allergy 99:138–154
Varricchi G, Bagnasco D, Borriello F, Heffler E, Canonica GW (2016) Interleukin-5 pathway inhibition in the treatment of eosinophilic respiratory disorders: evidence and unmet needs. Curr Opin Allergy Clin Immunol 16:186–200
Rosenberg HF, Dyer KD, Foster PS (2013) Eosinophils: changing perspectives in health and disease. Nat Rev Immunol 13:9–22
Furuta GT, Atkins FD, Lee NA, Lee JJ (2014) Changing roles of eosinophils in health and disease. Ann Allergy Asthma Immunol 113:3–8
Ehrenfeld P, Millan C, Matus CE, Figueroa JE, Burgos RA, Nualart F, Bhoola KD, Figueroa CD (2006) Activation of kinin B1 receptors induces chemotaxis of human neutrophils. J Leukoc Biol 80:117–124
Gleich GJ, Schroeter AL, Marcoux JP, Sachs MI, O’Connell EJ, Kohler PF (1984) Episodic angioedema associated with eosinophilia. N Engl J Med 310:1621–1626
Khoury P, Herold J, Alpaugh A, Dinerman E, Holland-Thomas N, Stoddard J, Gurprasad S, Maric I, Simakova O, Schwartz LB, Fong J, Lee CC, Xi L, Wang Z, Raffeld M, Klion AD (2015) Episodic angioedema with eosinophilia (Gleich syndrome) is a multilineage cell cycling disorder. Haematologica 100:300–307
Marone G, Borriello F, Varricchi G, Genovese A, Granata F (2014) Basophils: historical reflections and perspectives. Chem Immunol Allergy 100:172–192
de Paulis A, Prevete N, Fiorentino I, Walls AF, Curto M, Petraroli A, Castaldo V, Ceppa P, Fiocca R, Marone G (2004) Basophils infiltrate human gastric mucosa at sites of Helicobacter pylori infection, and exhibit chemotaxis in response to H. pylori-derived peptide Hp(2–20). J Immunol 172:7734–7743
Nouri-Aria KT, Irani AM, Jacobson MR, O’Brien F, Varga EM, Till SJ, Durham SR, Schwartz LB (2001) Basophil recruitment and IL-4 production during human allergen-induced late asthma. J Allergy Clin Immunol 108:205–211
de Paulis A, Prevete N, Fiorentino I, Rossi FW, Staibano S, Montuori N, Ragno P, Longobardi A, Liccardo B, Genovese A, Ribatti D, Walls AF, Marone G (2006) Expression and functions of the vascular endothelial growth factors and their receptors in human basophils. J Immunol 177:7322–7331
Varricchi G, Loffredo S, Galdiero MR, Marone G, Cristinziano L, Granata F (2018) Innate effector cells in angiogenesis and lymphangiogenesis. Curr Opin Immunol 53:152–160
Schroeder JT (2011) Basophils: emerging roles in the pathogenesis of allergic disease. Immunol Rev 242:144–160
Varricchi G, Marone G, Kovanen PT (2020) Cardiac mast cells: underappreciated immune cells in cardiovascular homeostasis and disease. Trends Immunol 41:734–746
Espinosa E, Valitutti S (2018) New roles and controls of mast cells. Curr Opin Immunol 50:39–47
Kirshenbaum AS, Goff JP, Semere T, Foster B, Scott LM, Metcalfe DD (1999) Demonstration that human mast cells arise from a progenitor cell population that is CD34(+), c-kit(+), and expresses aminopeptidase N (CD13). Blood 94:2333–2342
Galli SJ, Gaudenzio N, Tsai M (2020) Mast Cells in Inflammation and Disease: Recent Progress and Ongoing Concerns. Annu Rev Immunol 38:49–77
Nosbaum A, Augey F, Nicolas JF, Berard F (2014) Pathophysiology of urticaria. Ann Dermatol Venereol 141(Suppl 3):S559–S564
James C, Bernstein JA (2017) Current and future therapies for the treatment of histamine-induced angioedema. Expert Opin Pharmacother 18:253–262
Busse PJ, Smith T (2017) Histaminergic angioedema. Immunol Allergy Clin N Am 37:467–481
Duran WN, Breslin JW, Sanchez FA (2010) The NO cascade, eNOS location, and microvascular permeability. Cardiovasc Res 87:254–261
Yoshihiro Fukuoka MRH, a. L. B. S. (2016) Functional angiotensin and bradykinin receptors are expressed on primary human mast cells. J Immunol 196(1 Supplement):126–219
Cross LJ, Heaney LG, Ennis M (1997) Histamine release from human bronchoalveolar lavage mast cells by neurokinin A and bradykinin. Inflammation research : official journal of the European Histamine Research Society ... [et al.] 46:306–309
Lee PY, Pearce FL (1990) Histamine secretion from mast cells stimulated with bradykinin. Agents Actions 30:67–69
Bernstein JA, Moellman J (2012) Emerging concepts in the diagnosis and treatment of patients with undifferentiated angioedema. Int J Emerg Med 5:39
Moon TC, Befus AD, Kulka M (2014) Mast cell mediators: their differential release and the secretory pathways involved. Front Immunol 5:569
Varricchi G, Rossi FW, Galdiero MR, Granata F, Criscuolo G, Spadaro G, de Paulis A, Marone G (2019) Physiological Roles of Mast Cells: Collegium Internationale Allergologicum Update 2019. Int Arch Allergy Immunol 179:247–261
Imamura T, Dubin A, Moore W, Tanaka R, Travis J (1996) Induction of vascular permeability enhancement by human tryptase: dependence on activation of prekallikrein and direct release of bradykinin from kininogens. Laboratory investigation; a journal of technical methods and pathology 74:861–870
Sala-Cunill A, Bjorkqvist J, Senter R, Guilarte M, Cardona V, Labrador M, Nickel KF, Butler L, Luengo O, Kumar P, Labberton L, Long A, Di Gennaro A, Kenne E, Jamsa A, Krieger T, Schluter H, Fuchs T, Flohr S, Hassiepen U, Cumin F, McCrae K, Maas C, Stavrou E, Renne T (2015) Plasma contact system activation drives anaphylaxis in severe mast cell-mediated allergic reactions. J Allergy Clin Immunol 135:1031–1043
Triggiani M, Giannattasio G, Calabrese C, Loffredo S, Granata F, Fiorello A, Santini M, Gelb MH, Marone G (2009) Lung mast cells are a source of secreted phospholipases A2. J Allergy Clin Immunol 124:558–65
Galli SJ, Tsai M (2012) IgE and mast cells in allergic disease. Nat Med 18:693–704
Maas C, Oschatz C, Renne T (2011) The plasma contact system 2.0. Semin Thromb Hemost 37:375–381
Guilarte M, Sala-Cunill A, Luengo O, Labrador-Horrillo M, Cardona V (2017) The mast cell, contact, and coagulation system connection in anaphylaxis. Front Immunol 8:846
Capila I, Linhardt RJ (2002) Heparin-protein interactions. Angew Chem 41:391–412
Oschatz C, Maas C, Lecher B, Jansen T, Bjorkqvist J, Tradler T, Sedlmeier R, Burfeind P, Cichon S, Hammerschmidt S, Muller-Esterl W, Wuillemin WA, Nilsson G, Renne T (2011) Mast cells increase vascular permeability by heparin-initiated bradykinin formation in vivo. Immunity 34:258–268
Kleniewski J, Donaldson V (1988) Granulocyte elastase cleaves human high molecular weight kininogen and destroys its clot-promoting activity. J Exp Med 167:1895–1907
Blom B, Spits H (2006) Development of human lymphoid cells. Annu Rev Immunol 24:287–320
Larosa DF, Orange JS (2008) 1. Lymphocytes. J Allergy Clin Immunol 121, S364–9; quiz S412
Freud AG, Caligiuri MA (2006) Human natural killer cell development. Immunol Rev 214:56–72
Kalia V, Sarkar S, Gourley TS, Rouse BT, Ahmed R (2006) Differentiation of memory B and T cells. Curr Opin Immunol 18:255–264
Nemazee D (2017) Mechanisms of central tolerance for B cells. Nat Rev Immunol 17:281–294
Cillari E, Misiano G, Arico M, La Rocca E, Lio D, di Leonardo S, Brai M (1986) Modification of peripheral blood T-lymphocyte surface receptors and Langerhans cell numbers in hereditary angioedema. Am J Clin Pathol 85:305–311
Brickman CM, Tsokos GC, Balow JE, Lawley TJ, Santaella M, Hammer CH, Frank MM (1986) Immunoregulatory disorders associated with hereditary angioedema. I. Clinical manifestations of autoimmune disease. J Allergy Clin Immunol 77:749–757
Salemi M, Mandala V, Muggeo V, Misiano G, Milano S, Colonna-Romano G, Arcoleo F, Cillari E (2016) Growth factors and IL-17 in hereditary angioedema. Clin Exp Med 16:213–218
Longhurst H, Cicardi M (2012) Hereditary angio-oedema. Lancet 379:474–481
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201:233–240
Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W (2007) Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445:648–651
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ (2007) TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 8:1390–1397
Esplugues E, Huber S, Gagliani N, Hauser AE, Town T, Wan YY, O’Connor W Jr, Rongvaux A, Van Rooijen N, Haberman AM, Iwakura Y, Kuchroo VK, Kolls JK, Bluestone JA, Herold KC, Flavell RA (2011) Control of TH17 cells occurs in the small intestine. Nature 475:514–518
Lopez-Lera A, Cabo FS, Garrido S, Dopazo A, Lopez-Trascasa M (2013) Disease-modifying factors in hereditary angioedema: an RNA expression-based screening. Orphanet J Rare Dis 8:77
Castellano G, Divella C, Sallustio F, Montinaro V, Curci C, Zanichelli A, Bonanni E, Suffritti C, Caccia S, Bossi F, Gallone A, Schena FP, Gesualdo L, Cicardi M (2018) A transcriptomics study of hereditary angioedema attacks. J Allergy Clin Immunol 142:883–891
Mondino A, Blasi F (2004) uPA and uPAR in fibrinolysis, immunity and pathology. Trends Immunol 25:450–455
Navaratna D, Menicucci G, Maestas J, Srinivasan R, McGuire P, Das A (2008) A peptide inhibitor of the urokinase/urokinase receptor system inhibits alteration of the blood-retinal barrier in diabetes. FASEB J 22:3310–3317
Trimarchi H (2013) Primary focal and segmental glomerulosclerosis and soluble factor urokinase-type plasminogen activator receptor. World Journal of Nephrology 2:103–110
Kessel A, Peri R, Perricone R, Guarino MD, Vadasz Z, Novak R, Haj T, Kivity S, Toubi E (2012) The autoreactivity of B cells in hereditary angioedema due to C1 inhibitor deficiency. Clin Exp Immunol 167:422–428
Zotter Z, Csuka D, Szabo E, Czaller I, Nebenfuhrer Z, Temesszentandrasi G, Fust G, Varga L, Farkas H (2014) The influence of trigger factors on hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis 9:44
Farkas H, Fust G, Fekete B, Karadi I, Varga L (2001) Eradication of Helicobacter pylori and improvement of hereditary angioneurotic oedema. Lancet 358:1695–1696
Zotter Z, Veszeli N, Kohalmi KV, Varga L, Imreh E, Kovacs G, Nallbani M, Farkas H (2016) Bacteriuria increases the risk of edematous attacks in hereditary angioedema with C1-inhibitor deficiency. Allergy 71:1791–1793
Frank MM, Gelfand JA, Atkinson JP (1976) Hereditary angioedema: the clinical syndrome and its management. Ann Intern Med 84:580–593
Ozden G, Yanardag Acik D (2020) Can the neutrophil-lymphocyte ratio predict type 1 hereditary angioedema attacks? Scand J Clin Lab Invest, 1–5
Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, Drummert Johnson C, Xu K, Jiang X, Wang H, Yang XC (2020) Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol 40:e138–e152
Young MR (2012) Endothelial cells in the eyes of an immunologist. Cancer Immunol Immunother : CII 61:1609–1616
Kajdacsi E, Jani PK, Csuka D, Varga L, Prohaszka Z, Farkas H, Cervenak L (2016) Novel vasoregulatory aspects of hereditary angioedema: the role of arginine vasopressin, adrenomedullin and endothelin-1. J Clin Immunol 36:160–170
Prevete N, Staiano RI, Granata F, Detoraki A, Necchi V, Ricci V, Triggiani M, De Paulis A, Marone G, Genovese A (2013) Expression and function of angiopoietins and their tie receptors in human basophils and mast cells. J Biol Regul Homeost Agents 27:827–839
Katsanos GS, Anogeianaki A, Orso C, Tete S, Salini V, Antinolfi PL, Sabatino G (2008) Mast cells and chemokines. J Biol Regul Homeost Agents 22:145–151
Lin C, He H, Liu H, Li R, Chen Y, Qi Y, Jiang Q, Chen L, Zhang P, Zhang H, Li H, Zhang W, Sun Y, Xu J (2019) Tumour-associated macrophages-derived CXCL8 determines immune evasion through autonomous PD-L1 expression in gastric cancer. Gut 68:1764–1773
Tecchio C, Cassatella MA (2016) Neutrophil-derived chemokines on the road to immunity. Semin Immunol 28:119–128
Meier HL, Schulman ES, Heck LW, MacGlashan D, Newball HH, Kaplan AP (1989) Release of elastase from purified human lung mast cells and basophils. Identification as a Hageman factor cleaving enzyme. Inflammation 13:295–308
Aratani Y (2018) Myeloperoxidase: Its role for host defense, inflammation, and neutrophil function. Arch Biochem Biophys 640:47–52
Ferrara AL, Piscitelli F, Petraroli A, Parente R, Galdiero MR, Varricchi G, Marone G, Triggiani M, Di Marzo V, Loffredo S (2019) Altered metabolism of phospholipases, diacylglycerols, endocannabinoids, and N-acylethanolamines in patients with mastocytosis. J Immunol Res 2019:5836476
Imamura M, Kawasaki T, Savchenko AS, Ohashi R, Jiang S, Miyamoto K, Ito Y, Iwanari H, Sagara M, Tanaka T, Hamakubo T, Kodama T, Uchiyama M, Naito M (2007) Lipopolysaccharide induced expression of pentraxin 3 in human neutrophils and monocyte-derived macrophages. Cell Immunol 248:86–94
Razvina O, Jiang S, Matsubara K, Ohashi R, Hasegawa G, Aoyama T, Daigo K, Kodama T, Hamakubo T, Naito M (2015) Differential expression of pentraxin 3 in neutrophils. Exp Mol Pathol 98:33–40
Lim HS, Patel JV, Lip GY (2006) Reactive oxygen species production by circulating monocytes: insights from pathophysiology to clinical hypertension. J Hum Hypertens 20:307–309
Obtulowicz K, Goralska J, Bogdali A, Dyga W, Obtulowicz A, Myszkowska D, Ziemianin M, Gruca A, Solnica B, Czarnobilska E (2020) Bradykinin and oxidative stress in patients with hereditary angioedema due to C1 inhibitor deficiency. Polish archives of internal medicine 130:79–88
Swindle EJ, Metcalfe DD (2007) The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol Rev 217:186–205
Blom M, Tool AT, Wever PC, Wolbink GJ, Brouwer MC, Calafat J, Egesten A, Knol EF, Hack CE, Roos D, Verhoeven AJ (1998) Human eosinophils express, relative to other circulating leukocytes, large amounts of secretory 14-kD phospholipase A2. Blood 91:3037–3043
Ho IC, Arm JP, Bingham CO 3rd, Choi A, Austen KF, Glimcher LH (2001) A novel group of phospholipase A2s preferentially expressed in type 2 helper T cells. J Biol Chem 276:18321–18326
Loffredo S, Staiano RI, Granata F, Costantino V, Borriello F, Frattini A, Lepore MT, Mangoni A, Marone G, Triggiani M (2014) Simplexide induces CD1d-dependent cytokine and chemokine production from human monocytes. PLoS One 9:e111326
Parameswaran N, Patial S (2010) Tumor necrosis factor-alpha signaling in macrophages. Crit Rev Eukaryot Gene Expr 20:87–103
Mukai K, Tsai M, Saito H, Galli SJ (2018) Mast cells as sources of cytokines, chemokines, and growth factors. Immunol Rev 282:121–150
Li D, Xue W, Li M, Dong M, Wang J, Wang X, Li X, Chen K, Zhang W, Wu S, Zhang Y, Gao L, Chen Y, Chen J, Zhou BO, Zhou Y, Yao X, Li L, Wu D, Pan W (2018) VCAM-1(+) macrophages guide the homing of HSPCs to a vascular niche. Nature 564:119–124
Demirturk M, Akpinar TS, Kose M, Gelincik A, Colakoglu B, Buyukozturk S (2017) Endocan: a novel marker of endothelial dysfunction in C1-inhibitor-deficient hereditary angioedema. Int Arch Allergy Immunol 174:104–107
Detoraki A, Staiano RI, Granata F, Giannattasio G, Prevete N, de Paulis A, Ribatti D, Genovese A, Triggiani M, Marone G (2009) Vascular endothelial growth factors synthesized by human lung mast cells exert angiogenic effects. J Allergy Clin Immunol 123:1142–1149
Funding
Open access funding provided by Università degli Studi di Napoli Federico II within the CRUI-CARE Agreement. IIR-ITA-002138
Author information
Authors and Affiliations
Contributions
The idea for the article is attributable to Stefania Loffredo and Maria Bova. All authors contributed to the literature research and data analysis. The first draft of the manuscript was written by Stefania Loffredo, Anne Lise Ferrara, Maria Bova, Maria Celeste Gigliotti, and Angelica Petraroli. and all authors commented on previous versions of the manuscript. Stefania Loffredo, Maria Bova, and Giuseppe Spadaro critically revised the work. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no competing interests.
Ethical Approval
The Ethical Committee of the University of Naples Federico II (protocol number: 300/18) approved that plasma of C1-INH-HAE, and healthy controls obtained during routine diagnostics could be used for research investigating the physiopathology of hereditary angioedema.
Informed Consent
Written informed consent was obtained from patients in according to the principles expressed in the Declaration of Helsinki.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ferrara, A.L., Cristinziano, L., Petraroli, A. et al. Roles of Immune Cells in Hereditary Angioedema. Clinic Rev Allerg Immunol 60, 369–382 (2021). https://doi.org/10.1007/s12016-021-08842-9
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12016-021-08842-9